
Abstract
Background: Mitochondrial disorders are heterogeneous clinical syndromes caused by defective activity in the mitochondrial respiratory chain, resulting in a faulty oxidative phosphorylation system. These inherited disorders are individually rare, and furthermore they are phenotypic variables. The genetically characterized mitochondrial disorders are rarely associated with epileptic encephalopathies.
Case presentation: We present the clinical phenotype, biochemical analysis, and electrographic and neuro-radiological features of a 5-month-old girl with epileptic encephalopathy, microcephaly, severe psychomotor delay, hypertrophic cardiomyopathy, and abnormal MRI scan. Using whole-genome sequencing technique, compound heterozygous mutations of the VARS2 gene were revealed, with one previously unreported frameshift mutation.
Conclusion: Our report extends the phenotypic spectrum of VARS2-related disorders with an initial presentation of epileptic encephalopathy and early death due to malignant arrhythmia.
Introduction
Mitochondrial disorders are heterogeneous clinical syndromes caused by defective activity in the mitochondrial respiratory chain (MRC), resulting in a faulty oxidative phosphorylation system (OXPHOS) (Citation1), associated with a broad range of nuclear and mitochondrial gene mutations. These inherited disorders are rare and phenotypically variable, and thus generally unfamiliar to clinicians. Epileptic encephalopathies (EE) constitute a heterogeneous group of disorders characterized by intractable seizures with specific abnormalities on electroencephalography (EEG) and severe developmental delay or regression (Citation2); however, a clearly defined aetiology and a diagnostic algorithm for these clinical entities have not been fully established.
In our case study we present a clinical phenotype, a biochemical analysis, as well as electrographic and neuroradiological features of a patient with EE. A genetic analysis revealed an MRC defect.
Methods
Acquisition of a clinical case
We made a comprehensive evaluation of our patient, including molecular genetics investigations. The patient’s parents signed an informed consent in agreement with the Declaration of Helsinki.
Molecular genetics
Genomic DNA from the proband, father, and mother was isolated from peripheral blood samples, and whole-exome sequencing (WES) was completed. WES, bioinformatic analysis, variant confirmation, and segregation analysis was performed in the CeGaT GmbH laboratory (Tübingen, Germany). Coding and flanking intronic regions were enriched using the Agilent in solution technology and were sequenced using the Illumina HiSeq/NovaSeq system (Illumina Inc., San Diego, California, USA). Variants (SNVs/small indels) with a minor allele frequency (MAF) <1.5% were evaluated. Known disease-causing variants (according to HGMD) were evaluated up to 5% MAF. The MAF were taken from the 1000 Genomes, dsSNP, GnomAD, and in-house database. For the index case, 95.61% of the targeted regions were covered by a minimum of 30 high-quality sequencing reads per base. All filtered variants were further analysed using Mutation Taster, fathmm, Mutation Assessor, SIFT, fathmm-MKL coding, LRT, and PROVEAN for pathogenicity prediction. Two algorithms were applied (Citation3) for assessments of the consequences of variants on splicing. WES identified compound heterozygous mutations in the VARS2 gene (NM_001167734.1) in our patient. Both parents are heterozygous carriers of variants in VARS2.
Results
A female child was born at term after an uneventful pregnancy and delivery. She was the first child from the first pregnancy of healthy non-consanguineous parents. The family history was unremarkable. The birth weight, length, and head circumference were in accordance with the gestational age (4030 g, 51 cm, and 34 cm, respectively). Computed tomography (CT) was performed at one month of age because of failure of head growth (50th percentile at birth and 10th after neonatal period). Craniosynostosis was excluded, with no other pathological findings. Neurohabilitation was started due to delayed acquisition of milestones.
At 5 months of age she developed infantile spasms consistent with hypsarrhythmia on EEG studies. Clinical examinations showed microcephaly with ‘pear-shaped face’, hypertelorism, and a wide nasal bridge. Global motor development was delayed by 2–3 months. Language and social skills were in accordance with the patient’s age. Laboratory findings revealed an increased concentration of alanine in plasma (537 μmol/L, normal range 100–439 μmol/L) and cerebrospinal fluid (36.1 μmol/L, normal range 19.9–31.3 μmol/L), as well as mildly elevated concentrations of lactate in plasma (2.0–5.4 mmol/L, normal range 0.33–2.0 mmol/L) and cerebrospinal fluid (3.0 mmol/L, normal range 1.1–2.8 mmol/) indicative of mitochondrial disorder, while others were normal. Magnetic resonance imaging (MRI) performed at that time revealed diffuse cerebral atrophy with more prominent hypoplasia of the cerebellum (especially vermis), brainstem, and corpus callosum ().
Figure 1. (A) First MRI scan, age 5 months. Sagittal T1-weighted and coronal T2-weighted imaging showing diffuse white matter reduction, marked cerebral and cerebellar atrophy, corpus callosum thinning, and brainstem hypoplasia. (B) MRI scan, age 9 months. Coronal T2-weighted imaging showing marked progression of cerebral and cerebellar atrophy and ventriculomegaly (dominantly left occipital ventricle) due to diffuse white matter necrosis, while MRI spectroscopy showed markedly increased myoinositol peak, increased lactate peak, and reduced N-acetyl aspartate peak.
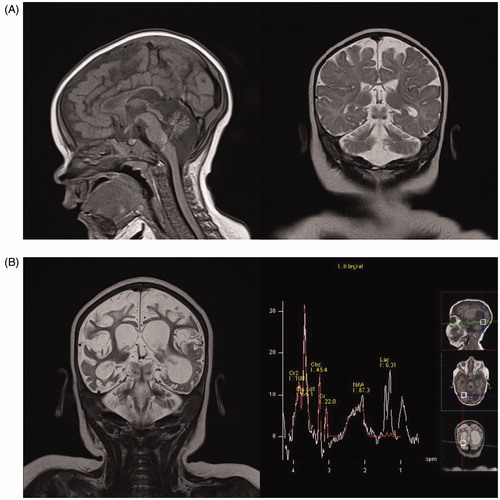
At the age of 6 months multiple seizure types (mastication, eyelid and lip twitching, myoclonic seizures of the trunk and extremities, tonic and clonic seizures) were noticed. Treatment attempts with several antiepileptic drugs including folic acid, cholecalciferol, pyridoxine and modified Atkins diet were not successful. EEG studies showed continuous burst–suppression pattern. All in all, the clinical status of the child developed into EE. Microcephaly, severe global hypotonia, failure to thrive, and feeding and breathing difficulties were more and more pronounced. Pendular nystagmus and oedema of hands and feet were periodically noticed. Echocardiography showed hypertrophic obstructive cardiomyopathy with fast progression, and atenolol treatment was initiated.
Genetic analysis was performed at the age of 9 months, and it revealed compound heterozygous mutations of the VARS2 gene: previously reported pathogenic variant (c.1100C>T; p.Thr367Ile) transmitted from the father and one unreported frameshift mutation (c.603_606dupGATG; p.Arg203Aspfs*37) transmitted from the mother. An additional MRI with spectroscopy showed a further significant loss of the brain parenchyma and a high lactate peak ().
A multidisciplinary treatment approach was applied to treat the neurologic, cardiac, respiratory, and gastrointestinal complications. In accordance with the established diagnosis, treatment with L-carnitine and coenzyme Q10 was initiated. Despite multiple supportive and symptomatic treatment regimens, the child died of malignant arrhythmia at the age of 10 months.
Discussion
Combined oxidative phosphorylation deficiency 20 (COXPD20) (OMIM #615917) is an autosomal recessive mitochondrial encephalocardiomyopathy caused by variants in the VARS2 gene (OMIM #612802) located on chromosome 6p21 (https://www.omim.org/entry/615917). The VARS2 gene contains 30 exons and encodes mitochondrial valyl tRNA-synthetase, which participates in mitochondrial protein synthesis (Citation4). Despite the estimated prevalence of MRC defects being 1 in 5000 live births, it is often difficult to find the molecular basis. The underlying genetic basis is not possible to identify in 40% to 70% of the patients, even though there is biochemical evidence of MRC defects (Citation5). Presently, there are 10 reported pathogenic variants in the VARS2 gene (https://www.ncbi.nlm.nih.gov/clinvar/?term=VARS2[all]).
When considering our patient in comparison with previously reported patients with VARS2 mutations (Citation5–10), it appears that the main phenotypic features are seizures, various degrees of developmental delay, facial dysmorphism, brain atrophy, and cardiomyopathy. The main clinical features, biochemical studies, and molecular findings are shown in .
Table 1. Clinical, biochemical and molecular findings in previously reported patients with VARS2 mutation.
Bruni et al. (Citation10) elaborated clinical, biochemical, and molecular features in 13 patients from 9 families diagnosed by WES. Two previously reported cases by Taylor et al. (Citation5) and by Pronicka et al. (Citation9) were also included. Bruni et al. (Citation10) presented a new homozygous (c.1258G>A; p.Ala420Thr) variant and four new compound heterozygous variants, along with already known pathogenic variants (). The study found the mutant VARS2 allele c.1100C>T; p.Thr367Ile the most common—in six patients in homozygous and in five patients in heterozygous states. Laboratory findings implied a mitochondrial disease, while the EEG and brain MRI of these patients did not show any disease-characteristic pattern. The common problem in these patients (n = 11/17) is hypertrophic cardiomyopathy, with only a few patients not being affected. The c.1100C>T; p.Thr367Ile variant is considered to have less of an effect on the heart (Citation10). Alsemari et al. (Citation11) published a study describing four patients with Angelman-like syndrome and mutation in the VARS2 gene, providing new insight into genotype–phenotype correlation.
To date, our case is one of the 17 described cases of severe infantile mitochondrial disorder associated with VARS2 pathogenic variants. The phenotypic spectrum of VARS2-related gene variants is complex, and no clear genotype–phenotype correlations have been established. Therefore, a detailed description of phenotype related to specific mutations is of great importance. Our report extends the phenotypic spectrum of VARS2-related disorders in the context of EE, severe neurodevelopmental delay, abnormal brain MRI scan, and one novel frameshift mutation (c.603_606dupGATG; p.Arg203Aspfs*37) in VARS2 (NM_001167734.1). With the exception of supportive care, currently no disease-modifying treatment exists for the disease.
The genetically characterized mitochondrial disorders are rarely associated with EE, with very few clinical reports available (Citation12). A timely evaluation of patients with EE resulting in a specific diagnosis is of great value. We want to emphasize the importance of genetic analysis in the early diagnostic efforts of these patients. WES is a very powerful, non-invasive, accessible, and not overly expensive diagnostic tool. The finding of a specific gene mutation is of exceptional importance to clinicians for further management, and for the family to cope with the diagnosis and prognosis. Genetic counselling about familial risk, as well as for potential prenatal diagnostics, is of great value to the family.
Although the field of mitochondrial encephalomyopathies is highly dynamic, the genetic diagnosis of these disorders, which may offer the possibility to design targeted therapy, remains challenging. In the near future, the WES approach will provide valuable information regarding the genes implicated in MRC defects, which are important in the differential diagnosis of EE.
Acknowledgements
The authors would like to thank our patient’s family for participating in the study. They thank Dr Saskia Biskup and CeGaT GmbH, who performed whole-exome analysis of the patient and parents. Also, they thank their colleagues from the Clinical Hospital Centre Rijeka for ensuring multidisciplinary treatment for their patient.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Notes on contributors
Lucija Ruzman
Lucija Ruzman is a Ph.D. student at the University of Rijeka, School of Medicine. She works as a pediatric resident at the Department of Pediatrics at the Clinical Hospital Center Rijeka. Her main topic of interests are genetic disorders.
Ivana Kolic
Ivana Kolić is a pediatrician and pediatric neurologist. She works at Child Neurology and Child Psychiatry Department at the Clinical Hospital Center Rijeka and is a Ph.D. student at the University of Rijeka, School of Medicine. Her main topic of interests interest are neurodevelopment disorders.
Jelena Radic Nisevic
Jelena Radic Nisevic, Ph.D., is a pediatrician and pediatric neurologist at Child Neurology and Child Psychiatry Department at the Clinical Hospital Center Rijeka and University of Rijeka, School of Medicine.
Antonija Ruzic Barsic
Antonija Ruzic Barsic is a radiologist, Ph.D. student of the University of Osijek. She is the Head of Radiology Department at the Thalassotherapia Opatija Hospital. Her main topic of interest is pediatric neuroradiology.
Ingrid Skarpa Prpic
Ingrid Skarpa Prpic, Ph.D., is an Assistant Professor of Neurology. She works at Clinical Hospital Center Rijeka and University of Rijeka, School of Medicine. Her main topics of interest are demyelinating disease and paroxysmal disorders.
Igor Prpic
Igor Prpic, Ph.D., is a full time professor of pediatrics-permanent election, and Head of Child Neurology and Child Psychiatry Department at the Clinical Hospital Center Rijeka and University of Rijeka, School of Medicine. He is subspecialist in pediatric neurology and in neonatology. Since 2009 he runs Croatian Referral Centre of Ministry of Health for childhood epilepsy and convulsive disorders.
References
- Ghezzi D, Zeviani M. Assembly factors of human mitochondrial respiratory chain complexes: physiology and pathophysiology. Adv Exp Med Biol. 2012;748:65–106.
- Scheffer IE, Berkovic S, Capovilla G, Connolly MB, French J, Guilhoto L, et al. ILAE classification of the epilepsies: position paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017;58:512–21.
- Jian X, Boerwinkle E, Liu X. In silico prediction of splice-altering single nucleotide variants in the human genome. Nucleic Acids Res. 2014;42:13534–44.
- Bonnefond L, Fender A, Rudinger-Thirion J, Giege R, Florentz C, Sissler M. Toward the full set of human mitochondrial aminoacyl-tRNA synthetases: characterization of AspRS and TyrRS. Biochemistry. 2005;44:4805–16.
- Taylor RW, Pyle A, Griffin H, Blakely EL, Duff J, He L, et al. Use of whole-exome sequencing to determine the genetic basis of multiple mitochondrial respiratory chain complex deficiencies. JAMA. 2014;312:68–77.
- Diodato D, Melchionda L, Haack TB, Dallabona C, Baruffini E, Donnini C, et al. VARS2 and TARS2 mutations in patients with mitochondrial encephalomyopathies. Hum Mutat. 2014;35:983–9.
- Pereira S, Adriao M, Sampaio M, Basto MA, Rodrigues E, Vilarinho L, et al. Mitochondrial encephalopathy: first Portuguese report of a VARS2 causative variant. JIMD Rep. 2018;42:113–9.
- Baertling F, Alhaddad B, Seibt A, Budaeus S, Meitinger T, Strom TM, et al. Neonatal encephalocardiomyopathy caused by mutations in VARS2. Metab Brain Dis. 2017;32:267–70.
- Pronicka E, Piekutowska-Abramczuk D, Ciara E, Trubicka J, Rokicki D, Karkucińska-Więckowska A, et al. New perspective in diagnostics of mitochondrial disorders: two years’ experience with whole-exome sequencing at a national paediatric centre. J Transl Med. 2016;14:174.
- Bruni F, Di Meo I, Bellacchio E, Webb BD, McFarland R, Chrzanowska-Lightowlers ZMA, et al. Clinical, biochemical, and genetic features associated with VARS2‐related mitochondrial disease. Hum Mutat. 2018;39:563–78.
- Alsemari A, Al-Younes B, Goljan E, Jaroudi D, BinHumaid F, Meyer BF, et al. Recessive VARS2 mutation underlies a novel syndrome with epilepsy, mental retardation, short stature, growth hormone deficiency, and hypogonadism. Hum Genomics. 2017;11:33.
- Paciorkowski AR, Thio LL, Dobyns WB. Genetic and biologic classification of infantile spasms. Pediatr Neurol. 2011;45:355–67.