Abstract
The aim of this study was to evaluate the sensitivity and the detection limit of a real-time polymerase chain reaction (Q-PCR) developed for the qualitative and quantitative detection of Mycoplasma gallisepticum. No cross-reactivity was observed with DNA from other important avian mycoplasmas, including Mycoplasma synoviae and Mycoplasma meleagridis. However, the Q-PCR could not distinguish between M. gallisepticum and Mycoplasma imitans. The Q-PCR had detection limits 10 to 1000 times lower than a conventional commercial PCR method and than culture. The Q-PCR was used quantitatively by incorporating a set of external M. gallisepticum DNA standards, derived from a M. gallisepticum log-phase culture of a known concentration. The number of colony-forming unit equivalents per millilitre in tracheal swabs from experimentally infected birds could be determined from a single sample. The method had good reproducibility and correlated well with standard counting techniques using culture. It can be concluded that the Q-PCR described is suitable for qualitative and quantitative detection of M. gallisepticum in clinical samples.
PCR en temps réel pour la détection qualitative et quantitative de Mycoplasma gallisepticum
Le but de cette étude a été d'évaluer la sensibilité et les limites de détection du test d'amplification en chaîne par polymérase en temps réel (Q-PCR) développé pour la détection qualitative et quantitative de Mycoplasma gallisepticum (Mg). Aucune réaction croisée n'a été observée avec l'ADN d'autres mycoplasmes aviaires importants, incluant M. synoviae et M. meleagridis . Cependant, le test Q-PCR n'a pas pu différencier Mg de M. imitans. Les limites de détection ont été de 10 à 1000 fois inférieures à celles d'un test PCR du commerce (cPCR) et à la culture. Le test Q-PCR a été utilisé quantitativement en incorporant un set de standards externes d'ADN de Mg dérivé d'une culture de Mg, en phase logarithmique, de concentration connue. Le nombre d'équivalents unité formant colonie/ml dans les écouvillons trachéaux des animaux infectés expérimentalement a pu être déterminé à partir d'un seul échantillon. La reproductibilité de la méthode a été bonne et une bonne corrélation a été observée avec les techniques standard de comptage après culture. Il peut être conclu que le test Q-PCR décrit est approprié à la détection qualitative et quantitative de Mg à partir d'échantillons cliniques.
Real-Time-PCR für den qualitativen und quantitativen Nachweis von Mycoplasma gallisepticum
Ziel dieser Studie war es, die Sensibilität und die Nachweisgrenze einer für die qualitative und quantitative Bestimmung von Mycoplasma gallisepticum (MG) entwickelten Real-Time-Polymerasekettenreaktion (Q-PCR) zu untersuchen. Mit der DNS anderer wichtiger aviärer Mykoplasmen einschließlich M. synoviae und M. meleagridis wurden keine Kreuzreaktionen beobachtet. Die Q-PCR konnte jedoch nicht zwischen MG und M. imitans unterscheiden. Die Q-PCR hatte eine 10-1000-mal niedrigere Nachweisgrenze als die konventionelle kommerzielle PCR (cPCR) und die kulturelle Nachweismethode. Die Q-PCR wurde zum quantitativen Nachweis genutzt, indem ein Satz externer MG-DNS-Standards aus MG-log-Phasen-Kulturen mit bekannter Konzentration inkorporiert wurde. Die Zahl der Kolonie bildenden Einheiten/ml in Trachealtupfern von experimentell infizierten Tieren konnte in einer einzigen Probe bestimmt werden. Die Methode weist eine gute Reproduzierbarkeit auf und korrelierte mit Standard-Zählverfahren bei der Kultivierungsmethode. Daraus kann geschlossen werden, dass die beschriebene Q-PCR für den qualitativen und quantitativen Nachweis von MG in Untersuchungsproben geeignet ist.
PCR a tiempo real par la detección cuantitativa y cualitativa de Mycoplasma gallisepticum
El objetivo de este estudio fue el de evaluar la sensibilidad y el límite de detección de una reacción en cadena de la polimerasa a tiempo real (Q-PCR) desarrollado para la detección cualitativa y cuantitativa de Mycoplasma gallisepticum (Mg). No se observó reactividad cruzada con el ADN de otros importantes micoplasmas aviares, incluidos M. synoviae y M. meleagridis. Aún así, la Q-PCR no pudo distinguir entre Mg y M. imitans. LA Q-PCR presentó límites de detección de 10 a 1,000 veces menores que la PCR comercial convencional (cPCR) y que el cultivo. La Q-PCR se utilizó como método cuantitativo mediante la incorporación de un grupo de ADN de Mg estándares externos, derivados de un cultivo de Mg en fase log, de concentración conocida. El número de de equivalentes de unidades formadoras de colonias/ml en hisopos traqueales de aves infectadas experimentalmente pudo ser determinado a partir de una sola muestra. El método presentó buena reproducibilidad y se correlacionó bien con las técnicas estándares de contaje usadas en cultivos. Se puede concluir que la Q-PCR descrita es útil para la detección cualitativa y cuantitativa de Mg en muestras clínicas.
Introduction
Mycoplasma gallisepticum is an important pathogen causing great economic losses in the poultry industry. The improvement of diagnostic tools for the direct detection of mycoplasmas has always been a subject for research as these tools play an important role in both eradication programmes and in experimental work. Culture methods, which are often labour-intensive and require specially formulated media, have increasingly been replaced by hybridization techniques, polymerase chain reaction (PCR), or combinations of both techniques, to detect the presence of M. gallisepticum-specific DNA sequences (Fernandèz et al., Citation1993; Johansson et al., Citation1993; Khan & Kleven, Citation1993; Bascuñana et al., Citation1994; Fan et al., Citation1995; García et al., Citation1995 Citation1996 Citation1997; Lauerman et al., Citation1995; Silveira et al., Citation1996; Kiss et al., Citation1997; Kojima et al., Citation1997; Lauerman, Citation1998; Salisch et al., Citation1998 Citation1999; Marois et al., Citation2002). More recently, real-time PCR methods have been described for qualitative and quantitative detection of a great variety of pathogens. In these methods the production of specific PCR products is monitored using fluorimetric detection of amplified products and identified using melting curve analysis or specific hybridization probes (Wittwer et al., Citation1997; Wittwer, Citation2001). A qualitative real-time PCR for the detection of M. gallisepticum was described by Carli & Eyigor (Citation2003), although no M. gallisepticum could be detected in swabs taken from serologically positive birds.
In the present study we describe the development and evaluation of a real-time PCR capable of both qualitative and quantitative detection of M. gallisepticum in experimental in vivo and in vitro samples. In this real-time PCR quantification of specific DNA in unknown samples, expressed as colony-forming unit (CFU) equivalents per millitre, was realized using external DNA standards (Rasmussen, Citation2001), derived from a culture with a known M. gallisepticum concentration. The limit of detection and cross-reactivity of the quantitative M. gallisepticum PCR (Q-PCR) were compared with those of a conventional commercial PCR (cPCR) and culture. The variability of the technique was also investigated.
Materials and Methods
Mycoplasma strains
All mycoplasma strains used () were cultured using mycoplasma agar (Avian Mycoplasma Solid Medium; Mycoplasma Experience, Reigate, UK) or mycoplasma broth (Avian Mycoplasma Liquid Medium; Mycoplasma Experience).
Table 1. Mycoplasma strains used in M. gallisepticum Q-PCR cross-reactivity testing
Preparation of samples and isolation of mycoplasma DNA
For all strains and samples tested DNA was isolated either from 1.0 ml mycoplasma broth culture or from tracheal swabs washed out in 1.0 ml mycoplasma broth (when Q-PCR and culture were carried out on the same sample), or from tracheal swabs washed in 1.0 ml sterile phosphate-buffered saline (when only Q-PCR was carried out). Fifty microlitres of this sample were used for preparing serial dilutions for colony counting as described later. For PCR, the remaining 950 µl were centrifuged for 10 min at 16 000×g and the pellet washed twice with sterile phosphate-buffered saline, resuspended in 25 µl sterile phosphate-buffered saline, incubated at 120°C for 15 min and cooled on ice. After a final centrifugation for 2 min at 16 000×g, supernatants were cooled to 4°C and used directly for PCR or stored at −70°C.
Colony counting
Twenty microlitre volumes of serial 10-fold dilutions were spread on mycoplasma agar plates in duplicate, incubated at 37±1°C under 5% CO2 and examined weekly for colonies. Final colony counts were carried out after 14 days of incubation and the number of CFU per millitre was calculated using only dilutions with less than 300 colonies per plate. All counts were expressed as log10 M. gallisepticum CFU per millilitre.
Preparation of in vitro standards and diluted samples
Four cultures of strain M. gallisepticum/chicken/NL/Dev-1608Vin/99 (coded as MgF1999) (Landman et al., Citation2004; Feberwee et al., Citation2005)—cultures A, B, C and D—were inoculated simultaneously from the same stock culture in order to determine the reproducibility of colony counts. All four cultures were inoculated at a starting concentration of 20 CFU/ml and sampled after 48 h of incubation. DNA was isolated from serial 10-fold dilutions (10−1 to 10−6) prepared from a 1.0 ml sample of culture A taken after 48 h of incubation. Isolated DNA was aliquotted in 100 µl volumes and stored at −70°C for use as standards in Q-PCR experiments to calculate M. gallisepticum concentrations (CFU equivalents/ml) in samples from cultures B, C and D, and samples from chickens. Results from five independent runs were used to determine interassay variability and to determine a standard curve, which was used in all subsequent Q-PCR experiments. Samples taken from culture B at 0, 12, 48, 72 and 144 h of incubation were used to determine the effect of non-viable mycoplasma cells on culture and Q-PCR results. The number of CFU/ml were determined and compared with estimates obtained using Q-PCR results and the DNA standard curve (in duplicate) and expressed as CFU equivalents/ml. The colony counts were calculated from dilutions with less than 300 CFU/plate. In order to determine detection limits of the Q-PCR, culture and cPCR, 1.0 ml samples were taken from culture C at 48 h and serial 10-fold dilutions were prepared in duplicate for colony counting, cPCR and Q-PCR. Detection limits were defined as the highest dilution(s) yielding a positive PCR result and expressed as CFU equivalents/ml needed for a positive PCR reaction, using the culture colony counts as a reference. Finally, to determine the reproducibility of the combination of DNA extraction and the measurement of crossing point (Cp) and melting temperature (Tm) by Q-PCR, culture D was used. M. gallisepticum DNA was isolated from 10 individual 2.0 ml samples taken 48 h after inoculation.
Animal experiments
Three different experiments, A, B and C, were designed to determine the correlation between Q-PCR and colony counts in vivo. In experiment A, three 66-week-old specific pathogen free White Layers were housed in a negative pressure isolator and infected at day 0 by intratracheal inoculation with 1.0 ml culture of MgF1999 containing 0.5×104 CFU/ml. At days 0, 2, 4, 10, 15, 20, 25, 30 and 35, tracheal swabs were taken from each bird and used for simultaneous Q-PCR and colony counting by culture. In this pilot experiment no control birds were used. In experiment B, tracheal samples were taken from five, and in experiment C from 10, 25-week-old specific pathogen free White Layer hens that had been inoculated intratracheally with 1.0 ml culture of MgF1999 containing 7.5×104 CFU/ml (experiment B) and 3.2×104 CFU/ml (experiment C). Additionally, in experiments B and C five uninfected control birds were sampled in order to exclude influence of sample handling on the results of Q-PCR. Tracheal samples were taken at days 0, 5, 14 and 21 (experiment B) and at day 0, 5, 14 and 28 (experiment C) after inoculation of the infected group. The tracheal samples from experiments A, B and C were examined to determine the number of CFU/ml by Q-PCR and colony counting. Additionally, DNA samples from experiment B were diluted 1:100 and 1:10 000 and re-tested by Q-PCR to check for inhibition. All tracheal samples were collected using sterile cotton swabs (Technical Service Consultants, Heywood, UK). Before sampling, the swab was dipped into sterile mycoplasma broth and gently squeezed against the side of the container to remove excess fluid. The samples were transported at 4°C and processed within 3 h of sample collection. The colony counts were calculated from dilutions yielding less than 300 CFU/plate.
PCR tests
For the Q-PCR, M. gallisepticum-specific primers Mg14F (5′-GAG CTA ATC TGT AAA GTT GGT C-3′, Tm=57.80°C) and Mg13R (5′-GCT TCC TTG CGG TTA GCA AC-3′, Tm=63.64°C) were used (Lauerman, Citation1998). These primers amplify a 186 base pair (bp) sequence from the 16S ribosomal RNA gene of M. gallisepticum. A BLAST search was performed online (http://www.ncbi.nlm.nih.gov/BLAST/) to confirm the theoretical specificity of the primers. The Tm of the 186 bp M. gallisepticum PCR product was calculated using Tm Utility version 1.51 (Idaho Technologies Inc., Salt Lake City, Utah, USA). For the cPCR, the M. gallisepticum DNA Test Kit (IDEXX Laboratories, Westbrook, Maine, USA) was used. The PCR was performed and results were interpreted according to the manufacturer's instructions. All Q-PCR reactions were performed on a LightCycler 1.0 system (Roche Applied Science, Mannheim, Germany) using the DNA Master SYBR Green I kit, 2 µl extracted sample DNA, M. gallisepticum standard or water (negative control), 20 pmol each of the forward and reverse primers per reaction and 6 mM MgCl2 in a final volume of 20 µl. The PCR reaction was incubated for 35 cycles and fluorescence was monitored during each cycle at 72°C. Following amplification, a melting curve analysis was performed starting at 65°C using a temperature transition rate of 0.2°C/sec to determine Tm values of PCR products. Cp values were defined as the cycle number yielding a maximum value of the second derivative of the amplification curve of the sample. Samples were regarded as positive when both a measurable Cp and the expected Tm (± 0.5°C) were seen.
For all samples tested, the concentration of M. gallisepticum in CFU equivalents/ml was calculated from a series of external M. gallisepticum DNA standards prepared as already described, using the second derivative method included in the LightCycler data analysis software. To facilitate this and at the same time validate individual runs, one of the standards and a negative control (water) were included in every run. To confirm the presence of a specific PCR product of 186 bp, one series of M. gallisepticum DNA standards was analysed after PCR in a 2% agarose gel.
Results
Cross-reactivity
All five M. gallisepticum strains tested were readily detected in the M. gallisepticum Q-PCR and in the conventional PCR (). Mycoplasma imitans strain Mim 4229 was detected by Q-PCR but not by cPCR. Both PCR tests detected none of the other mycoplasmas tested (). BLAST analysis revealed significant alignments of primers Mg14F and Mg13R with Genbank sequences AE016969.1, AE016968.1, L35043.3, L08897.1, M22441.1 (M. gallisepticum 16s rRNA) and L24103.1 (M. imitans 16s rRNA).
Limit of detection
For cPCR and Q-PCR, limits of detection were based on culture C at 48 h, when it contained 5.8×107 CFU/ml. Dilution of this culture showed that the limit of detection of the M. gallisepticum Q-PCR (highest positive dilution of 1010) was 10 to 1000 times lower than that of cPCR (highest positive dilution of 105) and that M. gallisepticum DNA was detected at concentrations comparable with between 0.01 and 0.06 CFU equivalents/ml. In this experiment, cPCR detected M. gallisepticum DNA at concentrations comparable with between 0.63 and 5.20 CFU equivalents/ml.
Reproducibility
The results from colony counts from cultures A, B, C and D were 7.1, 7.3, 7.8 and 8.1 log10 CFU/ml, respectively. The method was highly reproducible, giving a mean count of 7.6±0.4 log10 CFU/ml for these 48-h cultures. The Q-PCR results for the M. gallisepticum DNA standards derived from culture A that were tested in five independent runs on five different days showed a high reproducibility for calculated CFU equivalents/ml, Tm and Cp ( and ). Results of a typical Q-PCR run using M. gallisepticum DNA standards, compared with electrophoresis results for the same samples, are shown in . As for the results of samples taken from culture B at different time intervals, up to 72 h, there was a correlation between the number of M. gallisepticum CFU/ml determined by culture and CFU equivalents calculated by Q-PCR. However, after 144 h there were discrepancies between Q-PCR and culture, with no viable M. gallisepticum isolated from the culture, but Q-PCR still positive (). The reproducibility of the determinations of Cp, Tm and calculated CFU equivalents/ml for the 10 individual M. gallisepticum DNA samples (tested in duplicate) from culture D, with a concentration of 8.1 log10 CFU/ml, showed that in this set of samples, the Cp values varied between 9.8 and 12.0 (mean 11.25, standard error of the mean [SEM]±0.65) corresponding to a difference of between 7.0 and 7.7 log10 M. gallisepticum CFU equivalents/ml (mean 7.2, SEM±0.2). The predicted Tm, based on the sequence of the amplified 186 bp product, was 85.34°C. The melting temperatures measured for these 10 samples following Q-PCR varied between 85.45 and 85.60°C (mean 85.5, SEM±0.2). A similar range (85.49 to 85.74 °C) was observed for Tm determinations on M. gallisepticum DNA standards in five independent runs ().
Figure 1. Reproducibility of Q-PCR crossing points of DNA standards compared with log10 M. gallisepticum CFU/ml determined by culture. Mean of five different Q-PCR runs. Bars indicate SEM.
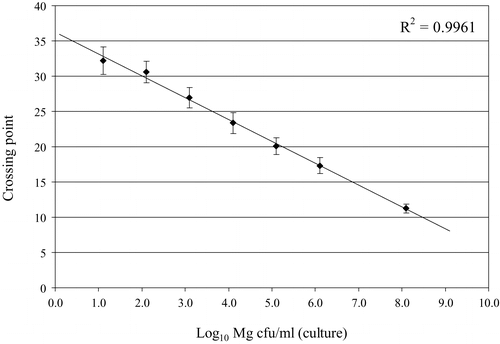
Figure 2. Results of M. gallisepticum DNA standard in Q-PCR and electrophoresis. 1 to 7, 10-fold dilutions from culture A, tested in duplicate, from undiluted (sample 1, 106.1 CFU equivalents/ml) to 10−6 (sample 7, 101.1 CFU equivalents/ml); 8, H2O; M, marker (DNA 50 bp Marker XIII; Roche Applied Science, Mannheim, Germany).
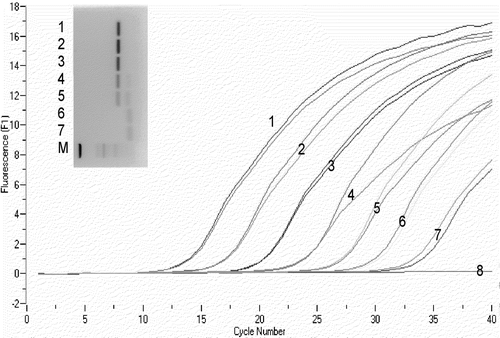
Table 2. Results of reproducibility of Q-PCR based on M. gallisepticum DNA standards derived from culture A
Table 3. Results of Q-PCR and culture on time-interval samples taken from culture B
In vivo experiments
presents the results of comparing colony counts with Q-PCR on swabs from experimentally infected birds. In experiment A, shortly after infection Q-PCR was positive and culture was negative, although the number of M. gallisepticum bacteria detected by Q-PCR was low. Samples taken 1 day later (experiments B and C, day 5) were both culture-positive and Q-PCR-positive. In all three experiments there was a discrepancy between culture and Q-PCR results, especially when the concentration of M. gallisepticum was high. However, statistical analysis revealed no significant differences between variances (F test, P>0.05) and means for the two tests (t test, P=0.539). Correlation between colony counts and Q-PCR over the three experiments was 0.88 (Pearson's correlation coefficient) when all data were included and 0.95 when culture-negative/Q-PCR-positive results were treated as false negatives and omitted from the calculation. A better correlation (0.97) was found when Q-PCR was performed on samples diluted 1:100. This dilution effect was less evident at day 21 (). Further dilution of Q-PCR samples to 1:10 000 reduced the correlation to 0.88.
Figure 3. Effect of dilution of samples on estimation of CFU equivalents/ml using Q-PCR (samples from experiment B). Bar indicates SEM.
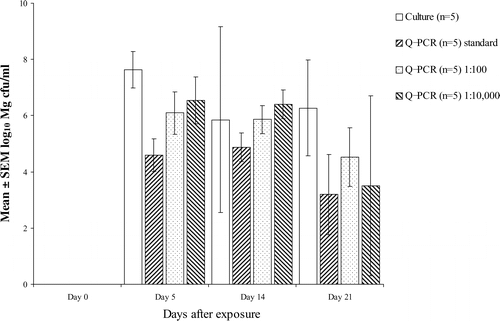
Table 4. Results of in vivo experiments
Discussion
The specificity of the M. gallisepticum Q-PCR test was shown by BLAST analysis and the results from assay of different mycoplasma strains. BLAST analysis showed good specificity for both the Mg14F and Mg13R primers. Analysis of the primers and of the amplified target sequence from the 16S rRNA genes of M. gallisepticum (GenBank accession number M22441) and M. imitans (GenBank accession number L24103) revealed that this sequence was 100% identical for both strains studied. This finding was confirmed in vitro. M. imitans was not detected by the cPCR used in this study. For all other mycoplasmas tested, the Q-PCR and cPCR results were negative. The Q-PCR detected 10 to 1000 times lower concentrations of M. gallisepticum DNA than the cPCR and culture, indicating that low numbers of M. gallisepticum may be missed by culture and cPCR but can be detected by Q-PCR. The greater sensitivity of Q-PCR may be partially attributable to the use of SYBR Green fluorescence for the detection of products rather than visualization of bands after conventional gel electrophoresis ().
The variability in the results obtained with the M. gallisepticum DNA standards derived from culture A (between runs) and the limited variability of determinations of Tm, Cp and estimated concentrations on 10 sample replicates suggest that reproducibility of both the sample preparation protocol and the Q-PCR technique is high. Although a log-phase culture was used for each experiment, both the cPCR used in this study and the Q-PCR could detect M. gallisepticum DNA in culture-negative dilutions. This might be explained by a real difference in limit of detection and by the fact that PCR, in general, also detects DNA from non-viable bacteria. This is supported by the observation that up to 72 h after incubation there was a relationship between results from colony counts and the Q-PCR, but that at 144 h Q-PCR was still positive while culture was negative. Plots of crossing points for standards agianst the colony counts yielded a linear regression with an R 2 value of 0.9961 (). Thus, a 10-fold difference in concentration should lead to a Cp difference of 3.32 if the PCR had a 100% efficiency. As the difference between sample A (Cp=17.33) and sample D (Cp=11.25) based on colony counts was 100-fold (108.1 to 106.1), the difference in Cp would be expected to be 6.64 rather than 6.08, suggesting that the efficiency of the PCR in the presence of large amounts of M. gallisepticum was less than 100%, and that this may result in underestimation of the concentration of M. gallisepticum. In the in vivo experiments, all Q-PCR determinations of concentrations between day 10 and day 30 after infection were lower than those determined by culture. In contrast, in the in vitro experiments, Q-PCR showed higher sensitivity than culture. The potential effect of inhibitory factors on the PCR reaction and the loss of M. gallisepticum cells during sample processing were also considered by Carli & Eyigor (Citation2003). Diluting samples prior to Q-PCR partially alleviated this problem (), probably because the inhibitory factors were reduced in concentration, but diluting samples too much could also lead to loss of sensitivity (, day 21). It is also possible that in the presence of high numbers of M. gallisepticum, the primer concentrations used may become a limiting factor in the Q-PCR, leading to reduced PCR efficiency. The Q-PCR yielded higher estimates than culture of the concentration of M. gallisepticum at days 4 and 35 after inoculation. At day 4, this can be explained by the lower sensitivity of culture when low concentrations are present; at day 35, by the detection of non-viable mycoplasmas by Q-PCR.
Although the Q-PCR could not distinguish between M. gallisepticum and M. imitans, M. imitans has not yet been isolated from commercial flocks of chickens or turkeys (Dupiellet, Citation1984; Buntz et al., Citation1986; Bradbury et al., Citation1993). Thus this Q-PCR is suitable for the quantitative detection of M. gallisepticum in clinical samples and can be used for the diagnosis of M. gallisepticum-infected flocks after initial diagnosis by serology (data not shown). The results of this study also show that the Q-PCR can be used for the quantification of M. gallisepticum in a single sample, using external DNA standards. This means that the assay could replace the labour-intensive and expensive cultural methods still used for quantitative studies (e.g. transmission studies), provided that factors that may influence the sensitivity of the test and the correlation with culture results, such as inhibitory factors in the sample or the simultaneous detection of viable and non-viable mycoplasmas, are taken into account.
Translations of the abstract in French, German and Spanish are available on the Avian Patholgy website.
Acknowledgments
The authors thank Dr A. L. J. Gielkens and Dr J. A. Stegeman for critical reviewing this work. This research was supported by a grant from the National Board for Poultry and Eggs (PPE) of The Netherlands.
Related Research Data
References
- Bascuñana , C.R. , Mattsson , J.G. , Bölske , G. and Johansson , K.E. 1994 . Characterization of the 16S rRNA genes from Mycoplasma sp. strain F38 and development of an identification system based on PCR . Journal of Bacteriology , 176 : 2577 – 2586 .
- Bradbury , J.M. , Abdul-Wahab , O.S.M. , Yavari , C.A. , Dupiellet , J.P. and Bové , J.M. 1993 . Mycoplasma imitans sp. nov. is related to Mycoplasma gallisepticum and found in birds . International Journal of systematic Bacteriology , 43 : 721 – 728 .
- Buntz , B. , Bradbury , J.M. , Vuillaume , A. and Rousselot-Paillet , D. 1986 . Isolation of Mycoplasma gallisepticum from geese . Avian Pathology , 15 : 615 – 617 .
- Carli , K.T. and Eyigor , A. 2003 . Real-time polymerase chain reaction for detection of Mycoplasma gallisepticum in chicken trachea. Research note . Avian Diseases , 47 : 712 – 717 .
- Dupiellet , J.P. (1984) . Mycoplasmes et acholeplasmes des palmipèdes a foie gras: isolement caracterisation, étude du rôle dans la pathologie. Rappport de D.E.A. de Pathologie , Université de Bordeaux II, Villenave d’ Ornon , France .
- Fan , H.H. , Kleven , S.H. , Jackwood , M.W. , Johansson , K.E. , Pettersson , B. and Levisohn , S. 1995 . Species identification of avian mycoplasmas by polymerase chain reaction and restriction fragment length polymorphism analysis . Avian Diseases , 39 : 398 – 407 .
- Feberwee , A. , Mekkes D.R. , De Wit , J.J. , Hartman , E.G. & Pijpers , A. (2005) . Comparison of culture, PCR and different serological tests for detection of Mycoplasma gallisepticum and Mycoplasma synoviae infections . Avian Diseases , 49 , 260 – 268 .
- Fernández , J. , Mattsson , J.G. and Bölske , G. 1993 . Species-specific oligonucleotide probes complementary to 16S rRNA of Mycoplasma gallisepticum and Mycoplasma synoviae . Research in Veterinary Science , 55 : 130 – 136 .
- García , M. , Jackwood , M.W. , Levisohn , S. and Kleven , S.H. 1995 . Detection of Mycoplasma gallisepticum, M. synoviae and M. iowae by multi-species polymerase chain reaction and restriction fragment length polymorphism . Avian Diseases , 39 : 606 – 616 .
- García , M. , Jackwood , M.W. , Head , M. , Levisohn , S. and Kleven , S.H. 1996 . Use of species-specific oligonucleotide probes to detect Mycoplasma gallisepticum, M. synoviae and M. iowae PCR amplification products . Journal of Veterinary Diagnostic Investigation , 8 : 56 – 63 .
- García , M. , Gerchman , I. , Meir , R. , Jackwood , M.W. , Kleven , S.H. and Levisohn , S. 1997 . Detection of Mycoplasma meleagridis from dead-in-shell turkey embryos by polymerase chain reaction and culture . Avian Pathology , 26 : 765 – 778 .
- Johansson , K.E. 1993 . “ Detection and identification of mycoplasmas with diagnostic DNA probes complementary to ribosomal RNA ” . In Rapid Diagnosis of Mycoplasmas , Edited by: Kahane , I. and Adoni , A. 139 – 154 . New York : Plenum Press .
- Khan , M.I. and Kleven , S.H. 1993 . Detection of Mycoplasma gallisepticum infection in field samples using a species-specific DNA probe . Avian Diseases , 37 : 880 – 883 .
- Kiss , I. , Matiz , K. , Kaszanyitzky , E. , Chávez , Y. and Johansson , K.E. 1997 . Detection and identification of avian mycoplasmas by polymerase chain reaction and restriction fragment length polymorphism assay . Veterinary Microbiology , 58 : 23 – 30 .
- Kojima , A. , Takahashi , T. , Kijima , M. , Ogikubo , Y. , Nishimura , M. , Nishimura , S. , Harasawa , R. and Tamura , Y. 1997 . Detection of mycoplasma in avian live virus vaccines by polymerase chain reaction . Biologicals , 25 : 365 – 371 .
- Landman , W.J.M. and Feberwee , A. 2001 . Field studies on the association between amyloid arthtopathy and Mycoplasma synoviae infection, and experimental reproduction of the condition in brown layers . Avian Pathology , 30 : 629 – 639 .
- Landman , W.J.M. , Corbanie , E.A. , Feberwee , A. and van Eck , J.H.H. 2004 . Aerosolization of Mycoplasma synoviae compared with Mycoplasma gallisepticum and Enterococcus faecalis . Avian Pathology , 33 : 210 – 215 .
- Lauerman , L.H. 1998 . “ Mycoplasma PCR assays ” . In Nucleic Acid Amplification Assays for Diagnosis of Animal Diseases , Edited by: Lauerman , L.H. 41 – 48 . Davis : American Association of Veterinary Laboratory Diagnosticians .
- Lauerman , L.H. , Chilina , A.R. , Closser , J.A. and Johansen , D. 1995 . Avian mycoplasma identification using polymerase chain reaction amplicon and restriction fragment length polymorphism analysis . Avian Diseases , 39 : 804 – 811 .
- Marois , C. , Dufour-Gesbert , F. and Kempf , I. 2002 . Polymerase chain reaction for detection of Mycoplasma gallisepticum in environmental samples . Avian Pathology , 31 : 163 – 168 .
- Rasmussen , R. 2001 . “ Quantification on the LightCycler ” . In Rapid Cycle Real-time PCR—Methods and Applications , Edited by: Meuer , S. , Wittwer , C. and Nakagawara , K. 21 – 34 . Heidelberg : Springer-Verlag .
- Salisch , H. , Hinz , K.-H. , Graack , H.-D. and Ryll , M. 1998 . A comparison of a commercial PCR-based test to culture methods for detection of Mycoplasma gallisepticum and Myoplasma synoviae in concurrently infected chickens . Avian Pathology , 27 : 142 – 147 .
- Salisch , H. , Ryll , M. , Hinz , K.-H. and Neumann , U. 1999 . Experiences with multispecies polymerase chain reaction and specific oligonucleotide probes for the detection of Mycoplasma gallisepticum and Mycoplasma synoviae . Avian Pathology , 28 : 337 – 344 .
- Silveira , R.M. , Fiorentin , L. and Marques , E.K. 1996 . Polymerase chain reaction optimization for Mycoplasma gallisepticum and M. synoviae diagnosis . Avian Diseases , 40 : 218 – 222 .
- Wittwer , C. 2001 . “ Rapid cycle real-time PCR: methods and applications ” . In Rapid Cycle Real-time PCR—Methods and Applications , Edited by: Meuer , S. , Wittwer , C. and Nakagawara , K. 1 – 8 . Heidelberg : Springer-Verlag .
- Wittwer , C.T. , Ririe , K.M. , Andrew , R.V. , David , D.A. , Gundry , R.A. and Balis , U.J. 1997 . The LightCycler: a microvolume multisample fluorimeter with rapid temperature control . BioTechniques , 22 : 176 – 181 .