
ABSTRACT
Live attenuated vaccines are extensively used worldwide to control the outbreak of infectious laryngotracheitis. Virulent field strains showing close genetic relationship with the infectious laryngotracheitis virus (ILTV) vaccines of chicken embryo origin have been detected in the poultry industry. Polymerase chain reaction-restriction fragment length polymorphism (PCR-RFLP) analysis, a reliable molecular epidemiological method, of multiple genomic regions was performed. The PCR-RFLP is a time-consuming method that requires considerable amount of intact viral genomic DNA to amplify genomic regions greater than 4 kb. In this study, six variable genomic regions were selected and amplified for sequencing. The multi-allelic PCR-sequence genotyping showed better discrimination power than that of previous PCR-sequencing schemes using single or two target regions. The allelic variation patterns yielded 16 strains of ILTV classified into 14 different genotypes. Three Korean field strains, 550/05/Ko, 0010/05/Ko and 40032/08/Ko, were found to have the same genotype as the commercial vaccine strain, Laryngo Vac (Zoetis, Florham Park, NJ, USA). Three other Korean field strains, 40798/10/Ko, 12/07/Ko, and 30678/14/Ko, showed recombined allelic patterns. The multi-allelic PCR-sequencing method was proved to be an efficient and practical procedure to classify the different strains of ILTV. The method could serve as an alternate diagnostic and differentiating tool for the classification of ILTV, and contribute to understanding of the epidemiology of the disease at a global level.
Introduction
Infectious laryngotracheitis (ILT) is an acute upper respiratory disease in chickens. ILT outbreaks caused by infectious laryngotracheitis virus (ILTV, gallid herpesvirus I) are responsible for considerable economic loss in the poultry industry worldwide (Bagust et al., Citation2000).
To control the disease, two types of live attenuated vaccines have been developed using serial passages in tissue cultures (tissue culture origin [TCO] vaccine) or embryonated chicken eggs (chicken embryo origin [CEO] vaccine). Since CEO vaccines have shown better efficacy than TCO vaccines, they have been intensively used in the field worldwide (Johnson et al., Citation2010; Vagnozzi et al., Citation2012). However, after bird-to-bird passages (Guy et al., Citation1991), increased virulence was observed for CEO vaccines and virulent field strains of ILTV frequently showed close genetic relationship with the CEO vaccines (Guy et al., Citation1989; Andreasen et al., Citation1990; Keeler et al., Citation1993). Therefore, the differentiation of field strains of ILTV and vaccine strains became important for epidemiological investigation of ILTV.
ILTV has a single serotype (Cover & Benton, Citation1958). Hence, genotyping using polymerase chain reaction-restriction fragment length polymorphism (PCR-RFLP) of multiple target regions has been used worldwide to differentiate field strains from the commercial vaccine strains of ILTV instead of serotyping (Chang et al., Citation1997; Han & Kim, Citation2001; Kirkpatrick et al., Citation2006; Oldoni et al., Citation2008).
In the USA, four genomic regions including the ORFB-TK (4.67 Kb), gM/UL9 (1.42 Kb), ICP4 (4.98 Kb), and UL47/gG (2.93 Kb) were amplified and digested with multiple endonucleases to differentiate CEO, TCO vaccines, and field isolates of ILTV (Oldoni et al., Citation2008). In Australia, three genomic regions including the TK (2.24 Kb), ICP18.5 (5.89 Kb), and ICP4 (4.98 Kb) were targeted for PCR reaction (Kirkpatrick et al., Citation2006; Blacker et al., Citation2011). The PCR-RFLP method can classify vaccine and field strains, including recombinants of ILTV, into different genotypes; however, it is hard to obtain sufficient amount of intact viral DNA from field swab samples to perform PCR-RFLP analysis. Additionally, PCR-RFLP is time consuming and multiple endonuclease reactions are expensive.
Commercial service for the Sanger sequencing is quick and less expensive. Moreover, sequencing results of multiple PCR products using Sanger sequencing can be obtained within 48 h in many countries. Due to an increasing number of ILTV full genome sequences available (Lee, et al., Citation2011a; Lee, et al., Citation2011b; Chandra et al., Citation2012; Lee et al., Citation2012; Spatz et al., Citation2012; Garcia et al., Citation2013; Kong et al., Citation2013), highly variable genomic regions between the strains of ILTV can be detected using comparative genome analysis. The sequences determined through PCR-sequencing of these variable regions might be enough to differentiate the strains of ILTV. Previously, one or two target regions, mainly the TK and ICP4 genes, were used for the PCR-sequencing method (Han & Kim, Citation2001; Creelan et al., Citation2006; Ojkic et al., Citation2006; Neff et al., Citation2008; Chacon & Ferreira, Citation2009; Moreno et al., Citation2010; Shehata et al., Citation2013), but genotyping using allelic variations from multiple target regions of the entire ILTV genome was never tested.
In this study, comparative genome alignment was performed using the available complete genomes of ILTV and variable genomic regions showing allelic variations were manually selected to achieve the best discrimination power. In silico genotypic patterns were generated, and the genotyping method was validated using Korean field strains and commercial vaccine strains of ILTV.
Materials and methods
Comparative genome analysis
Sixteen strains of ILTV that were involved in the nucleotide sequence alignment of complete genome using the MAFFT software (Katoh & Standley, Citation2013) are listed in . To achieve the best discrimination power for genotyping of ILTV, highly variable genomic regions shorter than 600 bp in length were manually selected in order to determine the nucleotide sequence of the selected regions with a single run of Sanger sequencing. In addition the genomic regions targeted were scattered throughout the entire genome to increase the probability of detecting recombinant strains of ILTV. Pairwise alignments of nucleotide sequences of the selected variable regions were performed and phylogenetic trees were generated with the alignments using the maximum likelihood method and 100 bootstrap runs.
Table 1. Strains of ILTV used for comparative genome analysis and their sequencing types according to patterns of allelic variations from the six target regions.
Genotyping of Korean field strains and vaccines using allelic variations
Primers that were designed to amplify the selected variable genomic regions are listed in . To test the designed primers, viral genomes were extracted using the Viral Gene-Spin™ Viral DNA/RNA extraction kit (iNtRON Biotechnology, Seongnam, Gyeonggi Province, Korea) from viral transport media containing trachea samples or samples from commercial vaccine vials. Selected genomic regions were amplified using PCR as follows: initial denaturation at 95°C for 5 min, followed by 35 cycles of 95°C for 30 s, 60°C for 45 s, and 72°C for 1 min, and a final extension at 72°C for 5 min. PCR products were cleaned and the PCR products were sequenced using Sanger sequencing. The sequencing results obtained were compared with known sequences using Blastn to determine the allelic sequence type of each fragment.
Table 2. List of primers designed to amplify variable genomic regions of ILTV.
Results
Six genomic regions were selected for genotyping of ILTV in nucleotide sequence alignment of the complete genomes of ILTV. The selected regions were located in UL54, UL52, gB, ICP18.5, ICP4, and gJ genes () and each was able to separate 16 strains of ILTV into four (UL52 and UL54 genes) up to eight patterns (ICP4 gene) (). Locations of all variations including single-nucleotide polymorphisms (SNPs) and indels between patterns are shown in . Although there were few sequence variations in genomic regions of the UL54 and UL52 genes, the SNPs allowed differentiation of the commercially available vaccines to multiple genotypes. In the phylogenetic analysis using nucleotide sequence alignments of the selected genomic regions, Australian origin strains of ILTV were mainly out-grouped and most of the field isolates were clustered with CEO vaccines (). A total of 14 genotypes were detected from 16 complete genome sequences of ILTV using the genotyping scheme depending on allelic variations (). The multi-allelic PCR-sequencing method showed better discrimination power than two PCR-RFLP methods described earlier (). The Laryngo Vac, Serva, CEO TRVX and LT Blen vaccines that classified in the same group using the two PCR-RFLP methods could be differentiated into sequence types 1, 2 and 3 in this study. Also the A20 and SA2, Australian origin CEO vaccines that could not be differentiated using either of the PCR-RFLP methods could be differentiated into sequence types 13 and 14 using the multi-allelic PCR-sequencing method. Furthermore, there was inconsistency of the results of PCR-RFLP between previous reports and in silico patterns. Six pairs of primers designed to amplify the selected genomic regions successfully amplified each target region of the viral genomic DNA of the Korean field strains or vaccine strains of ILTV. The genotyping results of the Korean field strains or commercial vaccine strains are summarized in . Commercial ILTV vaccines available in Korea, the Nobilis ILT (MSD Animal Health Korea, Mapo-gu, Seoul, Korea) and the LT Blen (Merial Korea, Seocho-gu, Seoul, Korea), produced the same allelic patterns as predicted using the complete genome sequences of the vaccines obtained from other countries ( and 4). Six Korean field strains of ILTV showed three different allelic patterns. Three of the Korean field strains, 550/05/Ko, 0010/05/Ko, and 40032/08/Ko, showed the same allelic patterns as that of the Laryngo Vac (Zoetis, USA) strain, which is another commercially available vaccine strain of ILTV in Korea (). Three of the Korean field strains, 40798/10/Ko, 12/07/Ko, and 30678/14/Ko, generated new allelic patterns for the gJ gene with a SNP and an insertion of 30 bp sequence. The 12/07/Ko and 30678/14/Ko showed new allelic pattern for the ICP4 gene which with exception of a SNP was very similar to that of the Serva and other CEO vaccines (). Furthermore, the three Korean field strains showed mixed allelic patterns to those of the global vaccines and the Australian origin vaccines (). Therefore, the three Korean field isolates might have been generated as a result of historical recombination between distinct ILTV genotypes.
Figure 1. Nucleotide sequence alignment for the complete genomes of ILTV and locations of the selected variable regions. Alignment of the complete genome sequences of 16 strains of ILTV was performed using the MAFFT software. Vertical lines indicate SNPs and dashes indicate sequence gaps. The locations of primer binding sites are depicted using triangles.

Figure 2. Nucleotide sequence alignments for amplified genomic regions between different sequence types of ILTV. Alignment of sequencing results of the polymerase chain reaction (PCR)-amplified specific genomic regions containing partial genes of ILTV was performed using the MAFFT. Locations of SNPs between different sequence types are indicated and dashes indicate sequence gaps.

Figure 3. Phylogenetic analyses for the selected variable genomic regions of ILTV. Maximum likelihood phylogenetic trees were generated using alignments of nucleotide sequences of the selected highly variable genomic regions. One hundred bootstrap replicates were used to assess the significance of the tree topology. Bars indicate nucleotide substitutions per site. The allelic clusters containing sequence gaps were denoted with asterisks.
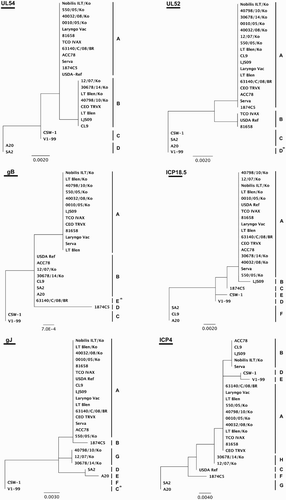
Table 3. Genotypes of strains of ILTV according to patterns of PCR-RFLP or multi-allelic PCR-sequencing.
Table 4. Genotypes of Korean field strains and vaccine strains of ILTV according to patterns of allelic variations from the six target regions.
Discussion
In this study, we tried to find a reliable method for ILTV classification. RFLP does not require extensive interpretation of results and therefore is less prone to misinterpretation. However, it is time consuming and expensive. The real cause of discrepancies detected between RFLP patterns reported in previous studies and those generated in silico as part of this study is unclear, although different clones/isolates of the ILTV strains may have been used in RFLP and whole genome sequencing experiments. For example, the CSW-1 strain used Kirkpatrick et al. (Citation2006) for PCR-RFLP shared the same origin as that used by Lee et al. (Citation2013) for whole genome sequencing, however, the latter had been passaged several times in cell culture and was perhaps cell adapted. The difference in the ICP4 gene from the CSW-4 clone used in these two studies is evident by the larger PCR product generated from ICP4 by Kirkpatrick et al. (Citation2006) than that predicted from genome sequence reported by Lee et al. (Citation2013). Whether ICP4 gene may have undergone changes (such as deletion of repeat sequences) during passage in vitro needs to be investigated. In this study, a combination of sequencing results from six different genomic regions showed a better discrimination power than PCR-RFLP methods and classified16 ILTV strains into 14 different genotypes. Especially most of commercially available vaccine strains could be differentiated with this method. These results suggest that an exact source of field strain derived from ILTV vaccine could be tracked with the multi-allelic PCR-sequencing method. In previous studies, sequencing of two different regions of the ICP4 differentiated Brazilian and Peruvian field strains from CEO and TCO vaccines. Moreover, genotypes obtained using PCR-DNA sequencing completely matched with that of PCR-RFLP patterns (Chacon & Ferreira, Citation2009). ILTV is a double-stranded DNA virus with a highly stable genome. In previous studies, nucleotide substitution rate in herpes simplex virus-1 genome was estimated to be 3.5 × 10–8/nucleotide/year (Sakaoka et al., Citation1994). Consistent with the estimation, only two non-synonymous SNPs were found from the comparative genome study between two Australian origin CEO vaccines (Lee et al., Citation2011b).
As some of strains of ILTV could not be imported into Korea, we could not validate the predicted genotypic patterns using all virus samples involved in the comparative genome analysis. We tested the PCR conditions using Korean field strains and vaccine strains of ILTV available in Korea. Multiple PCR reactions designed in this study successfully amplified target regions from all tested viral DNA. Sequencing results revealed that the commercial vaccines sold in Korea showed consistent allelic patterns of PCR products with those of the in silico predicted allelic patterns of the same vaccines sold overseas. Interestingly, three of the Korean field strains, 40798/10/Ko, 12/07/Ko, and 30678/14/Ko, showed mixed allelic patterns of PCR products. Nucleotide sequences of five genomic regions including partial genomic regions of the UL54, UL52, gB, ICP18.5, and ICP4 genes of the 40798/10/Ko strain of ILTV were identical to those of the global CEO vaccines, while the nucleotide sequence of the partial gJ gene was very similar to that of the CEO vaccines of Australian origin. In 12/07/Ko and 30768/14/Ko strains of ILTV, nucleotide sequences of three genomic regions including the partial gB, gJ, and ICP4 genes were very similar to those of the Australian origin CEO vaccines, while other regions were similar to those of the global CEO vaccines. These results suggested that the Korean field strains of ILTV might be natural recombinant strains between global CEO vaccines and a strain having very similar genotype to the Australian origin vaccines. This kind of recombinant has been detected in previous studies (Lee et al., Citation2013). Furthermore, the Australian origin vaccines have never been imported into Korea; therefore, a field strain of ILTV belonging to the genotype of the Australian origin vaccine might be present in Korea or both Korean and Australian vaccines may share a common ancestor.
In this study, we developed an ILTV classification method using allelic variations from multiple genomic regions. This method proved to be an efficient and practical tool in differentiating each strain of ILTV. The method can be an alternative tool for ILTV differentiation and contribute to the understanding of the epidemiology of the disease at the global level.
Additional information
Funding
References
- Andreasen, J.R. Jr., Glisson, J.R. & Villegas, P. (1990). Differentiation of vaccine strains and Georgia field isolates of infectious laryngotracheitis virus by their restriction endonuclease fragment patterns. Avian Diseases, 34, 646–656. doi: 10.2307/1591259
- Bagust, T.J., Jones, R.C. & Guy, J.S. (2000). Avian infectious laryngotracheitis. Revue scientifique et technique, 19, 483–492. doi: 10.20506/rst.19.2.1229
- Blacker, H., Kirkpatrick, N., Rubite, A., O’Rourke, D. & Noormohammadi, A. (2011). Epidemiology of recent outbreaks of infectious laryngotracheitis in poultry in Australia. Australian Veterinary Journal, 89, 89–94. doi: 10.1111/j.1751-0813.2010.00665.x
- Chacon, J.L. & Ferreira, A.J. (2009). Differentiation of field isolates and vaccine strains of infectious laryngotracheitis virus by DNA sequencing. Vaccine, 27, 6731–6738. doi: 10.1016/j.vaccine.2009.08.083
- Chandra, Y.G., Lee, J. & Kong, B.W. (2012). Genome sequence comparison of two United States live attenuated vaccines of infectious laryngotracheitis virus (ILTV). Virus Genes, 44, 470–474. doi: 10.1007/s11262-012-0728-7
- Chang, P.C., Lee, Y.L., Shien, J.H. & Shieh, H.K. (1997). Rapid differentiation of vaccine strains and field isolates of infectious laryngotracheitis virus by restriction fragment length polymorphism of PCR products. Journal of Virological Methods, 66, 179–186. doi: 10.1016/S0166-0934(97)00050-5
- Cover, M.S. & Benton, W.J. (1958). The biological variation of the infectious laryngotracheitis virus. Avian Diseases, 2, 375–383. doi: 10.2307/1587477
- Creelan, J.L., Calvert, V.M., Graham, D.A. & McCullough, S.J. (2006). Rapid detection and characterization from field cases of infectious laryngotracheitis virus by real-time polymerase chain reaction and restriction fragment length polymorphism. Avian Pathology, 35, 173–179. doi: 10.1080/03079450600598244
- Garcia, M., Volkening, J., Riblet, S. & Spatz, S. (2013). Genomic sequence analysis of the United States infectious laryngotracheitis vaccine strains chicken embryo origin (CEO) and tissue culture origin (TCO). Virology, 440, 64–74. doi: 10.1016/j.virol.2013.02.007
- Guy, J.S., Barnes, H.J., Munger, L.L. & Rose, L. (1989). Restriction endonuclease analysis of infectious laryngotracheitis viruses: comparison of modified-live vaccine viruses and North Carolina field isolates. Avian Diseases, 33, 316–323. doi: 10.2307/1590850
- Guy, J.S., Barnes, H.J. & Smith, L. (1991). Increased virulence of modified-live infectious laryngotracheitis vaccine virus following bird-to-bird passage. Avian Diseases, 35, 348–355. doi: 10.2307/1591188
- Han, M.G. & Kim, S.J. (2001). Analysis of Korean strains of infectious laryngotracheitis virus by nucleotide sequences and restriction fragment length polymorphism. Veterinary Microbiology, 83, 321–331. doi: 10.1016/S0378-1135(01)00423-0
- Johnson, D.I., Vagnozzi, A., Dorea, F., Riblet, S.M., Mundt, A., Zavala, G. & García, M. (2010). Protection against infectious laryngotracheitis by in ovo vaccination with commercially available viral vector recombinant vaccines. Avian Diseases, 54, 1251–1259. doi: 10.1637/9401-052310-Reg.1
- Katoh, K. & Standley, D.M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Molecular Biology and Evolution, 30, 772–780. doi: 10.1093/molbev/mst010
- Keeler, C.L. Jr., Hazel, J.W., Hastings, J.E. & Rosenberger, J.K. (1993). Restriction endonuclease analysis of Delmarva field isolates of infectious laryngotracheitis virus. Avian Diseases, 37, 418–426. doi: 10.2307/1591668
- Kirkpatrick, N.C., Mahmoudian, A., O’Rourke, D. & Noormohammadi, A.H. (2006). Differentiation of infectious laryngotracheitis virus isolates by restriction fragment length polymorphic analysis of polymerase chain reaction products amplified from multiple genes. Avian Diseases, 50, 28–33. doi: 10.1637/7414-072205R.1
- Kong, C., Zhao, Y., Cui, X., Zhang, X., Cui, H., Xue, M. & Wang, Y. (2013). Complete genome sequence of the first Chinese virulent infectious laryngotracheitis virus. PLoS One, 8, e70154. doi: 10.1371/journal.pone.0070154
- Lee, S.W., Devlin, J.M., Markham, J.F., Noormohammadi, A.H., Browning, G.F., Ficorilli, N.P., Hartley, C.A. & Markham, P.F. (2011a). Comparative analysis of the complete genome sequences of two Australian origin live attenuated vaccines of infectious laryngotracheitis virus. Vaccine, 29, 9583–9587. doi: 10.1016/j.vaccine.2011.10.055
- Lee, S.W., Devlin, J.M., Markham, J.F., Noormohammadi, A.H., Browning, G.F., Ficorilli, N.P., Hartley, C.A. & Markham, P.F. (2013). Phylogenetic and molecular epidemiological studies reveal evidence of multiple past recombination events between infectious laryngotracheitis viruses. PLoS One, 8, e55121. doi: 10.1371/journal.pone.0055121
- Lee, S.W., Markham, P.F., Coppo, M.J., Legione, A.R., Markham, J.F., Noormohammadi, A.H., Browning, G.F., Ficorilli, N., Hartley, C.A. & Devlin, J.M. (2012). Attenuated vaccines can recombine to form virulent field viruses. Science, 337, 188. doi: 10.1126/science.1217134
- Lee, S.W., Markham, P.F., Markham, J.F., Petermann, I., Noormohammadi, A.H., Browning, G.F., Ficorilli, N.P., Hartley, C.A. & Devlin, J.M. (2011b). First complete genome sequence of infectious laryngotracheitis virus. BMC Genomics, 12, 197. doi: 10.1186/1471-2164-12-197
- Moreno, A., Piccirillo, A., Mondin, A., Morandini, E., Gavazzi, L. & Cordioli, P. (2010). Epidemic of infectious laryngotracheitis in Italy: characterization of virus isolates by PCR-restriction fragment length polymorphism and sequence analysis. Avian Diseases, 54, 1172–1177. doi: 10.1637/9398-051910-Reg.1
- Neff, C., Sudler, C. & Hoop, R.K. (2008). Characterization of western European field isolates and vaccine strains of avian infectious laryngotracheitis virus by restriction fragment length polymorphism and sequence analysis. Avian Diseases, 52, 278–283. doi: 10.1637/8168-110107-Reg.1
- Ojkic, D., Swinton, J., Vallieres, M., Martin, E., Shapiro, J., Sanei, B. & Binnington, B. (2006). Characterization of field isolates of infectious laryngotracheitis virus from Ontario. Avian Pathology, 35, 286–292. doi: 10.1080/03079450600815481
- Oldoni, I. & Garcia, M. (2007). Characterization of infectious laryngotracheitis virus isolates from the US by polymerase chain reaction and restriction fragment length polymorphism of multiple genome regions. Avian Pathology, 36, 167–176. doi: 10.1080/03079450701216654
- Oldoni, I., Rodriguez-Avila, A., Riblet, S. & Garcia, M. (2008). Characterization of infectious laryngotracheitis virus (ILTV) isolates from commercial poultry by polymerase chain reaction and restriction fragment length polymorphism (PCR-RFLP). Avian Diseases, 52, 59–63. doi: 10.1637/8054-070607-Reg
- Sakaoka, H., Kurita, K., Iida, Y., Takada, S., Umene, K., Kim, Y.T., Ren, C.S. & Nahmias, A.J. (1994). Quantitative analysis of genomic polymorphism of herpes simplex virus type 1 strains from six countries: studies of molecular evolution and molecular epidemiology of the virus. Journal of General Virology, 75, 513–527. doi: 10.1099/0022-1317-75-3-513
- Shehata, A.A., Halami, M.Y., Sultan, H.H., Abd El-Razik, A.G. & Vahlenkamp, T.W. (2013). Chicken embryo origin-like strains are responsible for Infectious laryngotracheitis virus outbreaks in Egyptian cross-bred broiler chickens. Virus Genes, 46, 423–430. doi: 10.1007/s11262-012-0870-2
- Spatz, S.J., Volkening, J.D., Keeler, C.L., Kutish, G.F., Riblet, S.M., Boettger, C.M., Clark, K.F., Zsak, L., Afonso, C.L., Mundt, E.S., Rock, D.L. & Garcia, M. (2012). Comparative full genome analysis of four infectious laryngotracheitis virus (Gallid herpesvirus-1) virulent isolates from the United States. Virus Genes, 44, 273–285. doi: 10.1007/s11262-011-0696-3
- Vagnozzi, A., Zavala, G., Riblet, S.M., Mundt, A. & Garcia, M. (2012). Protection induced by commercially available live-attenuated and recombinant viral vector vaccines against infectious laryngotracheitis virus in broiler chickens. Avian Pathology, 41, 21–31. doi: 10.1080/03079457.2011.631983