Abstract
Viral population dynamics of very virulent infectious bursal disease virus (vvIBDV) field strains isolated in the Iberian Peninsula since the first outbreak in the 1990s have been analysed. Low levels of genetic variability and a global purification selection pattern were reported in 480 base pairs of the hypervariable region of the VP2 gene, indicating a lack of a selection-driven immune escape in the evolutive pathway of the virus. The viral population structure of vvIBDV strains in the Iberian Peninsula showed a strong relationship between geography and phylogeny, with two main groups observed. A global comparison among vvIBDV strains also showed an association with sequences from the same country. The low variability, the strong purifying selection and the geographical pattern observed point to a picture where the virus evolves slowly, occupying the same geographical niche for a long time. The scenario depicted fits well with the biological features of the virus: being able to remain viable for long periods of time due to a strong environmental resistance, and as an immunosuppressive agent, capable per se of annihilating temporally the immune system of the host.
Introduction
Infectious bursal disease (IBD) or “Gumboro disease” was reported for the first time nearly 50 years ago. However, nowadays it is considered to be endemic in most of the poultry-producing countries, and it is one of the major causes for economic losses in the poultry industry (Van den Berg et al., Citation2000). Infectious bursal disease virus (IBDV; genus Avibirnavirus, Fam. Birnaviridae) is the aetiological agent of this acute and highly contagious viral infection that only affects young chickens (Müller et al., Citation1979). Two serotypes (1 and 2) of IBDV have been recognized, but only serotype 1 has been demonstrated to be pathogenic (Lukert & Saif, Citation1997). IBDV particles have a non-enveloped, icosahedral capsid and present a bi-segmented (A and B) dsRNA genome. The larger segment A (around 3.4 kb) generates VP2, VP3, VP4 and VP5 proteins, and the smaller genome segment B (around 2.8 kb) encodes the viral RNA polymerase VP1 (Van den Berg, Citation2000). The external surface of the capsid is composed by trimeric sub-units formed by the VP2 protein that has been identified as the host-protective antigen (Brown et al., Citation1994; Cui et al., Citation2003; Müller et al., Citation2003; Van den Berg, Citation2000). Monoclonal neutralizing antibodies have been shown to bind to VP2, within a central hypervariable region comprising a highly hydrophobic region with two small hydrophilic peaks at each terminus, between amino acid residues 206 and 350 (Bayliss et al., Citation1990). Because it contains antigenic determinants and virulence markers, the VP2 protein has been commonly used in phylogenetic studies (Van den Berg et al., Citation2004).
In 1987, very virulent strains of infectious bursal disease virus (vvIBDV) were isolated in Europe, and quickly spread to Africa, Asia and South America (Lukert & Saif, Citation1997). Numerous reports of these acute IBD cases indicated that vvIBDV exhibited a high genetic and antigenic homogeneity (Van den Berg et al., Citation2004, and references cited therein). In the Iberian Peninsula, the first acute IBD outbreaks were reported in the early 1990s, and the pathologic and genetic studies of the isolated viruses demonstrated vvIBDV to be involved (Pagès-Mante et al., Citation1991; Majó et al., Citation2002). In 2002, a second outbreak of acute IBD started in the north-east region of Spain, and rapidly spread through the whole peninsula (Dolz et al., Citation2005). Genetically, the vvIBDV isolates of the 2002 outbreak were closely related with the ones isolated in the early 1990s. Since then, despite the intensive vaccination programmes and the strict biosecurity measures implemented, vvIBDV strains have become endemic in the Iberian Peninsula and acute IBD cases frequently occur.
Since the usage and availability of nucleic acid sequences have been extended in the veterinary field, numerous studies have characterized IBDV isolates all over the world (i.e., Brown et al., Citation1994; Etterradossi et al., Citation2000; Zierenberg et al., Citation2000; Liu et al., Citation2002; Rudd et al., Citation2002; Hernandez et al., Citation2006), generally following outbreak events. However, few studies have attempted to investigate population structure and viral dynamics both at a longitudinal and geographical scale seeking for clarification of IBDV evolution. Since 2002, an ongoing epidemiological surveillance has been carried out at the Centre de Recerca en Sanitat Animal (Bellaterra, Spain) by means of clinical samples submitted from suspicious IBD cases. The present work analyses the viral population dynamics of vvIBDV field strains isolated in the Iberian Peninsula since the first outbreak in the 1990s. A global comparison among vvIBDV strains reported worldwide is also assessed.
Materials and Methods
Isolates
vvIBDV strains detected from clinical samples (bursa of Fabricius) submitted to the Centre de Recerca en Sanitat Animal between 2002 and 2009 were included (n=77). In addition, sequences from vvIBDV strains isolated in the 1990s (n=3) and 2002 (n=22) outbreaks, published in previous studies (Majó et al., Citation2002; Dolz et al., Citation2005), were included in the comparison. Names, origins and accession numbers of the sequences included in the study are reported in (n=102). In addition, 65 sequences available at GenBank (Supplementary , online only) from different countries around the world were used to study global geographic patterns in vvIBDV.
Table 1. Origin, year of isolation and accession number for the 102 partial VP2 Iberian sequences used in the study.
Polymerase chain reaction and sequencing
For all suspicious cases, viral RNA was extracted with the NucleoSpin RNA Virus kit (Macherey-Nagel, Düren, Germany) and amplified using a reverse transcriptase-polymerase chain reaction assay for the VP2 gene, as previously described by Dolz et al. (Citation2005). Sequences of 480 base pairs from each of the hypervariable region in the VP2 gene were obtained. The analysed region included 159 amino acid residues, from positions 202 to 360, containing the hydrophilic peaks responsible for the IBDV antigenicity.
Phylogenetic and population studies
The phylogenetic relationships among the 102 Iberian IBDV sequences were estimated with a maximum parsimony network of the nucleotide sequences by the statistical parsimony method of Templeton et al. (Citation1992) using the TCS computer program (Clement et al., Citation2000). Patterns of nucleotide diversity distribution among regions were estimated by a hierarchical nested analysis of molecular variance (AMOVA; Excoffier et al., Citation1992) of the frequency distribution of sequences and their pairwise divergence at three hierarchical levels: within populations (ϕst), among populations within groups (ϕsc) and among groups (ϕct). Two analyses were performed using Arlequin software (version 3.0: Excoffier et al., Citation2005): one considering a temporal design and a second considering a geographical design. The temporal design separated sequences among outbreaks: during the 1990s, in 2002, and after 2002. In the geographic design, two large areas that contained different regions of the Iberian Peninsula were considered: the Mediterranean region (Catalunya, València, lles Balears, Murcia, Andalucía) together with the north-eastern region (Aragón, Navarra, Rioja; M/E region); and the Atlantic and central-western regions (Portugal, Galicia, Castilla-la-Mancha, Castilla-León; A/W region). A matrix of nucleotide divergence between regions was calculated using MEGA 4 (Tamura et al., Citation2007) and the gamma Tamura–Nei model. This distance matrix of nucleotide divergence was used to perform principal coordinate (PCO) analysis using the PCO program (Anderson, Citation2003). The PCO analysis determines a small set of synthetic variables (the coordinates) that explain the variability of the original dataset. The first principal coordinate accounts for as much variability in the data as possible, and each following coordinate accounts for as much of the remaining variability as possible. Finally, a neighbour-joining tree including Iberian and world vvIBDV nucleotide sequences was constructed using MEGA 4 (Tamura et al., Citation2007) and the gamma Tamura–Nei model. The confidence of internal branches was estimated by means of 1000 bootstrap replicates.
Selection pressure
The existence of selective pressures along partial VP2 sequences was assessed by calculating the difference between non-synonymous (dN) and synonymous (dS) rates (dN – dS) both globally and per codon using the modified Nei–Gojobori method by MEGA 4 software. These differences were calculated with the SNAP web utility (http://hivweb.lanl.gov/content/hivdb/SNAP/WEBSNAP/SNAP.html).
Results
Nucleotide and amino acid variability
Among the 102 sequences of 480 base pairs each, 68 variable nucleotide positions were reported, most of which (n=55) were synonymous and only 13 were non-synonymous. The majority of the sequence changes were point mutations; more than one-half of the nucleotide (53%, 36 out of 68) and amino acid (61%, eight out of 13) changes were detected in only one or two strains. Only five nucleotide mutations were present in a large number of sequences (35C → T, 71G → A, 116C → T, 295G → A, 362G → A); four were silent, and only the nucleotide change in position 295 (VP2 position 1027) caused an amino acid substitution in residue 98 (VP2 position 299) (S → N). The percentage of nucleotide identity ranged from 94.7 to 100%.
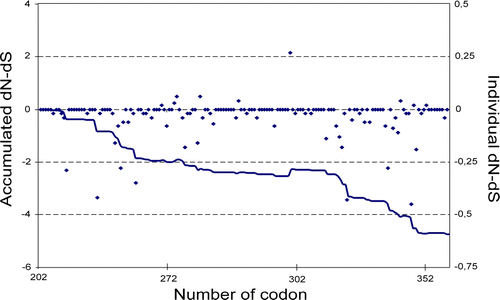
Most of the amino acid codons showed a neutral selection (). Codons identified under negative selection (55 residues, 34.6%) were mainly grouped in two regions of the VP2 protein (residues 212 to 272, and residues 311 to 351). Few positions (5.7%) showed a positive selection pressure; nearly all presented a value close to a zero, except position 299 where a frequent amino acid substitution was reported.
Figure 1. Accumulated (line, values referred to the left axis) and individual (points, values referred to the right axis) differences between non-synonymous and synonymous rates (dN – dS) for the VP2 gene. Numbers on the horizontal axis represent the amino acid position.
Temporal and geographical analyses of Iberian vvIBDV sequences
The statistical parsimony network of the IBDV () reflected the low number of mutations previously described in the nucleotide and amino acid variability: a maximum of 14 steps separated the most distant Iberian strains. Isolates from the first outbreak in the 1990s were placed closely in the network, whereas sequences from the second outbreak in 2002, and more recent ones, were distributed all over the network, indicating the lack of a temporal structure. In contrast, nearly all of the strains from the Atlantic and north-western Iberian Peninsula (Galicia, Castilla-León, Portugal) clustered together and separated from the Mediterranean and north-eastern ones (), which correlates with the nucleotide positions described above. This point was also supported by the AMOVA results (), where the temporal structure of the Iberian strains was not supported (Φct=0.0775, P=0.4909), whereas a significant percentage of variation (24.45%) was allocated between the M/E and A/W groups (Φct=0.2445, P=0.0091). In addition, the neighbour-joining tree among regions clearly separated the A/W regions from the M/E ones (data not shown). Finally, the first axis of the PCO analysis placed the A/W regions in the positive scores and the M/E regions in the negative scores ().
Figure 2. Statistical parsimony network based on the nucleotide sequences. Every branch represents a nucleotide mutation: black circles, mutations with no frequency; ovals, strains isolated during the 1990s; dashed squares, strains isolated in 2002; solid squares, strains isolated after 2002.
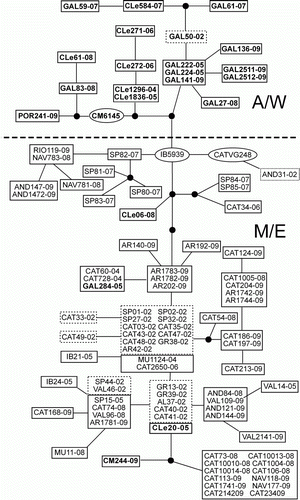
Figure 3. Iberian Peninsula map, showing the relative frequencies of viral strain group Atlantic/West (A/W, black) and Mediterranean/East (M/E, grey) in every geographical area.
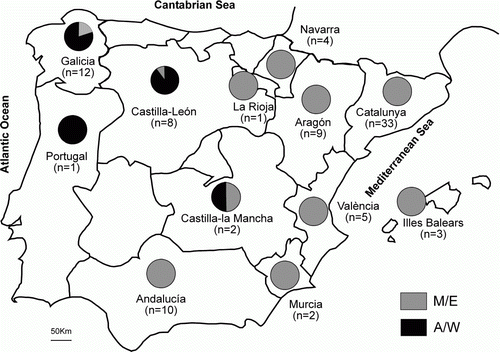
Figure 4. PCO analysis. Numbers between brackets indicate the percentage of variation explained by every coordinate.
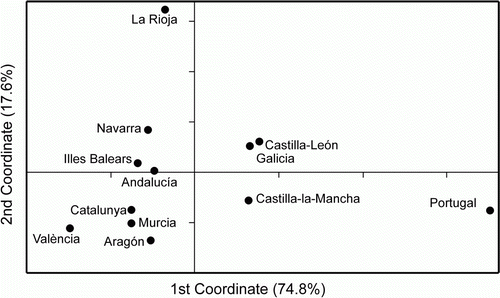
Table 2. AMOVA results.
Global geographic distribution patterns of vvIBDV sequences
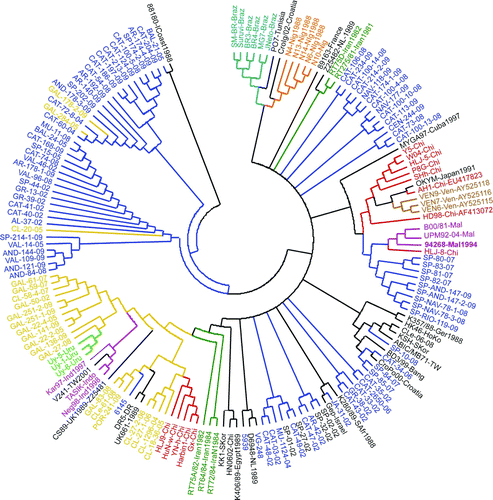
When Iberian sequences were compared with vvIBDV from other parts of the world, a geographic clustering tendency was observed () despite the Iberian sequences being split into distinct branches. Nearly all sequences from the A/W Iberian group clustered together in a single branch, except three sequences that, similarly to the statistical parsimony network, were placed in the M/E group. Sequences from the M/E group clustered into several branches along the tree, but one-half (36 out of 77) clustered into a single homogeneous branch. Several terminal branches showed a strong association with sequences from the same country. In several countries all of the sequences clustered into a single clade (Indonesia, Uruguay, Venezuela, Malaysia, Nigeria, Brazil), and in other cases were distributed in two clades (Iran, China).
Figure 5. Circular neighbour-joining tree based on 102 Iberian and 65 global vvIBDV nucleotide sequences. Colours indicate different geographic origins.
Discussion
In contrast with other dsRNA viruses, high nucleotide identity has characterized vvIBDV strains worldwide (>94%; Etterradossi et al., Citation2000). Indeed, the Iberian vvIBDV field strains showed low levels of genetic variability, alongside a global purification selection pattern in the studied fragment of the VP2 gene. Negative or purifying selection is reported when synonymous mutations (a nucleotide mutation that does not produce an amino acid change) outnumber non-synonymous mutations (a nucleotide mutation that produces an amino acid change) for a certain codon (Nei & Kumar, Citation2000). The negatively selected codons were mainly located in two regions roughly coincident with the major hydrophilic peaks (210 to 225 and 312 to 324) reported for the VP2 gene (Bayliss et al., Citation1990). Despite the regions under purifying selection being wider than the hydrophilic peaks, this is most probably a consequence of a hitchhiking effect commonly observed in regions under strong functional constraint (Barton, Citation2000). Considering that any change within these hydrophilic regions may potentially cause a change in the antigenic features of an isolate (Letzel et al., Citation2007; Jackwood & Sommer-Wagner, Citation2011), a strong purifying selection pattern as observed in this study could indicate a lack of a selection-driven immune escape in the evolutive pathway of the virus.
Partial VP2 sequences have been widely used in routine diagnosis and epidemiological studies worldwide (Jackwood & Sommer-Wagner, Citation2011). Nonetheless, in order to accurately infer an IBDV phenotype, genome fragments including segments A and B need to be analysed. Alternatively, in vivo assays should be carried out to confirm the very virulent phenotype. Keeping this limitation in mind, field isolates in the present study were classified as vvIBDV when clinical signs of acute IBD were observed in the birds and partial VP2 sequences clustered in the vvIBDV branch (data not shown). Many molecular studies in Gumboro have been restricted to the regional level: Spain (Pagès-Mante et al., Citation1991; Majó et al., Citation2002; Dolz et al., Citation2005), Uruguay (Hernandez et al., Citation2006), Croatia (Lojkic et al., Citation2008), People's Republic of China (Liu et al., Citation2002) or Brazil (Fernandes et al., Citation2009). In contrast, few studies seek for evolutive relationships on a global scale among vvIBDV strains (Eterradossi et al., Citation2000; Zierenberg et al., Citation2000). In order to determine the population structure present among vvIBDV strains, this study adopted two approaches: a general comparison among most (n=65) of the vvIBDV strains described worldwide, and a regional longitudinal analysis including 102 vvIBDV Iberian strains isolated during a 20-year period. According to all of the performed analyses—statistical parsimony network, AMOVA and PCO analysis—the viral population structure of vvIBDV strains in the Iberian Peninsula nowadays follows a geographic pattern, with two main groups observed: A/W and M/E. Nearly all of the strains isolated after 2002 clustered according to their geographical origin. When compared on a global scale, the vvIBDV strains clustered with a similar geographical-based pattern, despite some minor incongruence being shown, indicating a strong association between geography and viral diversity.
Very virulent IBDV exhibit a so-called vvIBDV genetic fingerprint in the VP2 hypervariable region at amino acid residues 222 (Ala), 242 (Ile), 256 (Ile), 294 (Ile) and 299 (Ser) (Van den Berg et al., Citation2004). However, the presence of a serine in the residue 299 was not a constant trait, although it is believed to be a common feature among European vvIBDV field strains (Domanska et al., Citation2004). Comparing the European and African vvIBDV strains, Etterradossi et al. (Citation2000) found an asparagine (N) for an African isolate at position 299 while the remaining French, British and Dutch isolates presented a serine (S) in this position. Interestingly, in our study, isolates from the A/W group presented the serine residue, while most isolates from the M/E group showed asparagine. Nevertheless, two of the M/E isolates that do not present the asparagine residue were isolated during the first vvIBDV reports in the 1990s. Whether a new strain arrived from Africa and spread only in the Mediterranean area after the 1990s, or a de novo mutation restricted to the Mediterranean region occurred between the first (1990) and the second (2002) outbreaks, remains an open question.
Taken together, the low variability, the strong purifying selection and the geographical pattern observed point to a picture where the virus evolves slowly, occupying the same geographical niche for a long time. The depicted scenario fits in well with the biological features of the virus: being able to remain viable for long periods of time due to a strong environmental resistance (Van den Berg, Citation2000); and as an immunosuppressive agent, being capable per se of annihilating temporally the immune system of the host. Due to the strong nature of IBDV and the widespread distribution of the virus, prevention and control of this disease may be achieved by a combination of biosecurity measures and the correct vaccination programme.
cavp_a_682562_sup_26380255.doc
Download MS Word (76.5 KB)Acknowledgements
K. Bertran held an FPU Pre-Doctoral grant from the Spanish Ministry of Education. M. Cortey received a grant from the Government of Catalonia.
References
- Anderson , M.J. 2003 . PCO: a FORTRAN computer program for principal coordinate analysis Department of Statistics . University of Auckland , New Zealand .
- Barton , N.H. 2000 . Genetic hitchhiking . Philosophical Transactions of the Royal Society B , 355 : 1553 – 1562 .
- Bayliss , C.D. , Spies , U. , Shaw , K. , Peters , R.W. , Papageorgiou , A. , Muller , H. and Boursnell , M.E. 1990 . A comparison of the sequences of segment A of four infectious bursal disease virus strains and identification of a variable region in VP2 . Journal of General Virology , 71 : 1303 – 1312 .
- Brown , M.D. , Green , P. and Skinner , M.A. 1994 . VP2 sequences of recent European “very virulent” isolates of infectious bursal disease virus are closely related to each other but are distinct from those of “classical” strains . Journal of General Virology , 75 : 675 – 680 .
- Clement , M. , Posada , D. and Crandall , K.A. 2000 . TCS: a computer program to estimate gene genealogies . Molecular Ecology , 9 : 1657 – 1660 .
- Cui , X. , Nagesha , H.S. and Holmes , I.H. 2003 . Identification of crucial residues of conformational epitopes on VP2 protein of infectious bursal disease virus by phage display . Journal of Virological Methods , 109 : 75 – 83 .
- Dolz , R. , Majó , N. , Ordoñez , G. and Porta , R. 2005 . Viral genotyping of infectious bursal disease viruses isolated from the 2002 acute outbreak in Spain and comparison with previous isolates . Avian Diseases , 49 : 332 – 339 .
- Domanska , K. , Mato , T. , Rivallan , G. , Smietanka , K. , Minta , Z. , de Boisseson , C. , Toquin , D. , Lomniczi , B. , Palya , V. and Eterradossi , N. 2004 . Antigenic and genetic diversity of early European isolates of Infectious bursal disease virus prior to the emergence of the very virulent viruses: early European epidemiology of Infectious bursal disease virus revisited? . Archives of Virology , 149 : 465 – 480 .
- Etterradossi , N. , Arnauld , C. , Tekaia , F. , Toquin , D. , Le Coq , H. , Rivallan , G. , Guittet , M. , Domenech , J. , van den Berg , T.P. and Skinner , M.A. 2000 . Antigenic and genetic relationship between European very virulent infectious bursal disease viruses and an early West-African isolate . Avian Pathology , 28 : 36 – 46 .
- Excoffier , L. , Smouse , P.E. and Quattro , J.M. 1992 . Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data . Genetics , 131 : 479 – 491 .
- Excoffier , L. , Laval , G. and Schneider , S. 2005 . Arlequin ver. 3.0: An integrated software package for population genetics data analysis . Evolutionary Bioinformatics Online , 1 : 47 – 50 .
- Fernandes , M.J. , Simoni , I.C. , Vogel , M.G. , Harakava , R. , Rivas , E.B. , Oliveira , M.B. , Kanashiro , A.M. , Tessari , E.N. , Gama , N.M. and Arns , C.W. 2009 . Molecular characterization of Brazilian infectious bursal disease virus isolated from 1997 to 2005 . Avian Diseases , 53 : 449 – 454 .
- Hernandez , M. , Banda , A. , Hernandez , D. , Panzera , F. and Perez , R. 2006 . Detection of very virulent strains of infectious bursal disease virus (vvIBDV) in commercial broilers from Uruguay . Avian Diseases , 50 : 624 – 631 .
- Jackwood , D.J. and Sommer-Wagner , S.E. 2011 . Amino acids contributing to antigenic drift in the infectious bursal disease Birnavirus (IBDV) . Virology , 409 : 33 – 37 .
- Letzel , T. , Coulibaly , F. , Rey , F.A. , Delmas , B. , Jagt , E. , Van Loon , A.A.M.W. and Mundt , E. 2007 . Molecular and structural bases for the antigenicity of VP2 of infectious bursal disease virus . Journal of Virology , 81 : 12827 – 12835 .
- Liu , J. , Zhou , J. and Kwang , J. 2002 . Antigenic and molecular characterization of recent infectious bursal disease virus isolates in China . Virus Genes , 24 : 135 – 147 .
- Lojkic , I. , Bidin , Z. and Pokric , B. 2008 . Sequence analysis of both genome segments of three Croatian infectious bursal disease field viruses . Avian Diseases , 52 : 513 – 519 .
- Lukert , P.D. and Saif , Y.M. 1997 . “ Infectious bursal disease ” . In Diseases of Poultry 10th , Edited by: Calnek , B.W. , Barnes , H.J. , Beard , C.W. , McDougald , L.R. and Saif , Y.M. 721 – 738 . Ames : Iowa State University Press .
- Majó , N. , El-Attrache , J. , Banda , A. , Villegas , P. , Ramis , A. , Pagès , A. and Ikuta , N. 2002 . Molecular characterization of Spanish infectious bursal disease virus field isolates . Avian Diseases , 46 : 859 – 868 .
- Mardassi , H. , Khabouchi , N. , Ghram , A. , Namouchi , A. and Karboul , A. 2004 . A very virulent genotype of infectious bursal disease virus predominantly associated with recurrent infectious bursal disease outbreaks in Tunisian vaccinated flocks . Avian Diseases , 48 : 829 – 840 .
- Muller , H. , Scholtissek , C. and Becht , H. 1979 . The genome of infectious bursal disease virus consists of two segments of double-stranded RNA . Journal of Virology , 3 : 584 – 589 .
- Müller , H. , Islam , M.d.R. and Raue , R. 2003 . Research on infectious bursal disease-the past, the present and the future . Veterinary Microbiology , 97 : 153 – 165 .
- Nei , M . & Kumar , S . 2000 . Molecular Evolution and Phylogenetics . Oxford University Press , New York , 333 pp.
- Pagès-Mante , A. , Pujol , P. , Duran , D. , Fernandez , F. and Hernando , A. 1991 . Estudios clinicos y laboratoriales de una cepa de la enfermedad de Gumboro (IBD) aislada en Baleares . Medicina Veterinaria , 9 : 476 – 480 .
- Rudd , M.F. , Heine , H.G. , Sapats , S.I. , Parede , L. and Ignjatovic , J. 2002 . Characterisation of an Indonesian very virulent strain of infectious bursal disease virus . Archives of Virology , 147 : 1303 – 1322 .
- Tamura , K. , Dudley , J. , Nei , M. and Kumar , S. 2007 . MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0 . Molecular Biology and Evolution , 24 : 1596 – 1599 .
- Templeton , A.R. , Crandall , K.A. and Sing , C.F. 1992 . A cladistic analyisis of phenotypic association with haplotypes inferred from restriction endonucleases mapping and DNA sequence data. III. Cladogram estimation . Genetics , 132 : 619 – 633 .
- Van den Berg , T.P. 2000 . Acute infectious bursal disease in poultry: a review . Avian Pathology , 29 : 175 – 194 .
- Van den Berg , T.P. , Eterradossi , N. , Toquin , D. & Meulemans , G. 2000 . Infectious Bursal Disease (Gumboro Disease) . In Diseases of Poultry: World Trade and Public Health Implications, OIE Scientific and Technical Review (pp. 509 – 543 ). Paris , , France .
- Van den Berg , T.P. , Morales , D. , Eterradossi , N. , Rivallan , G. , Toquin , D. , Raue , R. , Zierenberg , K. , Zhang , M.F. , Zhu , Y.P. , Wang , C.Q. , Zheng , H.J. , Wang , X. , Chen , G.C. , Lim , B.L. and Müller , H. 2004 . Assessment of genetic, antigenic and pathotypic criteria for the characterization of IBDV strains . Avian Pathology , 33 : 470 – 476 .
- Zierenberg , K. , Nieper , H. , Van den Berg , T.P. , Ezeokoli , C.D. , Voss , M. and Muller , H. 2000 . The VP2 variable region of African and German isolates of infectious bursal disease virus: comparison with very virulent, ‘classical’ virulent, and attenuated tissue culture-adapted strains . Archives of Virology , 145 : 113 – 125 .