
Abstract
During investigations into the outbreak of duck Tembusu virus (DTMUV) infection in 2011 in China, a DTMUV strain (DTMUV-AH2011) was isolated from the affected ducks. The length of the genome of the DTMUV-AH2011 strain was found to be 11,064 nucleotides and to possess 10,278 nucleotides of one open reading frame (ORF), flanked by 94 nucleotides of the 5′ non-translated region (NTR) and 692 nucleotides of the 3′ NTR. In comparison with five fully sequenced TMUV genomes, the genome of DTMUV-AH2011 had a 74 nucleotide insertion in the 3′ NTR. Comparison of the DTMUV-AH2011 fully deduced amino acid sequences with those of other Tembusu virus strains reported recently in China showed they had a highly conserved polyprotein precursor, sharing 98.9% amino acid identities, at least. The overall divergences of amino acid substitutions were randomly distributed among viral proteins except for the protein NS4B, the protein NS4B was unchanged. Knowledge of the biological characters of DTMUV and the potential role of the insertion in the 3′ NTR in RNA replication will be useful for further studies of the mechanisms of virus replication and pathogenesis.
Introduction
Since April 2010, a novel infectious agent emerged in China and caused extensive epidemics in egg-laying ducks. The affected ducks (including Pekin duck, Cherry Valley Pekin duck, Shaoxing duck, Jinyun duck, Longyan duck, Jinding duck and Khaki-Campbell duck) showed severe drops in egg production, declines in feed uptake, paralysis, severe oophoritis and regression and mortality (Su et al., Citation2011; Tang et al., Citation2012; Yun et al., Citation2012a). Hyperaemia, haemorrhage, degeneration, distortion and lymphocyte infiltration in ovaries and interstitial inflammation in the portal areas of livers were the main pathological changes observed consistently in almost all diseased ducks. Based on these changes and biological characters, the disease was named duck haemorrhagic oophoritis (DHO), and the emerging agent, a novel virus was shown to belong to a new genotype of Tembusu virus (TMUV) and was designated duck Tembusu virus (DTMUV) (Cao et al., Citation2011b; Yun et al., Citation2012b).
TMUV is a mosquito-borne flavivirus. It is a member of the Ntaya virus serogroup of the genus Flavivirus, family Flaviviridae (Lindenbach & Rice, Citation2003). TMUV was first described in 1957 in Kuala Lumpur, Malaysia. A chick-origin TMUV strain, originally designated as Sitiawan virus, was reported later causing encephalitis and retarded growth in broiler chicks (Kono et al., Citation2000). Clinical investigations demonstrated the DTMUV could also affect geese. A report about TMUV-SDHS isolated from house sparrows showed a close relationship with the YY5 strain, and this latter strain was highly pathogenic to ducks. This finding may play an important role in transmitting the virus among different species (Tang et al., Citation2012). In view of the epidemic characteristics of the DTMUV, as well as the hazard and losses that it caused in the duck industry, more attention to the virus is needed.
Like other flaviviruses, DTMUVs are spherical, enveloped viruses of approximately 40–60 nm in diameter. Flavivirus genome RNA has a type I cap structure at its 5′ terminus but lacks a polyadenylated tail at its 3′ terminus (Dong et al., Citation2007). The genome consists of single-stranded, positive-sense RNA of approximately 11,000 nt with a long open reading frame (ORF) encoding one large putative polyprotein. The putative polyprotein is co- and post-translationally cleaved by viral and cellular proteases into three structural proteins [capsid (C), membrane (M) and envelope (E)] and seven nonstructural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B and NS5) that function in virus replication, proteolysis and virus maturation (Lindenbach & Rice, Citation2003). The ORF is flanked by 5′ and 3′ non-translated regions (NTRs). Both NTRs form specific secondary stem-loop structures and functional domains that are essential for virus replication and assembly (Charlier et al., Citation2002; Markoff, Citation2003; Gritsun & Gould, Citation2006).
We examined the genomic traits of DTMUV to provide the basic information for improving disease surveillance, phylogenetic classification and development of sensitive and specific detection methods based on conserved sequences. Taken together, our results showed that the overall genome organization of DTMUV-AH2011 was similar to that of known flaviviruses, but it also had its unique characteristic.
Materials and Methods
Clinical and pathological investigations.
We were notified by the duck farmers of an unknown egg-drop disease. Clinical examination and the production records were checked for several affected flocks in Anhui province in China since February 2011. To exclude avian influenza virus infection, serum samples were collected from the affected flocks, and antibody titres against H5 and H9 subtype avian influenza A virus were tested with hemagglutination inhibition assays according to the standard method. Ducks in the affected flocks showing morbidity or death within 24 hours were taken to our laboratory for necropsy.
Virus isolation.
Specimens of spleen and ovary tissues with apparent clinical signs from the affected flocks were collected and processed for virus isolation. Briefly, the tissues were homogenized in sterile phosphate-buffered saline (pH 7.2) to give a 20% suspension (w/v) (Cao et al., Citation2011b; Yun et al., Citation2012a). The samples were first tested by reverse transcription polymerase chain reaction (RT-PCR) using primers ED3F/ED3R (DTMUV-ED3F: ACTCCATGGATCAGGGTTTGAAGCTGAAA, DTMUV-ED3R: CGGACTCGAGTGTGCTCCCACTTCTATG). Subsequently, positive samples were used for virus isolation. The filtered homogenates was inoculated into 9-day-old specific pathogen-free (SPF) embryonated chicken eggs (0.2 mL/embryo) via the yolk sac. The embryos were carefully monitored. When embryos died between 24 and 120 hours post-inoculation, the allantoic fluids were harvested and stored at −20°C until they were used for another round of inoculations or for molecular analyses. The isolated virus was cultured for at least three passages for amplification and sequencing. At the eighth passage, virus titre of the allantoic fluid was evaluated by the 50% embyro lethal dose (ELD50) on 9-day-old SPF embryonated chicken eggs.
Virus titre of the cell adapted DTMUV.
For cell culture passage, the virus was cultured in baby hamster kidney (BHK-21) cell line. BHK-21 cells were grown in Dulbecco's modified Eagle's medium (Invitrogen, Grand Island, NY, USA) supplemented with 10% heat inactivated foetal calf serum, 100 U penicillin/mL and 100 µg streptomycin/mL at 37°C under 5% CO2 atmosphere. Cell monolayers were infected with allantoic fluids from the fourth passage of the SPF embryonated chicken eggs (0.1 mL of a 1:10 dilution in medium), and the cells were observed daily to check for development of cytopathic effects (CPE). The cells were freeze–thawed three times after 72 h post-inoculation, cellular debris was removed by low-speed centrifugation and the supernatant fluid was re-inoculated onto fresh BHK-21 cells until the typical CPE of DTMUV appeared. At the fifth passage, virus titre of the cell culture supernatants was determined by plaque assay as described by others (Okuno et al., Citation1984; Yun et al., Citation2009). Cells were then fixed with formaldehyde and stained with crystal violet to visualize the plaques. The infected cells were examined by electron microscopy (data not shown).
RNA extraction and RT-PCR.
The viral RNA was extracted from 200 μL allantoic fluids from the fourth passage of the SPF embryonated chicken eggs by using the MiniBEST viral RNA Extraction kit (TaKaRa Biotechnology, Dalian, China) according to the manufacturer's protocol. The RNA was eluted in a final volume of 50 μL of elution buffer and stored at −80°C until further use. RNA was used as the template for first-strand cDNA synthesis using PrimeScript® reverse transcriptase (TaKaRa Biotechnology, Dalian, China) with random hexanucleotide primer according to the manufacturer's instructions. The cDNA was subsequently stored at −20°C.
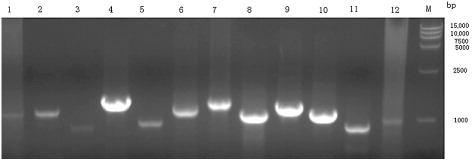
In order to amplify the completed genome, 12 pairs of primers () were designed according to the BYDV and the YY5 strain genome sequences (GenBank access number: JF312912 and JF270480, respectively). PCR was carried out with 2~3μL cDNA template. Products of amplification were evaluated by electrophoresis in a 1% agarose gel. DNA bands visualized by GoldViewTM Nucleic acid staining and UV transillumination were excised from the agarose gel and purified using the Takara Agarose Gel DNA Purification kit according to manufacturer's protocol (TaKaRa Biotechnology, Dalian, China).
Table 1. Primers used for synthesis and amplification of cDNA from DTMUV genome.
Sequencing.
Purified DNA products were ligated into the pMD18-T Easy vector according to manufacturer's instructions (TaKaRa Biotechnology), and ligated plasmids were introduced into DH5α competent Escherichia coli through chemical transformation. Transformed cells were spread onto LB/ampicillin plates. Individual colonies were used to inoculate 5 mL LB/ ampicillin broth. After overnight incubation at 37°C with shaking, plasmids were extracted from 1 to 2 mL bacterio-fluids using the Takara MiniBEST Plasmid Purification kit (TaKaRa Biotechnology). To verify the presence of DNA inserts, extracted plasmids were digested with EcoRI and Sal I, and examined by agarose gel electrophoresis.
The entire genome sequence of DTMUV-AH2011 was generated from 12 overlapping fragments. Overlapping consensus sequences were assembled using the SeqMan programs to generate contiguous full-genome sequences. Analysis of nucleotide and deduced amino acid sequence identities were performed by LASERGENE analysis software (DNASTAR Inc., Madison, WI, USA). This software was also used to predict the entire ORF for the polyprotein of TMUV as well as to determine the 5′ and 3′ NTR. Deduced amino acid sequences were analyzed and potential cleavage sites were predicated by using the software program SignalP 3.0.
RNA secondary structure analysis.
The RNA secondary structural model of the viral 3′ NTR of the DTMUV genomic RNA was constructed by the mfold program, and the energy minimization program of Mfold website server (http://mfold.rna.albany.edu/?q=mfold/RNA-Folding-Form) under default folding conditions (37°C, 1 M NaCl, no divalent ions and no limit on distance between paired bases) was used for constructing secondary structural model (Zuker, Citation2003). To predict the RNA secondary structures more accurately, the genomic RNA of two DTMUV strains (DTMUV-AH2011 and YY5) was used. The portable, windows-type RNAviz version 2.0 software (http://rnaviz.sourceforge.net/) was used to draw or modify the RNA secondary structure for the visualization or publication quality of RNA secondary structure (De Rijk et al., Citation2003).
Multiple alignments and sequence analyses.
The complete genome of the DTMUV-AH2011 strain (GenBank access number: KJ958533) was analyzed for searching ORF and translating to amino acid sequence using Lasergene software package (DNASTAR). Percent sequences similarity/divergence was calculated using the MegAlign program found in the Lasergene analysis software (Thompson et al., Citation1997; Tamura et al., Citation2011). The nucleotides and deduced amino acid sequences of the individual viral proteins of the DTMUV-AH2011 strain were compared with those of 12 flaviviruses in Ntaya virus serogroup, including some TMUV strains reported recently in China and some Bagaza virus strains ().
Table 2. Detailed background information of 9 TMUV strains reported recently in China and three Bagaza virus strains used in this study.
Phylogenetic analysis.
To better understand the phylogenetic relationship between DTMUV-AH2011 and other flaviviruses, full-length genome sequences of other previously published flavivirus strains of varying serogroup isolated from different locations and sources, and across a number of years, were downloaded from GenBank, including sequences from Murray Valley encephalitis virus (MVEV), West Nile virus (WNV), Japanese encephalitis virus (JEV), Kunjin virus (KUNV), St. Louis encephalitis virus (SLEV), Dengue virus (DENV), Yellow fever virus (YFV), Powassan virus (POWV), Langat virus (LANV), Louping ill virus (LIV), Tick-borne encephalitis virus (TBEV), Culex flavivirus () and some other Tembusu-related strains reported recently in China. Neighbour-joining phylogenetic trees based on nucleotide sequences were constructed using Clustal program. The robustness of phylogenetic construction was evaluated by bootstrapping using 1000 replicates. In Ntaya virus serogroup, to study the phylogeny of the TMUV E and NS5 protein, the nucleotides sequences of the E protein and NS5 protein of TMUV and other flaviviruses were aligned and then analyzed by Neighbour-joining method with 1000 bootstrap replicates in the Clustal W program to produce phylogenetic trees (Larkin et al., Citation2007).
Table 3. Background information of 20 selected strains of flaviviruses used in this study.
Results
Clinical and pathological investigations.
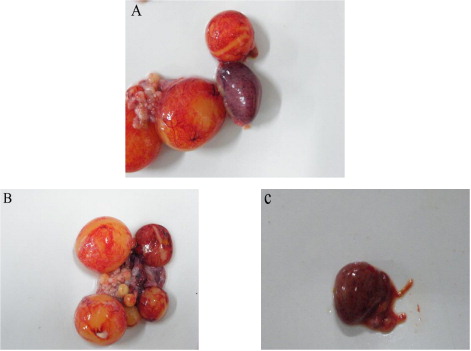
Since February 2011, many ducks developed the signs of DHO and died in several duck farms in Anhui province in China. The disease spread fast around the duck-producing regions and resulted in serious economic losses for the duck farms. The affected ducks manifested clinical signs of heavy egg drop, retarded growth, high fever and loss of appetite. As the disease progressed, some ducks exhibited an uncoordinated gait, and were reluctant or unable to walk, or even died. At necropsy, severe ovarian haemorrhage, oophoritis and regression were consistently observed, and ruptured ovarian follicles were also found. Enlarged spleens were occasionally found ( and ). All clinical signs and the pathologic changes of the affected ducks were similar to the clinical cases described by Su et al. (Citation2011).
Virus isolation and virus titre of the cell adapted DTMUV.
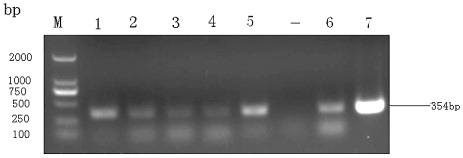
To investigate whether the disease was caused by DTMUV, specimens of spleen and ovary tissues from the affected flocks were collected, viral RNA was extracted from homogenates of the tissues, and tested by RT-PCR using primers specific for DTMUV. All the suspect clinical specimens were positive when primers specific for DTMUV (ED3F/ED3R) were used, and nucleotide sequencing confirmed that the infectious agent was the DTMUV. RT-PCR with the primers ED3F/ED3R resulted in the amplification of a 354 bp band, and some of the results are shown in .
In order to isolate the DTMUV, one of the viral positive filtered homogenates was inoculated into 9-day-old SPF embryonated chicken eggs (0.2 mL/embryo) via the yolk sac. All of the inoculated embryos died during 2–6 days post-inoculation with severely cutaneous haemorrhages. The allantoic fluids did not have haemagglutinating activity. The isolated virus was designated as DTMUV-AH2011. At the eighth passage, the 50% embyro lethal dose (ELD50) of the allantoic fluid was 10−3.84/0.2mL. In addition, most embryos died at the third day post-inoculation.
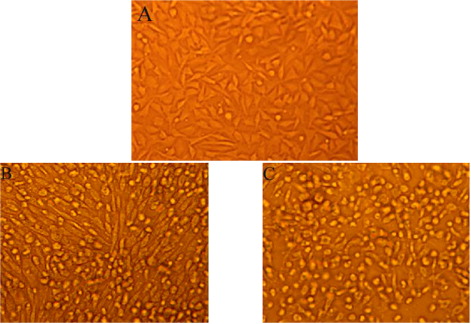
For cell culture identification, the allantoic fluids of the fourth passage of the SPF embryonated chicken eggs were collected and serially passaged in BHK-21 monolayer cells as described in Materials and Methods. At the fifth passage, the virus produced a marked CPE in the BHK-21 cells at 72 hours after infection, focal CPE appeared with the cells rounding up and floating free from the surface of the flask ( and ). The supernatant fluid had an infectivity titre of 2.8 × 105PFU/mL ().
Full-length genome and deduced amino acid sequence analysis.
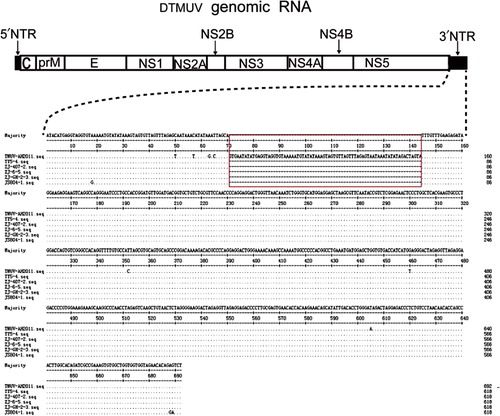
To further investigate the molecular characters of the DTMUV-AH2011 strain, the complete genome was amplified with several pairs of primers listed in (). On assembling these sequences, the length of the genome of the DTMUV-AH2011 strain was found to be 11,064 nucleotides and to possess 10,278 nucleotides of one ORF, flanked by 94 nucleotides of the 5′ NTR and 692 nucleotides of the 3′ NTR. The ORF encoded 3425 amino acids (aa) including three structural and seven non-structural proteins.
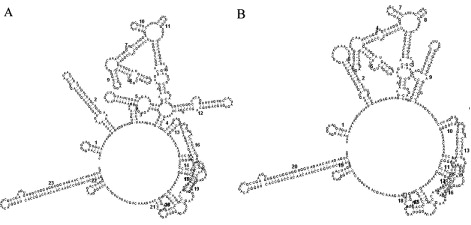
The phylogenetic analysis of the flavivirus genome showed that the isolated strain, DTMUV-AH2011, was genetically more similar to the Ntaya virus serogroup, especially some TMUV strains reported recently in China. The nucleotide sequences of the DTMUV-AH2011 strain were compared with those of 12 flaviviruses in Ntaya virus serogroup, including some TMUV strains reported recently in China and some Bagaza virus strains. In comparison with five fully sequenced DTMUV genomes currently available, we found that they had a high nucleotide sequence identity, all sharing approximately 97.7% nucleotide identities. In the case of 3′ NTR, the DTMUV-AH2011 strain had a 74 nucleotide insertion in the 3′ NTR. This insertion contained 69-nt duplicated nucleotides and was located at the 5′-proximal part of the 3′ NTR, which was located 69-nt downstream of the translation stop codon in the forward direction (). To exclude whether the insertion was an artefact, viral RNA was extracted from the homogenates of corresponding tissues, and the 3′ NTR was amplified by RT-PCR and the amplicon was sequenced directly. As the results described above, the insertion was also detected. At the same time, the 3′-distal part of the 3′ NTR exhibited relatively high sequence identity among different strains of the flaviviruses. However, through RNA secondary structure prediction, we found this inserted sequence formed two small stem-loop structures (). The two small stem-loop structures principally modified the structure of the 5′-proximal part of the 3′ NTR. The newly inserted sequence does not significantly interfere the RNA secondary structure of the 3′ NTR of the DTMUV-AH2011 strain, especially, the 3′-distal part of the 3′ NTR.
The nucleotides of the individual viral proteins of the DTMUV-AH2011 strain were also compared with those of other 12 flaviviruses. All of the results are shown in . Briefly, in comparison with nine TMUV strains, the nucleotides of the individual viral proteins revealed high levels of similarity (all nucleotide sequence identities >96.0%). Nucleotide changes were scattered throughout all the individual viral genes and no obvious difference occurred between the viral proteins. When compared with some Bagaza virus strains in the Ntaya group, the NS3 gene of DTMUV-AH2011 shared 74.9%, 75.0% and 75.1% nucleotide identity with that of Spain-H2010, DakAr-B209 and 96363, respectively. It was more conserved among the viral protein encoding regions. In contrast, the NS2A gene had a relatively higher rate of nucleotide change. In addition, the sequence similarities were very high between DTMUV-AH2011 and the other TMUV strains reported recently in China.
Table 4. Nucleotide sequence homology between DTMUV-AH2011 and twelve flaviviruses in Ntaya virus serogroup in individual viral proteins gene.
At the level of amino acids, the individual viral proteins of the DTMUV-AH2011 strain were also compared with those of the other 12 flaviviruses. As shown in , they had highly conserved polyprotein precursor and had high amino acid sequence identity between DTMUV-AH2011 strain and nine TMUV strains reported recently in China (amino acid identity ≥98.9%); however, the amino acid sequence identities between DTMUV-AH2011 strain and three Bagaza virus strains were relatively lower, ranging from 80.7% (DakAr-B209) to 81.2% (Spain-H2010) for the corresponding amino acid sequences. In comparison with the BYD-1 (the first reported duck-origin TMUV strain), the ORF of DTMUV-AH2011 had a total of 30 amino acid substitutions. The overall divergence at the level of amino acid substitution was 0.9%, these changes were randomly distributed among the viral proteins except the protein NS4B. The protein NS4B was most conservative among all of the currently available TMUV strains reported recently in China, there was no amino acid substitution detected. However, compared with three Bagaza virus strains in the Ntaya group, the protein NS4B was not most conservative among the viral proteins, the most conservative protein was protein NS3 (the amino acid identities among them being approximately 86.0%), this was consistent with the corresponding nucleotides sequence homology. Besides, between DTMUV-AH2011 and BYD-1, proteins NS4A, NS3 and NS2A were largely conservative, with only 0.3%, 0.4% and 0.4% amino acid substitution, respectively. In contrast, capsid and NS1 proteins had 4.2% and 1.4% amino acid divergence, respectively, with a relatively higher rate of amino acid change among the structural proteins and nonstructural proteins, respectively. Overall, according to our multiple alignments and sequence analyses, E protein was relatively conservative among all the structural proteins, and protein NS4B (between DTMUV-AH2011 and other TMUV strains) or protein NS3 (between DTMUV-AH2011 and Bagaza virus strains) was relatively conservative among all the nonstructural proteins. However, capsid proteins were relatively variable among all the structural proteins, and NS1 protein (between DTMUV-AH2011 and other TMUV strains) or protein NS2A (between DTMUV-AH2011 and Bagaza virus strains) was relatively variable among all the nonstructural proteins.
Table 5. Amino acid sequence homology between DTMUV-AH2011 and twelve flaviviruses in Ntaya virus serogroup in individual viral proteins.
Multiple alignments and phylogenetic analysis.
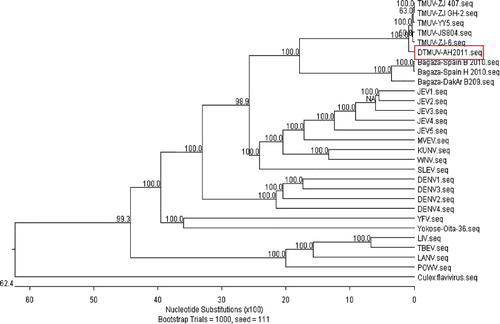
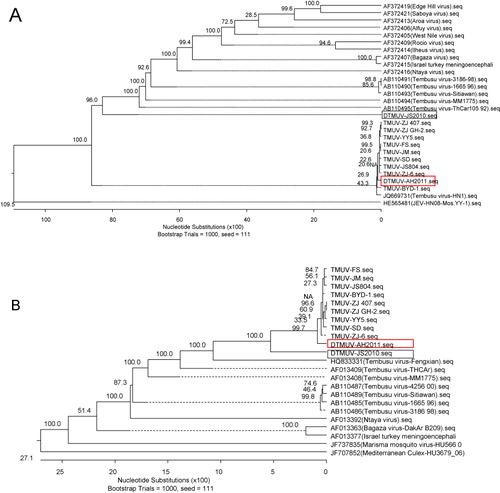
To establish the phylogenetic relationship between DTMUV-AH2011 and other flaviviruses, a phylogenetic tree was constructed using the complete genome sequences of DTMUV-AH2011 and 28 reference strains of flavivirus. The DTMUV-AH2011 strain, which was isolated in 2011, together with other Tembusu strains reported recently in China were grouped into a branch divergent on the phylogenetic trees (). All strains of Tembusu-related viruses were strongly clustered together and closely related to Bagaza virus. Much evidence has suggested that the flavivirus E protein is the major determinant of viral neurovirulence and neuroinvasiveness (McMinn, Citation1997; Cao et al., Citation2011a). To study the phylogenetic relationship of Ntaya virus group, a phylogenetic tree was constructed using the reported structural gene (partial E gene) and non-structural gene (partial NS5 gene) of some viruses. The E gene phylogenetic tree, produced by the NJ method and comprising a total of 28 virus strains, is shown in . For comparison with the E gene phylogeny, phylogenetic analysis using cognate partial NS5 gene sequence information was carried out. The NS5 phylogenetic tree produced here () was virtually consistent with those produced in previous studies using the NS5 gene. These analyses showed that the phylogenetic tree based on the partial E gene and partial NS5 gene corresponded well with the tree based on the full-length genome, with a minor difference.
Discussion
Since April 2010, a serious disease of acute infectious DHO in layer ducks had quickly spread in Southeast China (Cao et al., Citation2011b; Su et al., Citation2011; Li et al., Citation2012). It had significantly affected the duck industry and resulted in serious economic losses in Southeast China. The same clinical sign of egg drop syndrome was also found in several duck farms in Anhui province in China since February 2011. In this study, we attempted to isolate and identify the causative pathogen(s) responsible for this acute infectious disease. The causative pathogen was isolated from the diseased ducks and designated as DTMUV-AH2011, a newly emerged duck-origin TMUV (Yun et al., Citation2012a; Tang et al., Citation2013).
The gene sequence analysis showed that the DTMUV-AH2011 shared high similarities to all recently reported strains of TMUV except for the 3′ NTR. The 3′ NTR was found to be 692 nucleotides, containing a 74 nucleotide insertion, which was a specific tandem repeat sequence in DTMUV-AH2011 genome compared with all TMUV strains for which the 3′-terminal sequences were available. Interestingly, this insertion was also detected in five other samples collected from the same flocks of the same area whereas this insertion was not detected in virus strains detected in other regions in November 2012 suggesting that the DTMUV/AH/2011 strain was specific for that region.
Both NTRs of flavivirus form specific secondary stem-loop structures and functional domains that are essential for virus replication and assembly (Charlier et al., Citation2002; Markoff, Citation2003; Gritsun & Gould, Citation2006). Previous research showed that the presence of the 3′-proximal domains of the JEV 3′ NTR was sufficient to maintain the replication competency of the genomic RNA, whereas the presence of the remaining 5′-proximal domains displayed an enhancer-like function in promoting RNA replication efficiency (Alvarez et al., Citation2005; Yun et al., Citation2009). Through RNA secondary structure prediction, the inserted sequences formed two small stem-loop structures, and the structure of the proximal part of the 3′ NTR was modified. The newly inserted sequences did not significantly change the RNA secondary structure of the 3′ NTR of the DTMUV-AH2011 strain, especially in the 3′-distal part of the 3′ NTR, where most of the important elements in viral translation, replication and assembly are concentrated. Mutational analysis of the 3′ NTR using recombinant TBEV cDNA clones and a Kunjin virus replicon indicated that the deletions in the 5′-proximal domains of 3′ NTR may not affect the growth properties in vitro or virulence in mice (Khromykh & Westaway, Citation1997; Mandl et al., Citation1998). Therefore, only a relatively short region of the flavivirus 3′ NTR was required for genomic RNA replication, and nucleotide insertion or deletion at the 5′-proximal part of 3′ NTR would not cause a lethal mutant.
There is a report that foot-and-mouth disease virus (FMDV) strains often differ in the presence or absence of certain deletions in the 5′ NTR, suggesting that recombination events generating insertions or deletions in the noncoding regions appear to be very frequent during evolution of FMDV (Escarmís et al., Citation1995). Under the natural positive selection pressure, the viral replicase may be able to exercise the genome plasticity by deleting or adding genomic RNA stretches when structural and functional constraints allow (Hahn et al., Citation1987). The biological significance of the presence of the insertion in the 3′ NTR in the DTMUV-AH2011 strain is unknown. The occurrence of insertions in DTMUV may be similar to that of FMDV. However, the identification of the major determinants of viral replication within this region would require further investigation. Whether the direct repeat sequences regulated DTMUV-AH2011 genome replication in connection with other cis-acting replication elements present within the genome is worth exploring.
In the present study, the individual viral proteins of the DTMUV-AH2011 strain were also compared with other flaviviruses at the level of amino acid. As described above, the protein NS4B was the most conservative among the viral proteins, there were no amino acid substitutions between them. In particular, there was evidence to suggest that the central region of NS4B plays a role in the virulence phenotype of flaviviruses (Hanley et al., Citation2003; McArthur et al., Citation2003; Wicker et al., Citation2006). Previous studies have described mutations in the NS4B protein of attenuated or passage-adapted mosquito-borne flaviviruses, such as WNV suggesting that this protein plays an important role in replication and pathogenesis (Puig-Basagoiti et al., Citation2007; Wicker et al., Citation2012). However, flavivirus NS4B was also found to inhibit the interferon-signalling cascade at the level of nuclear STAT phosphorylation, and it also had been implicated in modulation of the stress-induced unfolded protein response (Munoz-Jordan et al., Citation2003; Ambrose & Mackenzie, Citation2011). This might be responsible for extensive epidemics of DTMUV in egg-laying and breeder ducks since April 2010 in China, with all strains being highly pathogenic to affected ducks.
The function of the various non-structural proteins of flavivirus in virus replication was more and more clear. NS3 and NS5 form components of the viral replicase responsible for replicating RNA. Their sequences demonstrated a high degree of conservation among different flaviviruses (Hahn et al., Citation1987). Whether amino acid changes of NS3 and NS5 protein among different DTMUV strains lead to function alteration needs further investigation. E protein is the major structural protein of flavivirus, contains virus neutralizing epitopes, and plays an important role in virus receptor-binding, entry and fusion (Kuhn et al., Citation2002; Modis et al., Citation2004). High identity of E protein between TMUV-AH2011 and other DTMUV strains may be due to low immune pressure at the early epidemic stage of the pathogen.
In conclusion, the DTMUV-AH2011 is highly likely responsible for the acute infectious disease that occurred in Anhui in China in 2011. The detailed sequence analysis between DTMUV-AH2011 and strains reported recently in China showed that these new DTMUV strains were highly conserved except that DTMUV-AH2011 has 74 nucleotides insertion in the 3′ NTR. The biological function of the direct repeat insertion needs further research. Considering that TMUV continues to spread across the duck-producing regions in China, and many strains of the virus have been isolated, the research for selecting vaccine strains to better control this newly emerging infectious disease is urgent. TMUV is a vector-borne virus, and is also isolated from naturally infected house sparrows. Further studies are needed to elucidate the molecular mechanism of interspecies transmission of the virus and the mechanisms of pathogenesis, to remove the gaps in our knowledge regarding the biology of vectors and vertebrates as well as the natural transmission mechanism.
Additional information
Funding
References
- Alvarez, D.E., De Lella Ezcurra, A.L., Fucito, S. & Gamarnik, A.V. (2005). Role of RNA structures present at the 3′UTR of dengue virus on translation, RNA synthesis, and viral replication. Virology, 339, 200–212. 10.1016/j.virol.2005.06.009
- Ambrose, R.L. & Mackenzie, J.M. (2011). West Nile virus differentially modulates the unfolded protein response to facilitate replication and immune evasion. Journal of Virology, 85, 2723–2732. 10.1128/JVI.02050-10
- Cao, Q.S., Li, X.M., Zhu, Q.Y., Wang, D.D., Chen, H.C. & Qian, P. (2011a). Isolation and molecular characterization of genotype 1 Japanese encephalitis virus, SX09S-01, from pigs in China. Virology Journal, 8, 472. 10.1099/vir.0.79797-0
- Cao, Z., Zhang, C., Liu, Y., Liu, Y., Ye, W., Han, J., Zhang, D., Xu, F., Gao, X., Tang, Y., Shi, S., Wan, C., Zhang, C., He, B., Yang, M., Lu, X., Huang, Y., Diao, Y., Ma, X. & Zhang, D. (2011b). Tembusu virus in ducks, China. Emerging Infectious Diseases, 17, 1873–1875. 10.3201/eid1710.101890
- Charlier, N., Leyssen, P., Pleij, C.W., Lemey, P., Billoir, F., Van Laethem, K., Vandamme, A.M., De Clercq, E., de Lamballerie, X. & Neyts, J. (2002). Complete genome sequence of Montana Myotis leukoencephalitis virus, phylogenetic analysis and comparative study of the 3′ untranslated region of flaviviruses with no known vector. Journal of General Virology, 83, 1875–1885.
- De Rijk, P., Wuyts, J. & De Wachter, R. (2003). RnaViz 2: an improved representation of RNA secondary structure. Bioinformatics, 19, 299–300. 10.1093/bioinformatics/19.2.299
- Dong, H., Ray, D., Ren, S., Zhang, B., Puig-Basagoiti, F., Takagi, Y., Ho, C.K., Li, H. & Shi, P.-Y. (2007). Distinct RNA elements confer specificity to flavivirus RNA cap methylation events. Journal of Virology, 81, 4412–4421. 10.1128/JVI.02455-06
- Escarmís, C., Dopazo, J., Dávila, M., Palma, E.L. & Domingo, E. (1995). Large deletions in the 5′-untranslated region of foot-and-mouth disease virus of serotype C. Virus Research, 35, 155–167. 10.1016/0168-1702(94)00091-P
- Gritsun, T.S. & Gould, E.A. (2006). The 3′ untranslated regions of Kamiti River virus and Cell fusing agent virus originated by self-duplication. Journal of General Virology, 87, 2615–2619. 10.1099/vir.0.81950-0
- Hahn, C.S., Dalrymple, J.M., Strauss, J.H. & Rice, C.M. (1987). Comparison of the virulent Asibi strain of yellow fever virus with the 17D vaccine strain derived from it. Proceedings of the National Academy of Sciences, 84, 2019–2023. 10.1073/pnas.84.7.2019
- Hanley, K.A., Manlucu, L.R., Gilmore, L.E., Blaney, J.E., Jr., Hanson, C.T., Murphy, B.R., Whitehead, S.S. (2003). A trade-off in replication in mosquito versus mammalian systems conferred by a point mutation in the NS4B protein of dengue virus type 4. Virology, 312, 222–232. 10.1016/S0042-6822(03)00197-1
- Khromykh, A.A. & Westaway, E.G. (1997). Subgenomic replicons of the flavivirus Kunjin: construction and applications. Journal of Virology, 71, 1497–1505.
- Kono, Y., Tsukamoto, K., Abd Hamid, M., Darus, A., Lian, T.C., Sam, L.S., Yok, C.N., Di, K.B., Lim, K.T., Yamaguchi, S. & Narita, M. (2000). Encephalitis and retarded growth of chicks caused by Sitiawan virus, a new isolate belonging to the genus Flavivirus. The American Journal of Tropical Medicine and Hygiene, 63, 94–101.
- Kuhn, R.J., Zhang, W., Rossmann, M.G., Pletnev, S.V., Corver, J., Lenches, E., Jones, C.T., Mukhopadhyay, S., Chipman, P.R. & Strauss, E.G. (2002). Structure of dengue virus: implications for flavivirus organization, maturation, and fusion. Cell, 108, 717–725. 10.1016/S0092-8674(02)00660-8
- Larkin, M.A., Blackshields, G., Brown, N.P., Chenna, R., McGettigan, P.A., McWilliam, H., Valentin, F., Wallace, I.M., Wilm, A., Lopez, R., Thompson, J.D., Gibson, T.J. & Higgins, D.G. (2007). Clustal W and Clustal X version 2.0. Bioinformatics, 23, 2947–2948. 10.1093/bioinformatics/btm404
- Li, L., An, H., Sun, M., Dong, J., Yuan, J. & Hu, Q. (2012). Identification and genomic analysis of two duck-origin Tembusu virus strains in southern China. Virus Genes, 45, 105–112. 10.1007/s11262-012-0753-6
- Lindenbach, B.D. & Rice, C.M. (2003). Molecular biology of flaviviruses. Advances in Virus Research, 59, 23–61. 10.1016/S0065-3527(03)59002-9
- Mandl, C.W., Holzmann, H., Meixner, T., Rauscher, S., Stadler, P.F., Allison, S.L. & Heinz, F.X. (1998). Spontaneous and engineered deletions in the 3′ noncoding region of tick-borne encephalitis virus: construction of highly attenuated mutants of a flavivirus. Journal of Virology, 72, 2132–2140.
- Markoff, L. (2003). 5′- and 3′-noncoding regions in flavivirus RNA. Advances in Virus Research, 59, 177–228. 10.1016/S0065-3527(03)59006-6
- McArthur, M.A., Suderman, M.T., Mutebi, J-P., Xiao, S-Y. & Barrett, A.D.T. (2003). Molecular characterization of a hamster viscerotropic strain of yellow fever virus. Journal of Virology, 77, 1462–1468. 10.1128/JVI.77.2.1462-1468.2003
- McMinn, P.C. (1997). The molecular basis of virulence of the encephalitogenic flaviviruses. Journal of General Virology, 78, 2711–2722.
- Modis, Y., Ogata, S., Clements, D. & Harrison, S.C. (2004). Structure of the dengue virus envelope protein after membrane fusion. Nature, 427, 313–319. 10.1038/nature02165
- Munoz-Jordan, J.L., Sanchez-Burgos, G.G., Laurent-Rolle, M. & Garcia-Sastre, A. (2003). Inhibition of interferon signaling by dengue virus. Proceedings of the National Academy of Sciences, 100, 14333–14338. 10.1073/pnas.2335168100
- Okuno, Y., Igarashi, A., Fukunaga, T., Tadano, M. & Fukai, K. (1984). Electron microscopic observation of a newly isolated flavivirus-like virus from field-caught mosquitoes. Journal of General Virology, 65, 803–807. 10.1099/0022-1317-65-4-803
- Puig-Basagoiti, F., Tilgner, M., Bennett, C.J., Zhou, Y., Muñoz-Jordán, J.L., García-Sastre, A., Bernard, K.A. & Shi, P-Y. (2007). A mouse cell-adapted NS4B mutation attenuates West Nile virus RNA synthesis. Virology, 361, 229–241. 10.1016/j.virol.2006.11.012
- Su, J., Li, S., Hu, X., Yu, X., Wang, Y., Liu, P., Lu, X., Zhang, G., Hu, X., Liu, D., Li, X., Su, W., Lu, H., Mok, N.S., Wang, P., Wang, M., Tian, K., Gao, G.F. & Qiu, J. (2011). Duck egg-drop syndrome caused by BYD virus, a new Tembusu-related flavivirus. PloS One, 6, e18106. 10.1371/journal.pone.0018106.t002
- Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M. & Kumar, S. (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Molecular Biology and Evolution, 28, 2731–2739. 10.1093/molbev/msr121
- Tang, Y., Diao, Y., Gao, X., Yu, C., Chen, L. & Zhang, D. (2012). Analysis of the complete genome of Tembusu virus, a flavivirus isolated from ducks in China. Transboundary and Emerging Diseases, 59, 336–343. 10.1111/j.1865-1682.2011.01275.x
- Tang, Y., Diao, Y., Yu, C., Gao, X., Ju, X., Xue, C., Liu, X., Ge, P., Qu, J. & Zhang, D. (2013). Characterization of a Tembusu virus isolated from naturally infected house sparrows (Passer domesticus) in Northern China. Transboundary and Emerging Diseases, 60, 152–158. 10.1111/j.1865-1682.2012.01328.x
- Thompson, J.D., Gibson, T.J., Plewniak, F., Jeanmougin, F. & Higgins, D.G. (1997). The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Research, 25, 4876–4882. 10.1093/nar/25.24.4876
- Wicker, J.A., Whiteman, M.C., Beasley, D.W.C., Davis, C.T., McGee, C.E., Lee, J.C., Higgs, S., Kinney, R.M., Huang, C.Y-H. & Barrett, A.D.T. (2012). Mutational analysis of the West Nile virus NS4B protein. Virology, 426, 22–33. 10.1016/j.virol.2011.11.022
- Wicker, J.A., Whiteman, M.C., Beasley, D.W.C., Davis, C.T., Zhang, S., Schneider, B.S., Higgs, S., Kinney, R.M. & Barrett, A.D.T. (2006). A single amino acid substitution in the central portion of the West Nile virus NS4B protein confers a highly attenuated phenotype in mice. Virology, 349, 245–253. 10.1016/j.virol.2006.03.007
- Yun, S-I., Choi, Y-J., Song, B-H. & Lee, Y-M. (2009). 3′ cis-acting elements that contribute to the competence and efficiency of Japanese encephalitis virus genome replication: functional importance of sequence duplications, deletions, and substitutions. Journal of Virology, 83, 7909–7930. 10.1128/JVI.02541-08
- Yun, T., Ye, W., Ni, Z., Zhang, D. & Zhang, C. (2012a). Identification and molecular characterization of a novel flavivirus isolated from Pekin ducklings in China. Veterinary Microbiology, 157, 311–319. 10.1016/j.vetmic.2012.01.013
- Yun, T., Zhang, D., Ma, X., Cao, Z., Chen, L., Ni, Z., Ye, W., Yu, B., Hua, J., Zhang, Y. & Zhang, C. (2012b). Complete genome sequence of a novel flavivirus, duck Tembusu virus, isolated from ducks and geese in China. Journal of Virology, 86, 3406–3407. 10.1128/JVI.07132-11
- Zuker, M. (2003). Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Research, 31, 3406–3415. 10.1093/nar/gkg595