
ABSTRACT
Taxonomic information for New Zealand mosquito species is predominantly morphological with very few molecular data available to date. In this study, the mitochondrial COI gene and nuclear ITS1 were amplified and sequenced from DNA templates representing 17 species; 15 previously known New Zealand species, a newly discovered undescribed Aedes species from the Chatham Islands and a recently eradicated invader, Ae. camptorhynchus. This paper provides DNA barcoding sequences for the entire known New Zealand mosquito fauna, the first for the majority of these species. Phylogenetic analysis of COI and ITS1 indicated that the endemic species are all genetically distinct from the exotic species examined including vector species of pathogens of human diseases. The genus Opifex is distant from the genus Aedes based on analysis of ITS1 sequences, and Ae. chathamicus is more closely related to species within Aedes than to the genus Opifex. Results show Culex asteliae to be a valid species but Cx. rotoruae is not necessarily so. The Aedes species appears to be closely related to Ae. subalbirostris. The introduced Cx. quinquefasciatus was clearly shown to be related to Cx. pipiens; however, the endemic Cx. pervigilans was not. No evidence of population variation based on geographic location was detected.
KEYWORDS:
Introduction
The status of mosquito species in New Zealand
With just 13 endemic species (including one yet to be described), three introduced species and one recently eradicated invader, New Zealand has a very small mosquito fauna (Belkin Citation1962, Citation1968; Kay and Russell Citation2013). Most of the endemic species and the introduced Aedes australis have minimal contact with humans, feeding primarily on birds (Weinstein et al. Citation1997; Holder et al. Citation1999). The other two introduced species, Culex quinquefasciatus and Ae. notoscriptus are often found in aquatic habitats near human populations and have been shown to vector exotic arboviruses overseas (Kramer et al. Citation2011), as has the eradicated Australian southern saltmarsh mosquito Ae. camptorhynchus (Ballard and Marshall Citation1986).
Taxonomic work describing New Zealand mosquito species was predominantly conducted during the 1960s with two significant works that comprise the bulk of the knowledge obtained (Belkin Citation1962, Citation1968). Identification of the 15 species then recorded was based primarily on morphology and the classification in use then has remained largely unchanged. The number of domestic species is currently just 16 (i.e. Belkin’s original 15 plus Aedes sp. nov. – see later) following the successful eradication of the recently introduced Ae. camptorhynchus, from several sites around the country (Kay and Russell Citation2013).
The evolutionary history of these insects is of biological interest and potentially of medical significance too (Laird Citation1990; Derraik Citation2004; Buckley et al. Citation2015). Either they are derived in situ from one or more ancient Gondwanan lineages (vicariance) or they arrived later by dispersal and diverged to a greater or lesser extent. This latter scenario looks likely as several of these endemic taxa belong (or have been placed in) a variety of genera which are also found overseas. The identities of their closest relatives and the extent to which they have diverged from them post-introduction are questions best assessed by using molecular data (especially if they can be supported by a well-calibrated evolutionary clock). This would allow estimation of the relative likelihood that one or more of these New Zealand species might become vector competent or that related already competent species from overseas might become established in New Zealand.
Arbovirus situation and threat
New Zealand is an island nation which has historically been free of arboviral activity, with the exception of the endemic Whataroa virus which is established in bird populations on the West Coast of the South Island and has been isolated from the endemic mosquito species Culiseta tonnoiri and Cx. pervigilans (Ross et al. Citation1963). The introduced Cx. quinquefasciatus is an important vector of filariasisis in some regions of the world (Belkin Citation1968) as well as a number of viruses, including West Nile Virus (Goddard et al. Citation2002). Cx. quinquefasciatus is closely related to Cx. pipiens and part of the Cx. pipiens complex, which are important disease vectors (Fonseca et al. Citation2004). This situation is threatened by increasing international trade and tourism, global warming and invasive vector competent mosquito species (Derraik Citation2004; Sherif et al. Citation2019). The likelihood of medically significant arboviral agents of diseases arriving here is very high. Good knowledge and rapid, accurate identification of the species that are already in country can assist in the prevention of exotic vector species becoming established here.
Morphological identification of problem species
Traditional methods for the identification of mosquito specimens all tend to be based on morphology. Hence, they can be problematic, even for trained entomologists, and an extremely difficult, if not, impossible task with damaged specimens and for some life stages (e.g. eggs and pupae). There are no identification keys which can distinguish all New Zealand species and often several are required in combination to confidently identify a single specimen. There are very few keys which include information about adult males, making species determination particularly difficult for these specimens. Increasing numbers of cryptic (morphologically similar) species, taxonomic species groups and complexes are being discovered and traditional identification methods are now insufficient to definitively identify some specimens. Having access to molecular data in the form of a molecular taxonomic key which includes all mosquito species known to occur in New Zealand would allow many problem specimens to be identified. Additionally, such a key would allow border control officers to ascertain whether a specimen was of exotic origin.
Unsolved questions for New Zealand mosquitoes
There are a number of unresolved questions regarding the classification of New Zealand mosquitoes. First, it has been suggested that the endemic New Zealand genus Opifex is primitive within the subfamily Culicinae, possessing morphological features unique to the tribe Aedini (Dumbleton Citation1962; Belkin Citation1968). Second, Reinert et al. (Citation2004) proposed the transfer of Ae. chathamicus to Opifex based on morphological characteristics as part of a large review of Aedini. However, many of the suggested changes in this 2004 revision (including elevating many of the subgenera of Aedes to genus level) have not been universally accepted, pending further evidence (Harbach Citation2018). Next, there has been some scepticism regarding the validity of Cx. rotoruae and Cx. asteliae as distinct species from Cx. pervigilans, due to the similarity of many of their morphological features within all life stages. Belkin (Citation1968) considered them to comprise a Cx. pervigilans species complex but indicated that further work would be needed to support the recognition of Cx. asteliae as a distinct species. Finally, there is an undescribed Aedes species, Aedes sp. nov., believed to be endemic to the Chatham Islands (Cane and Courtney Citation2009), which is yet to be formally described and named, either morphologically and, or also perhaps, utilising molecular techniques.
Coquillettidia mosquitoes are unusual in that they are one of only three genera whose larvae are known not to be free-living within their aquatic habitat but have evolved specialised structures which enable them to survive underwater attached to submerged vegetation. A direct result of this is that the larvae are particularly hard to collect and the larval stages of the two New Zealand species are yet to be morphologically described. Specimens have been collected but whether they are one species or two is not known. It is anticipated that once the two species can be distinguished using molecular techniques, it will be possible to identify their larvae and thus their morphological characteristics can be attributed to the correct species. Interestingly, the adult specimens are morphologically distinct, and these species have been placed in separate subgenera based on their characteristics (Belkin Citation1962, Citation1968).
Advantages of molecular identification
Molecular techniques are being increasingly utilised for accurate mosquito identification as they can be applied with equal reliability to all life stages (e.g. Beebe et al. Citation2007). Genetic analyses can accurately separate species, determine synonymies, and help to describe new taxa. They have also proven especially useful in clarifying subspecies status and resolving cryptic species groups (e.g. Brust et al. Citation1998; Beebe et al. Citation2002; Vink et al. Citation2003; Batovska et al. Citation2016). Molecular identification can be used to separate the morphological indistinguishable species, damaged specimens or those life stages with less morphological features. This is an important point as prompt responses are required to deal with novel invaders and certainty is vital as costs involved in dealing with invasions can be prohibitive. To date, very little molecular work has been undertaken on New Zealand mosquitoes other than two investigations on the phylogenetic analysis of Ae. australis using COII and ITS2 sequences (Brust et al. Citation1998) and the identification of the introduced Cx. quinquefasciatus from the endemic Cx. pervigilans using the ACE2 gene (Smith and Fonseca Citation2004).
Application of molecular analysis
Phylogenetic analysis of mitochondrial DNA (mtDNA) sequences has become a preferred tool for taxonomic, population and evolutionary investigations of animals, including insects (Folmer et al. Citation1994; Simon et al. Citation1994; Lunt et al. Citation1996). The cytochrome oxidase subunit I (COI) gene region contains both conserved and variable regions which make it particularly useful for evolutionary studies (Lunt et al. Citation1996; Simon et al. Citation2006). Here we test the hypothesis that COI DNA sequences can be used to elucidate relationships within the Aedini and provide evidence on the correct placement of Ae. chathamicus, as well as for testing presently accepted relationships between endemic New Zealand species belonging to the genera Culex and Culiseta. Overall, this study will provide a complete COI barcoding library for the identification of the 17 New Zealand mosquito species (i.e. including Ae. camptorhynchus).
The nuclear ribosomal DNA (rDNA) cassette of the mosquito genome has been extensively studied in regard to mosquito taxonomy and systematics as the highly polymorphic nature of rDNA spacers often allows population structures to be examined as well as the identification of individual species (Beebe et al. Citation2002). The internal transcribed spacer (ITS) sequences are highly variable regions which have been shown to effectively distinguish species, species groups and cryptic species of mosquitoes (Brust et al. Citation1998; Beebe et al. Citation2000, Citation2002, Citation2007). We thus propose that examination of ITS1 will clarify any residual uncertainty surrounding the New Zealand endemic species of Culex and Culiseta and may also provide additional information regarding population variation within species (where it exists), if sufficient sequences from throughout the geographic range can be analysed.
Materials and methods
Specimen collection
Adult, pupal and/or larval specimens of 13 endemic and three introduced New Zealand mosquito species, as well as the recently eradicated Australian Ae. camptorhynchus, were obtained from the New Zealand BioSecure Entomology Laboratory (NZBEL) Mosquito Reference collection (). The specimens were collected from locations throughout New Zealand from 2003 to 2008. Adult specimens were collected using light traps baited with carbon dioxide (sometimes also with octenol), while larvae and pupae were sampled from natural and artificial containers and groundwater habitats using mosquito larvae dippers, plastic turkey basters and plastic Pasteur pipettes.
Table 1. New Zealand mosquito species sequenced for ITS1 and COI regions.
All specimens (apart from the new undescribed species) were identified using morphological characteristics and the dichotomous keys of Belkin (Citation1962, Citation1968), Pillai (Citation1966) and Russell (Citation1993). After identification, dry adult specimens and larval and pupal specimens preserved in 70% ethanol were stored at −20°C.
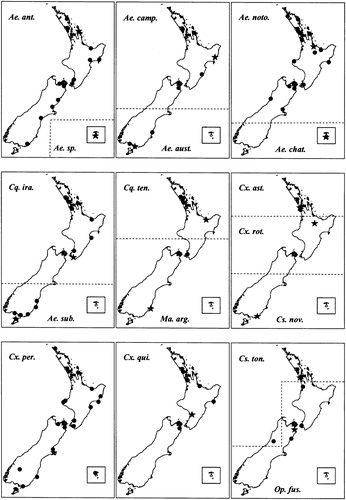
Single specimens were chosen to represent each species and used for COI sequencing analyses. But for ITS1, specimens of each species from as many distinct locations as possible were selected to represent the geographic range of the species, i.e. more specimens were obtained for widespread species than for localised species. When specimens were only available from a single location, two individuals were selected from that location wherever possible. presents maps showing the locations where each of the specimens used for obtaining the COI and ITS1 sequences were sourced.
Figure 1. Maps showing the source locations for the specimens of each species sequenced for ITS1. Specimens also sequenced for COI are represented by a star. Note: abbreviation for the species name used in the figure are: Ae. ant. = Aedes antipodeus; Ae. sp. = Aedes sp. nov.; Ae. aust. = Ae. australis; Ae. camp. = Ae. camptorhynchus; Ae. noto. = Ae. notoscriptus; Ae. chat. = Ae. chathamicus; Ae. sub. = Ae. subalbirostris; Cq. ira. = Coquillettidia iracunda; Cq. ten. = Cq tenuipalpus; Cx. ast. = Culex asteliae; Cx. rot. = Cx. rotoruae; Cx. per. = Cx. pervigilans; Cx. qui. = Cx. quinquefasciatus, Cs. ton. = Culiseta tonnoiri; Cs. nov. = Cs. novaezealandiae; Op. fus. = Opifex fuscus; Ma. arg. = Maorigoeldia argyropus.
DNA extraction
DNA was isolated from entire adult, pupal or larval mosquitoes via one of three methods. For the first method (used on the majority of specimens), individual specimens were thoroughly homogenised using a 1.5 mL plastic grinding pestle. The High Pure PCR Template Preparation Kit (Roche) was then used on the homogenate, following the manufacturer’s instructions for isolation of nucleic acids from mammalian tissue. Briefly, the mosquito fragments were lysed overnight with Tissue Lysis Buffer and Proteinase K at 55°C and incubated in a Hybaid hybridisation oven. The second method employed the ZR Insect/Tissue DNA Kit-5 (Zymo Research), again used according to the manufacturer’s instructions. A third DNA extraction and isolation method was employed for a single leg from a pinned specimen of the very rare Cs. novaezealandiae, a pinned Cs. tonnoiri both from the South Island and a dried specimen of the currently undescribed Aedes sp. nov. from the Chatham Islands (). Each of these samples was homogenised in 180 µL of ATL Buffer with 20 µL of Proteinase K (QIAGEN), then incubated overnight at 56°C. Total DNA was extracted from the lysates using a DNeasy Blood & Tissue Kit (QIAGEN) according to the manufacturer’s instructions, including two elution steps with 50 µL of AE buffer each time. All purified DNA templates were stored in elution buffer at 4°C prior to immediate use. For longer term storage, extracts were kept at −20°C and later archived at −80°C.
PCR amplification of target regions
For the bulk of the DNA extracted, each 25 µL reaction consisted of 1× Taq buffer (Roche), 0.2 mM dNTPs, 0.8 µM of each primer, 1 unit of Taq polymerase (Roche) and 1 µL of template DNA. Amplification was carried out in a Mastercycler Gradient S (Eppendorf AG) with a cycling profile including an initial denaturation at 94°C for 4 min, followed by 25–30 cycles of 94°C for 30 s, 45–51°C for 40 s, 72°C for 60 s and 7 min at 72°C as a final extension step.
For PCR using DNA template from the pinned or dried specimens of Cs. novaezealandiae, Cs. tonnoiri and Aedes sp. nov., each 20 µL reaction consisted of 1× GoTaq master mix (Promega, Madison, WI, USA), 250 nM of each primer, 0.04 µg/µL Bovine Serum Albumin (BSA, Sigma-Aldrich Co.), and 2 µL of DNA extract. Cycling conditions were initial denaturation at 94°C for 2 min, 40 cycles of 94°C for 15 s, 51°C for 30 s and 72°C for 45 s, followed by final extension step of 7 min at 72°C.
The 5′ end of the COI region was amplified using a modified version (5′-TCAACAAATCATAAAGATATTGG-3′) of the universal primer LCO1490 from Folmer et al. (Citation1994) and the (5′-ACTGTAAATATATGATGAGCTCA-3′) C1-N-2329 (mtd11) primer from Simon et al. (Citation1994). One aged specimen, Cs. novaezealandiae, produced low quality DNA sequences from the initial PCR product obtained using this system. To ensure high quality DNA sequences for this specimen, the primer pairs CI-J-1751 (5′- GGATCACCTGATATAGCATTCCC-3′) of Simon et al. (Citation1994) and C1-N-2329 were also used to amplify the DNA and products sequenced using the CI-J-1751 primer.
Primers used for amplification of ITS1 were those designed by Beebe et al. (Citation2000). The ITS1A forward primer (5′-CCTTTGTACACACCGCCCGTCG-3′) is located in the flanking segment of the 18S rRNA gene and the ITS1B is the full-length reverse complement (5′-TGTGAACTGCAGGACACTA-3′) of the original ITS2A primer (5′-ATGTGTCCTGCAGTTCACA-3′) located in the 5.8S gene (Beebe et al. Citation2000).
The majority of PCR products were tested for quality and quantity using 1% agarose gel electrophoresis, stained with ethidium bromide and visualised under ultraviolet light. Culiseta novaezealandiae and Aedes sp. nov. amplicons were electrophoresed on 1.2% agarose (in 1× TAE buffer) gel stained with SYBR® safe (Life Technologies™). Gel images were captured using digital recording equipment (Gel Doc Software system, BioRad, Hercules, CA, USA).
DNA sequencing
PCR products were purified using a High Pure PCR Product Purification Kit (Roche) and the ExoSAP-IT for PCR Product Kit (Affymetrix Inc., Santa Clara, CA, USA) following the manufacturer’s instructions. PCR products of ITS1 from Coquillettidia iracunda were cut directly from the gel prior to purification as more than one product had amplified. Purified products were sent to the Allan Wilson Centre, Massey University and EcoGene® (Auckland, New Zealand) for sequencing using Big Dye Terminator technology on a capillary ABI3730 Genetic Analyzer (Applied Biosystems Inc., Waltham, Massachusetts, USA).
The nucleotide sequences obtained were edited and assembled using Geneious Pro 9.1.5 (Biomatters Ltd., Auckland, New Zealand) or later versions. Then the new sequences were run against GenBank entries using BLASTN 2.2.23 (Zhang et al. Citation2000) and checked against published sequences in both the GenBank database using a BLAST procedure as described in Altschul et al. (Citation1990) or the BOLD database (Ratnasingham and Hebert Citation2007) to determine if they matched any sequences already submitted. Newly generated sequence data were deposited in GenBank under accession numbers, MG385730–MG385747 for COI and MG546009–MG546079 for ITS1. contains a full list of their source species and collection locations.
Phylogenetic analyses
The sequences obtained in this study were trimmed to remove primers and uninformative terminal regions, aligned using CLUSTAL X (Thompson et al. Citation1997) and any identical sequences were combined. The related species from different genera were also downloaded from GenBank for comparison. All the DNA sequences were then aligned with Geneious aligner in Geneious Pro 10.2.6 (http://www.geneious.com, Kearse et al. Citation2012) using default parameter values. The initial sequence alignments were examined and re-aligned using the MUSCLE Alignment software tool (Edgar Citation2004), and then manually rechecked and edited if necessary. Initially one alignment each from the available COI sequences and the ITS1 sequences were conducted, respectively, and the corresponding trees were constructed. However, the resultant trees were too big and confusing. The phylogenetic analysis of the COI sequences from the 17 species according to different genera were performed. As a result, four alignments were conducted and used for phylogenetic tree construction, namely, Aedes (Aedes and Opifex – A and Supplemental figure – fig. S1A), Culiseta (B), (Coquillettidia and Maorigoeldia– C and supplemental figure – fig. S1B) and Culex (D). For the phylogenetic analysis based on the ITS1 sequence, two alignments were conducted from the species of Aedes and Culex, respectively, and used for phylogenetic tree construction, for the species from Aedes (A) and the species of Culex (B). All the sequence alignment files used for the phylogenetic tree construction were uploaded to Zenodo at http://doi.org/10.5281/zenodo.3685292. In addition, the two alignments for the ITS1 sequences were also run in GBlocks Server online (Castresana Citation2000) to remove ambiguously aligned nucleotide positions. The resulted alignments were also used for the phylogenetic tree construction in Geneious 10.2.6 using the above setting to see whether the phylogenetic trees will differ.
Figure 2A and 2B. Maximum Likelihood trees showing the phylogenetic relationships of the New Zealand mosquito species plus the recently eradicated Aedes camptorhynchus based on COI sequences. Bootstrap support greater than 50% are shown on the tree, the accession numbers for the taxa obtained in this study and downloaded from GenBank are next to the species names. The sequences derived from the New Zealand mosquito species in this study are highlighted in bold letters. A. Aedes and Opifex species; B. Culiseta species.
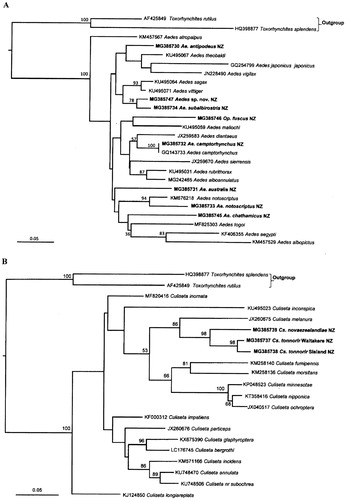
Figure 2C and 2D. Maximum Likelihood trees showing the phylogenetic relationships of the New Zealand mosquito species based on COI sequences. Bootstrap support greater than 50% are shown on the tree, the accession numbers for the taxa obtained in this study and downloaded from GenBank are next to the species names. The sequences derived from the New Zealand mosquito species in this study are highlighted in bold letters. C. Coquillettidia and Maorigoeldia species; D. Culex species.
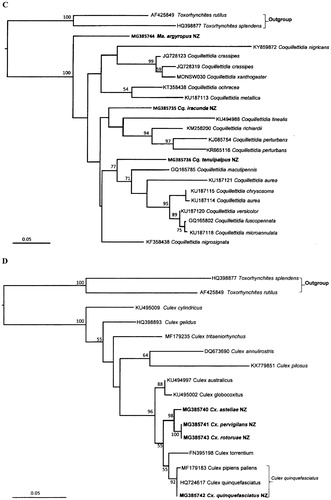
Figure 3. Bayesian trees of ITS1 sequences showing the phylogenetic relationships of New Zealand mosquito species of Aedes and Culex. Posterior probabilities (pp) over 0.5 are included in the tree, the accession numbers for the taxa obtained in this study and downloaded from GenBank are next to the species names. The sequences derived from the New Zealand mosquito species in this study are highlighted in bold letters. A. Aedes species; B. Culex species.
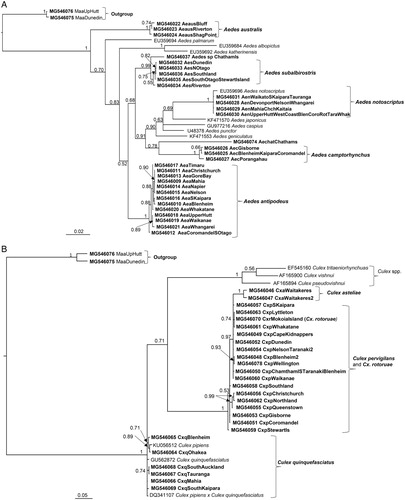
All phylogenies built were constructed using Neighbour-Joining (NJ), Maximum Likelihood (ML) and Bayesian (MB) methods in Geneious 10.2.6 (http://www.geneious.com, Kearse et al. Citation2012). ModelTest 3.04 (Posada and Crandall Citation1998), in conjunction with PAUP*4.0b10 (Swofford Citation2003), was used to select the best model using the Akaike Information Criterion. The analysis showed that the substitution model GTR (general time-reversible) + I (proportion of invariable sites) + G (gamma distribution) is the best-fit model for all four alignments. The phylogenetic trees generated using MrBayes were constructed under the model GTR + I + G (Huelsenbeck and Ronquist Citation2001). The trees were run with chain length of 1,100,000 and burn-in length of 100,000. The Maximum Likelihood analyses were conducted with PhyML 3.0 under the substitution model GTR, the NNI (default, fast) topology search and 1000 bootstrap replicate (Guindon and Gascuel Citation2003). Multiple runs were performed and the resulting parameter files were inspected for chain convergence in Tracer 1.5 (Rambaut and Drummond Citation2007), edited in FigTree v1.4.2 (Rambaut Citation2014) and PowerPoint. The trees were rooted using Toxorhynchites splendens (HQ398877) and Tx. rutilus (AF425849) as the outgroup for COI as this genus does not occur in New Zealand and is well separated phylogenetically from the genera being studied. Maorigoeldia argyropus was selected as the outgroup for the ITS1 region as its ITS1 sequences differed considerably from the rest of the New Zealand species.
All p-distances for the COI and ITS1 sequences were calculated in MEGA X (Kumar et al. Citation2018) using the above sequences. The intra- and inter-specific distances were compared, and the distance estimations were conducted using the p-distance model. There is a total of 775 for COI and 807 nucleotide positions for ITS1 in the final dataset for the New Zealand mosquito species examined. The p-distance for the ITS1 sequences from all the individual specimens sequenced and each genus were also calculated in MEGA X (Kumar et al. Citation2018). The results were shown in a supplemental table as excel format (table S1), in which there are five separate worksheets, which covered the sequence comparison for all the sequences from the New Zealand individuals (1-all), Aedes and Opifex (2-Aedes); Coquillettidia and Maorigoeldia (3- Coquillettidia); Culiseta (4-Culiseta) and Culex (5-Culex).
Results
All DNA yields were higher from whole insects processed using the High Pure PCR Template Preparation Kit (the first of our three extraction methods) and later specimens were extracted using only this method. High quality DNA templates were also obtained from the three dried specimens, Cs. tonnoiri, Cs. novaezealandiae and Aedes sp. nov., using the Qiagen method.
COI barcoding
Amplification of ∼840 bp fragments from the 5′ end of the COI gene was routinely obtained using the primer pair LCO1490 and CI-N-2329 and high-quality DNA sequences of ∼775 nt obtained from all 17 mosquito species. For the Cs. novaezealandiae specimen, an additional PCR using CI-J-1751 and CI-N-2329 was carried out and the full-length sequence was assembled from the individual sequences obtained from the two PCR products. The COI sequences for the New Zealand mosquitoes obtained in this study are ∼200 nt longer than the commonly used DNA barcoding region (Herbert et al. Citation2003). All the species in this study were morphologically identified and sequenced, the GenBank accession numbers of the sequence from the respective specimen were listed in . None of the 17 COI sequences obtained contain indels or stop codons. All the sequences are AT-rich, in a range from 67.4% to 70.8%, with mean value of 69.2%. Here, COI of Cs. novaezealandiae has the lowest value while COI of Ae. australis has the highest. The COI sequence comparisons showed that all the named species except for Cx. rotoruae, Cx. pervigilans and Cx. asteliae were separated from each other by >2% divergence (). Two of these species, Cx. rotoruae and Cx. pervigilans have identical COI sequences, and both share a 98.7% (10/775 nt) match to Cx. asteliae, with a 0.013 p-distance (). No previous COI sequences were available for most of the 17 species studied, the exceptions being Ae. camptorhynchus, Ae. notoscriptus and Cx. quinquefasciatus. The BLAST search showed that the COI sequences of Ae. camptorhynchus shared around 99%–100% identity while Ae. notoscriptus shared up to 99% identity with the corresponding species in the databases. The COI of Cx. quinquefasciatus shared up to 100% identity with Cx. pipiens and Cx. quinquefasciatus. In comparison, the COI sequences of the New Zealand Cx. pervigilans shared up to 97% identity Cx. pipiens, Cx. quinquefasciatus and Cx. torrentium.
Table 2. Estimates of evolutionary divergence (p-distance) between the New Zealand mosquito species based on COI sequences (775nt).
Phylogenetic analysis of COI gene data
Phylogenetic trees were constructed using the aligned COI sequences for the mosquito species obtained in this study and those from various closely related species as well as vector competent species available for download from GenBank (www.ncbi.nlm.nih.gov/genbank/). Initially the sequences for the 17 species and the available sequences from related and vector species were aligned together to construct a combined phylogenetic tree, however, the resulting tree did not provide additional information for the New Zealand species, but were overlarge and confusing. To better reflect the relationships among the New Zealand mosquito species, phylogenetic trees of different genera were constructed, the results are shown in (A, B, C and D).
Major groups cluster as expected, but there is an insufficient signal to clarify deeper divergences between them. Specifically, the relationships of the New Zealand Aedes species, Ae. chathamicus, Ae. australis and Ae. antipodeus, could not be resolved from other Aedes species examined using COI sequences in the Bayesian analysis (fig. S1A). However, Maximum Likelihood analysis using PHYML placed Ae. chathamicus and Ae. australis in the same clade with Ae. notoscriptus and three exotic Aedes species. Aedes antipodeus also clustered together with three exotic Aedes species, with weak support (<50% bootstrap support) (A). The eradicated species Ae. camptorhynchus was in a sub-clade with Australian collected Ae. camptorhynchus and linked to another sub-clade containing other exotic aedine species (Australian Ae. alboannulatus, Ae. rubrithorax and Holarctic Ae. diantaeus). The undescribed Aedes sp. nov. from New Zealand showed a close relationship with the endemic Ae. subalbirostris (A). Interestingly, Op. fuscus did not appear to be closely related to any of the Aedes species although it formed a weakly-supported clade with the Australian Ae. mallochi (A).
Next, the phylogenetic analysis revealed that Cs. tonnoiri is closely related to Cs. novaezealandiae, the pair of which formed a sub-clade (B). The two species showed a close relationship with Cs. melanura, with bootstrap support of 86%, but were distant from the other Culiseta species (B).
Maorigoeldia argyropus and the two Coquillettidia species were analysed together and formed a clade (C). Bayesian analysis showed that the relationship of Cq. iracunda could not be resolved whereas Cq. tenuipalpus appears to be more closely related to Ma. argyropus, with posterior probability (pp) of 0.70 (fig. S1B). Slightly different relationships among the three species were observed in the Maximum Likelihood tree (C), where Ma. argyropus is well separated from the Coquillettidia species, and Cq. iracunda and Cq. tenuipalpus are also distant from each other (C), but, are included in the same clade with other exotic Coquillettidia species (C).
The three endemic Culex species, Cx. asteliae, Cx. pervigilans and Cx. rotoruae, are recovered together in a single clade, with bootstrap support of 98%. Culex pervigilans and Cx. rotoruae are clustered together with bootstrap support of 100% (D). The New Zealand Cx. quinquefasciatus lies within a sub-clade with exotic Cx. quinquefasciatus and Cx. pipiens pallens (D).
ITS1
ITS1 fragments ranging from 388 to 798 bp were amplified using the primer pair ITS1A and ITS1B from all 17 mosquito species. All species amplified a single fragment with the exception of Cq. iracunda which amplified two PCR fragments at ∼500 and ∼800 bp. Both were examined to determine the true ITS1 sequence. After a BLAST search was run for our ITS1 sequences in the GenBank database, there were no close hits for most of the species from the current study except Ae. notoscriptus and Cx. quinquefasciatus. Aedes notoscriptus and Cx. quinquefasciatus shared about 98% identity with those of their corresponding species. The ITS1 sequences of the New Zealand Cx. pervigilans were quite divergent from those of Cx. pipiens and Cx. quinquefasciatus, about 25% different.
Phylogenetic analysis of the ITS1 region
The entire ITS1 region was sequenced for all 17 New Zealand mosquito species. The number of individual specimens studied for each species varied from 1 to 19 depending on the geographic range and specimen availability (). The alignment of the ITS1 sequences showed many indels among different individuals, even for those belonging to the same species. The sequence comparison showed that the New Zealand species are quite diverse with high p-distance values between different species, except Cx. pervigilans and Cx. rotoruae ( and table S1). The p-distances within each species are very low, the mean p-distance is less than 0.01 for all species examined, except Cx. quinquefasciatus with p-distance of 0.013 (). Besides the ITS1 sequences obtained in this study, there are no ITS1 sequences for other Culiseta, Coquillettidia, Opifex and Maorigoeldia available for comparison, therefore phylogenetic trees were constructed for Aedes and Culex only ( (A,B)). Similar topology was obtained for each of these ITS1 trees using either Neighbour-Joining, Bayesian or Maximum Likelihood methods, thus only Bayesian trees are shown in (A,B). Furthermore, the ITS1 phylogenetic trees obtained using the GBlocks alignment showed similar clusters for the New Zealand species, thus the analyses were not included in the current study.
Table 3. Genetic distance (p-distance) of ITS1 sequences from the New Zealand mosquito species.
The ITS1 sequences of Ma. argyropus are very different from those of all the other taxa examined. The sequence shares only 36%–59% identity with other New Zealand mosquito species and no close matches were found in GenBank. The two Ma. argyropus from Dunedin and Upper Hutt were used as the outgroup for the ITS1 trees ( (A,B)).
The phylogenetic analysis indicated that the undescribed Aedes species is most closely related to Ae. subalbirostris rather than any other species (A), consistent with the analysis of COI sequences (A). The analysis also suggested that Ae. chathamicus is most closely related to Ae. camptorhynchus of New Zealand, falling in the same sub-clade with that species, but only with a weak pp support of 0.78 (A). No ITS1 sequences for Ae. camptorhynchus were available for inclusion in our phylogenetic analysis.
Three variant forms of ITS1 (769–772 nt) were obtained for Op. fuscus. The size differences between the three individual sequences were caused by homopolymer runs of the base A, in which, there were 19, 17 and 16 As from the position 571–589 region of the ITS1 sequences for the three individuals (i.e. where 1–3 A indel difference among them), the rest sequences are identical in the ITS1 region for the three individuals. The sequence difference might be caused by the sequencing run. The ITS1 sequences of Op. fuscus are much longer than those of Aedes (∼ 500 nt) and diverse (∼50%, and table S1) from the Aedes species. When the ITS1 sequences of Op. fuscus were analysed together with those of Aedes species as for COI (A), the alignment produced too many gaps and more ambiguous positions, thus they were not included in the phylogenetic tree constructed for the Aedes species. These ITS1 results indicate that Op. fuscus is quite distant from Aedes species.
The ITS1 sequences of Cq. iracunda and Cq. tenuipalpus are distant from those of the other New Zealand species (table S1). The two shorter sequences examined shared ∼98% identity and the four longer fragments were ∼99% identical to each other. Further analysis showed that the shorter fragments are closely related to Cq. tenuipalpus while the longer fragments were well separated from the rest of the mosquitoes studied. Therefore, it more likely that the long fragments are not true ITS1 sequences for the species, thus they were not included in the analysis. No sequences for potentially related foreign species were available in GenBank for comparison.
The species, Culex pervigilans, Cx. rotoruae and Cx. asteliae, form a close grouping (B), similar to the results of the COI analysis shown in D. Culex asteliae is separated from Cx. pervigilans and Cx. rotoruae (B) with p-distance values above 0.02 for most of the individual specimen sequenced (table S1). Sequence comparison also showed that there is about a 12% difference between the ITS1 sequences from the New Zealand Cx. asteliae and Cx. pervigilans, thus ITS1 sequences can be used to separate them. A total of 20 Cx. pervigilans sequences were obtained from various locations. These showed identity values ranging from 95% to 99% (corresponding to 2–32 nt differences out of 646 nt). However, the ITS1 of Cx. rotoruae is identical with that of Cx. pervigilans from Lyttleton and has high similarity with ITS1 of Cx. pervigilans from other locations, with 95.4%–98.8% identity (8–29 nt differences out of 646 nt). In the phylogenetic analysis of ITS1, Cx. rotoruae clusters tightly (B) with Cx. pervigilans as might be expected.
The sequences of Cx. quinquefasciatus from different locations in New Zealand are grouped within the same sub-clade as the sequences of Cx. quinquefasciatus and Cx. pipiens from other countries, and all are well separated from those of Cx. pervigilans (B), with 1.00 pp support. The ITS1 sequence differences between the 17 individuals of Cx. pervigilans from various locations are small, with p-distances from 0 to 0.0085 (). In comparison, the intra-specific variation between six Cx. quinquefasciatus individuals is slightly greater, with a mean p-distance of 0.013, than the intra-specific variation between individuals of the other species studied (), e.g. the mean p-distance for Cx. pervigilans is only 0.0028 ().
The two New Zealand Culiseta species, Cs. novaezealandiae and Cs. tonnoiri, are closely related with a p-distance ∼0.03, but are separated from other species, which have p-distances of 0.3–0.5 (table S1). The intra-specific p-distance for the two Cs. tonnoiri is low, 0.0056 (). The phylogenetic analysis of ITS1 sequences () did not reveal clear location-associations for any of the species studied.
Discussion
This study is the first to produce molecular data for the entire New Zealand mosquito fauna. We collected 17 species from 44 locations and successfully amplified COI and ITS1 sequences from their genomes. The resulting sequence data allowed the construction of several well-resolved phylogenetic trees. In addition to providing valuable data, which was lacking for many of these species, the analyses have provided some clear insights into the relationships of these mosquitoes to one another and non-endemic species.
The genetic distances between the New Zealand mosquito taxa and those found overseas ( and ) are all relatively small. Our initial dataset presented here is probably too small to allow reliable molecular dating of divergence events. However, it is probably reasonable to erect a provisional working hypothesis that those species in New Zealand which also occur other countries represent relatively recent arrivals by dispersal from nearby landmasses. Much wider taxon sampling and more extensive genetic data from mosquitoes in New Zealand, Australia and New Caledonia are required before this proposition can be fully tested. Nonetheless, it is apparent that our endemic taxa have become morphologically distinct from their relatives elsewhere. Taking an optimistic view, one may be drawn to the conclusion that these evolutionary changes mean that endemic species are unlikely to become vector competent in the short term. Equally, considerable adaptive change seems to have taken place as they accommodated to the challenges of particular niches in the New Zealand environment. Thus, it may be difficult for existing overseas vector competent species to settle here. These ideas can only be ranked as speculative at this stage and fairly extensive further study will certainly be necessary to provide rigorous tests of our suggestions above.
Table 4. Ranges of COI DNA sequence differences between mosquitoes from overseas and New Zealand (Aedes and Opifex).
Table 5. Range of DNA sequence differences between mosquitoes from overseas and New Zealand (Culex and Opifex).
In general, the analyses of ITS1 sequences produced well-resolved phylogenetic trees (A, B) for most of the New Zealand species; however, those obtained in the analysis COI sequences (A–D, S1A, B) also provided valuable information. Additional sequences of exotic mosquito species for both gene regions would be beneficial for further resolving some relationships.
Invasive vector competent species with global medical significance due to their ability to successfully transmit arboviruses that were included in this study do not appear to be closely related to New Zealand mosquitoes. The introduced Cx. quinquefasciatus was clearly shown to be related to Cx. pipiens; however, the endemic Cx. pervigilans (previously also thought to be part of the group that includes Cx. pipiens and Cx. quinquefasciatus) was not. Therefore, domestic species including Cx. pervigilans are unlikely to become vector species of medical significance. In the future, however, the likelihood of one or more exotic species becoming established in New Zealand remains a very real possibility as such species continue to be intercepted at the country’s borders each year. Therefore, the molecular data can be used to assist in the identification of potential future incursions and associated incursion response management decisions.
In addition, the data from the two loci can already be said to provide a basis for entomological forensics in New Zealand. There are sufficient DNA sequence differences to unambiguously separate the domestic species from one another and from exotic species. The exception to this is Cx. rotoruae, which cannot be distinguished from Cx. pervigilans based on COI and ITS1 sequences. This is an important finding as specimens intercepted at borders may be cryptic larval forms or morphologically unrecognisable as adults. This study represents the first step to improving New Zealand’s border security.
The Aedes species are not entirely resolved based on the COI data (A–D, S1A, B). The analysis of ITS1 sequences produced better resolved relationships for most species. However, both loci indicated that Opifex is distant from Aedes and other New Zealand species, and is quite possibly ancestral to Aedes, as indicated previously in other studies (Dumbleton Citation1962; Belkin Citation1968). Additionally, the ITS1 data showed that Ae. chathamicus is most closely related to Ae. camptorhynchus within the aedine clade but not closely related to any of the other New Zealand species, including Op. fuscus. This does not provide support for Ae. chathamicus being placed in the genus Opifex as proposed by Reinert et al. (Citation2004). However, additional research involving more extensive sampling and exploring more molecular markers could provide more evidence on the appropriate placement of Ae. chathamicus.
The species Ae. camptorhynchus (New Zealand incursion and Australian resident) was most closely related to the exotic species Ae. diantaeus, a fellow member of the subgenus Ochlerotatus, based on the analysis of COI sequences. Interestingly, these were not closely related to the New Zealand species also in the same subgenus (Ae. subalbirostris, Ae. antipodeus and Aedes sp. nov.) based on analyses of either gene region. Further investigation is required here to better elucidate their relationships and determine if these species should remain within the same subgenus.
Both gene regions showed that Aedes sp. nov. is closely related to the endemic Ae. subalbirostris, whereas in the analysis of COI sequences it was placed in a clade with two exotic species of the subgenus Ochlerotatus. A taxonomic description of the adults of this species based on morphological characters has commenced (Kasper, pers. com., 2019). It will be interesting to see if the results of that study will agree with the inclusion of the species in the subgenus Ochlerotatus.
Both the COI and ITS1 sequence data presented here provide strong support for the species of Culex as being distinct from all other mosquito species in New Zealand. Culex asteliae was shown to be closely related to, but separate from, Cx. pervigilans, validating the thoughts of Belkin (Citation1968). Additional work may clarify the recognition of Cx. asteliae as a separate species. The data for Cx. rotoruae, however, does not support this mosquito taxon as a separate species from Cx. pervigilans (, ). Further work using additional gene regions is required to confirm this.
The sequences obtained from the two Coquillettidia species are significantly different; hence, distinguishing unknown immature life stages should be straightforward with molecular techniques. The COI and ITS1 sequence data for the two New Zealand Coquillettidia species concurs with previous observations (Belkin Citation1962, Citation1968) regarding the dissimilarity of the two species based on morphology, which has placed them in separate subgenera. The COI sequences clearly show that Cq. iracunda is more closely related to the three exotic species of the same subgenus (Coquillettidia) than it is to Cq. tenuipalpus (). Coquillettidia tenuipalpus remains the sole member of the subgenus Austromansonia.
No evidence was found for population variation within ITS1 for the commonly occurring Ae. antipodeus, Ae. notoscriptus and Cx. pervigilans despite collections from throughout their geographic ranges.
This study provides valuable data which will serve as a baseline for future taxonomic studies of New Zealand mosquitoes. The evolutionary trees are the reflections of relationships between the New Zealand mosquito species and the molecular data can help to clarify cases of disputed species identity. While addressing a number of interesting and long-standing questions around the status of various New Zealand mosquitoes, the data has to provide some intriguing new ones which future works will most likely focus on.
Table S1
Download MS Excel (45.4 KB)Fig S1B
Download TIFF Image (443 KB)Supplemental materials Table and Figure
Download MS Word (12.5 KB)Acknowledgements
The authors would like to thank Southern Monitoring Services Ltd., Victoria University of Wellington and the Ministry of Primary Industries for providing resources used by this project. Thanks also to Cor Vink, Ann Wood, Mathew Chan, Sherly George, Mark Disbury and Darryl McGinn for their invaluable assistance during this work. Geoff Chambers thanks Victoria University of Wellington for alumnus support. We are very much indebted to the editor and two anonymous reviewers who made extensive constructive suggestions to improve the text of our paper.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data that support the findings of this study are openly available in Zenodo at http://doi.org/10.5281/zenodo.3685292.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Related Research Data
References
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. Journal of Molecular Biology. 215:403–410.
- Ballard JWO, Marshall ID. 1986. An investigation of the potential of Aedes camptorhynchus (Thom.) as a vector of Ross River virus. Australian Journal of Experimental Biology and Medical Science. 64(2):197–200.
- Batovska J, Blacket MJ, Brown K, Lynch SE. 2016. Molecular identification of mosquitoes (Diptera: Culicidae) in southeastern Australia. Ecology and Evolution. 6:3001–3011. doi:10.1002/ece3.2095.
- Beebe NW, Cooper RD, Foley DH, Ellis JT. 2000. Populations of the south-west Pacific malaria vector Anopheles farauti s.s. revealed by ribosomal DNA transcribed spacer polymorphisms. Heredity. 84:244–253.
- Beebe NW, van den Hurk AF, Chapman HF, Frances SP, Williams CR, Cooper RD. 2002. Development and evaluation of a species diagnostic polymerase chain reaction-restriction fragment-length polymorphism procedure for cryptic members of the Culex sitiens (Diptera: Culicidae) subgroup in Australia and the Southwest Pacific. Journal of Medical Entomology. 39(2):362–369.
- Beebe NW, Whelan PI, van den Hurk AF, Ritchie SA, Corcoran S, Cooper RD. 2007. A polymerase chain reaction-based diagnostic to identify larvae and eggs of container mosquito species from the Australian region. Journal of Medical Entomology. 44(2):376–380.
- Belkin JN. 1962. The mosquitoes of the South Pacific (Diptera, Culicidae). Berkeley: University of California Press. 2 vols. Pp. 620 for vol. 1 and pp. 412 for vol. 2.
- Belkin JN. 1968. Mosquito studies (Diptera, Culicidae). VII. The Culicidae of New Zealand. Contributions of the American Entomological Institute. 3(1):1–182.
- Brust RA, Ballard JWO, Driver F, Hartley DM, Galway NJ, Curran J. 1998. Molecular systematics, morphological analysis, and hybrid crossing identify a third taxon, Aedes (Halaedes) wardangensis sp. nov., of the Aedes (Halaedes) australis species-group (Diptera: Culicidae). Canadian Journal of Zoology. 76(7):1236–1246.
- Buckley T, Krosch M, Leschen R. 2015. Evolution of New Zealand insects: summary and prospectus for future research. Austral Entomology. 54.
- Cane RP, Courtney RJ. 2009. But wait, there’s more … an update on the Chatham Island mosquitoes. The Weta. 38:24–30.
- Castresana J. 2000. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Molecular Biology and Evolution. 17:540–552.
- Derraik JGB. 2004. Exotic mosquitoes in New Zealand: a review of species intercepted, their pathways and ports of entry. Australian and New Zealand Journal of Public Health. 28(5):433–444.
- Dumbleton LJ. 1962. A new species and new sub-genus of Aedes (Diptera: Culicidae) from New Zealand. New Zealand Journal of Science. 5(1):17–27.
- Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research. 32:1792–1797.
- Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R. 1994. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Molecular Marine Biology and Biotechnology. 3(5):294–299.
- Fonseca D, Keyghobadi N, Malcolm C, Mehmet C, Schaffner F, Mogi M, Fleischer R, Wilkerson R. 2004. Emerging vectors in the Culex pipiens complex. Science. 303:1535–1538. doi:10.1126/science.1094247.
- Goddard LB, Roth AE, Reisen WK, Scott TW. 2002. Vector competence of California mosquitoes for West Nile virus. Emerging Infectious Diseases. 8(12):1385–1391.
- Guindon S, Gascuel O. 2003. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Systematic Biology. 52:696–704.
- Harbach RE. 2018. Culicipedia: Species-group, genus-group and family-group names in Culicidae (Diptera). CABI. Pp. 396.
- Herbert PD, Cywinska A, Ball SL, deWaard JR. 2003. Biological identifications through DNA barcodes. Proceedings of the Royal Society of London. Series B: Biological Sciences. 270(1512):313–321.
- Holder P, Browne G, Bullians M. 1999. The mosquitoes of New Zealand and their animal disease significance. Surveillance. 26(4):12–15.
- Huelsenbeck JP, Ronquist F. 2001. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 17:754.
- Kay BK, Russell RC. 2013. Mosquito eradication: the story of killing campto. Collingwood, Victoria: CSIRO. P. 255.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28(12):1647–1649.
- Kramer L, Chin P, Cane RP, Kauffman E, Mackereth G. 2011. Vector competence of New Zealand mosquitoes for selected arboviruses. The American Journal of Tropical Medicine and Hygiene. 85(1):182–189. doi:10.4269/ajtmh.2011.11-0078.
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K. 2018. MEGA x: molecular evolutionary genetics analysis across computing platforms. Molecular Biology and Evolution. 35:1547–1549.
- Laird M. 1990. New Zealand’s northern mosquito survey, 1988-89. Journal of the American Mosquito Control Association. 6:287–299.
- Lunt DH, Zhang D-X, Szymura JM, Hewitt GM. 1996. The insect cytochrome oxidase I gene: evolutionary patterns and conserved primers for phylogenetic studies. Insect Molecular Biology. 5(3):153–165.
- Pillai JS. 1966. Culiseta novaezealandiae, a new species of the subgenus Climacura Felt (Diptera, Culicidae: Culisetini), with notes on its ecology and development. Transactions of the Royal Society of New Zealand (Zoology). 8(11):125–133.
- Posada D, Crandall KA. 1998. MODELTEST: testing the model of DNA substitution. Bioinformatics. 14:817–818.
- Rambaut A. 2014. Figtree, a graphical viewer of phylogenetic trees [Internet]. Doi: http://tree. bio. ed. ac. uk/software/figtree.
- Rambaut A, Drummond AJ. 2007. Tracer v 1.4: MCMC trace analyses tool. http://beast.bio.ed.ac.uk/Tracer.
- Ratnasingham S, Hebert PDN. 2007. BARCODING: bold: the barcode of life data system. . Molecular Ecology Notes. 7:355–364. http://www.barcodinglife.org
- Reinert JF, Harbach RE, Kitching IJ. 2004. Phylogeny and classification of Aedini (Diptera: Culicidae), based on morphological characters of all life stages. Zoological Journal of the Linnean Society. 142:289–368.
- Ross RW, Austin FJ, Miles JAR, Maguire T. 1963. An arbovirus isolated in New Zealand. Australian Journal of Science. 26:20–21.
- Russell RC. 1993. Mosquitoes and mosquito-borne disease in southeastern Australia: a guide to the biology, relation to disease, surveillance, control and the identification of mosquitoes in southeastern Australia. Sydney, Department of Medical Entomology, Westmead Hospital, Westmead, NSW 2145, Australia and Department of Medicine, University of Sydney. p. 310.
- Sherif AE, Mclntyre M, Swan T, Kasper J, Derraik JGB, Baker MG, Hales S. 2019. Intercepted mosquitoes at New Zealand’s Ports of Entry, 2001 to 2018: current status and future concerns. Tropical Medicine and Infectious Disease. 4(3):101. doi:10.3390/tropicalmed4030101.
- Simon C, Buckley TR, Frati F, Stewart JB, Beckenbach AT. 2006. Incorporating molecular evolution into phylogenetic analysis, and a new compilation of conserved polymerase chain reaction primers for animal mitochondrial DNA. Annual Review of Ecology, Evolution, and Systematics. 37:545–579.
- Simon C, Frati F, Beckenbach A, Crespi B, Liu H, Flook P. 1994. Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Annals of the Entomological Society of America. 87:651–701.
- Smith JL, Fonseca DM. 2004. Rapid assays for identification of members of the Culex (Culex) pipiens complex, their hybrids, and other sibling species (Diptera: Culicidae). The American Journal of Tropical Medicine and Hygiene. 70(4):339–345.
- Swofford DL. 2003. PAUP*. phylogenetic analysis using parsimony (*and 0ther methods). Version 4.0b10. Sinauer Associates, Sunderland, MA.
- Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. 1997. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Research. 25:4876–4882.
- Vink CJ, Evans AM, Phillips CB, Murdoch TC, Tubbs MB. 2003. Molecular phylogenetic analysis supports the synonymy of Prodontria modesta (Broun) and Prodontria bicolorata Given (Coleoptera: Scarabaeidae: Melolonthinae). Journal of Insect Conservation. 7:215–221.
- Weinstein P, Laird M, Browne GN. 1997. Exotic and endemic mosquitoes in New Zealand as potential arbovirus vectors. Wellington: Ministry of Health. p. 16.
- Zhang Z, Schwartz S, Wagner L, Miller W. 2000. A greedy algorithm for aligning DNA sequences. Journal of Computational Biology. 7(1–2):203–214.