
ABSTRACT
Infectious bursal disease virus (IBDV) is an immunosuppressive pathogen with a bi-segmented, double-stranded RNA genome. Two serotypes, named 1 and 2, are recognized, and serotype 1 is traditionally divided into classical, variant and very virulent strains. Despite having great practical relevance, this classification lacks standardization and may therefore generate confusion. The widespread adoption of molecular techniques, which allow direct assessment of either pathogenicity or antigenicity, further complicates this scenario. Recently, two phylogeny-based classification methods have been proposed to obtain informative and standardized results. The first is based on the analysis of the capsid protein VP2 gene, considered as the main determinant of virulence, while the other one considers both VP2 and RNA polymerase VP1, located in different genome segments. The latter approach is therefore better suited to detect reassortment events, which play a significant role in IBDV evolution. This work reports the detection of eight strains in Portuguese broiler farms that diverged from all available IBDV sequences. Subsequent analyses of VP1 and VP2 sequences confirmed that these strains form a new genotype having segment A of a new genogroup and segment B of classical-like genogroup. No recombination events were identified, while several unique amino acid changes were found in key positions within the VP2. This discovery emphasizes the importance of steady and attentive monitoring of IBDV epidemiology. Further research efforts will be devoted to characterizing the novel genotype, by isolating new strains, assessing their pathogenicity and determining the level of protection ensured by commonly administered IBDV vaccines.
Eight IBDV strains with unique VP2 features were detected in Portugal.
Based on two distinct classification methods, the strains belong to a new genotype.
RESEARCH HIGHLIGHTS
Introduction
Infectious bursal disease (IBD) is an immunosuppressive disease with a severe impact on the worldwide poultry industry. It is caused by infectious bursal diseases virus (IBDV), a double-stranded RNA virus belonging to the family Birnaviridae, genus Avibirnavirus (https://talk.ictvonline.org/ictv-reports/ictv_online_report/dsrna-viruses/w/birnaviridae). By targeting immature B-lymphocytes, mainly resident in the bursa of Fabricius (BF), IBDV causes immunosuppression in young chicks, resulting in overt clinical outbreaks, sometimes characterized by high mortality, or subclinical infections (Alkie & Rautenschlein, Citation2016).
The IBDV genome is divided into two segments, named A and B. Segment B encodes for viral protein 1 (VP1), a viral RNA-dependent RNA polymerase, while segment A encodes for a non-structural protein (VP5) and a polyprotein which is then cleaved into a capsid protein, a scaffolding protein and a protease, known respectively as VP2, VP3 and VP4 (Maraver et al., Citation2003).
Two different IBDV serotypes, named 1 and 2, are recognized, but only the former is pathogenic. Further classifications have been proposed based on different criteria. Historically, the first distinction was made in the 1980s based on antigenic differences, allowing discrimination between classical and variant isolates. Another strain category, antigenically similar to classical strains but characterized by greater pathogenicity, was recognized shortly thereafter and denominated very virulent (Lasher & Davis, Citation1997).
Despite its indisputable practical value, this classification may be misleading, since it is based on different properties evaluated by different, unstandardized assays, and it does not fully capture IBDV variability. These issues are further complicated by the routine use of molecular assays for IBDV diagnosis. The comparative advantages of molecular assays, especially PCR-based assays, include their cost-effectiveness, rapid turnaround and possibility for standardization. However, IBDV genetic variability does not fully correlate with antigenic relatedness or virulence.
Recently, multiple IBDV phylogeny-based classification systems have been proposed, thus providing robust and standardized tools to obtain informative results from molecular data. Michel and Jackwood (Citation2017) developed a classification based on the sequence of the VP2 hypervariable region (hvVP2) and identified seven different genogroups (from G1 to G7) within serotype 1. This classification method was further elaborated in another work, which described the subdivision of genogroups 1, 2, 3 and 7 into different lineages and proposed a new systematic strain nomenclature (Jackwood et al., Citation2018).
Since VP2 is the main determinant of pathogenicity (Brandt et al., Citation2001), it makes perfect sense that its sequence is considered for a phylogeny-based IBDV classification. Additionally, the majority of IBDV sequences available in GenBank are partial VP2 sequences. However, regions belonging to both genome segments have been recognized to contribute to IBDV virulence (Boot et al., Citation2005; Escaffre et al., Citation2013) and the number of detected reassortant strains has increased in recent years (Lee et al., Citation2017; Stoute et al., Citation2019; Mato et al., Citation2020; Pikuka et al., Citation2020), highlighting the limits of relying only on the VP2 sequence and the inherent risk of misclassifications.
An increasingly adopted alternative, which represents an acceptable trade-off between practical constraints and the informativeness of results, is sequencing a portion of both segments A and B, particularly of VP2 and VP1. This approach is also recommended by the OIE guidelines (OIE, Citation2018). Following this criterion, a second classification system was recently proposed by Islam et al. (Citation2021), who established nine VP2 genogroups (A1-A8 within serotype 1, and A0 which corresponds to serotype 2) and five VP1 genogroups (B1-B5). Taking into consideration the existing combinations between the two segments (A1B1, A1B2, A1B3, A2B1, etc.), a total of 15 genotypes are currently recognized.
Despite considering two different but overlapping portions of the hvVP2 and adopting different criteria for genogroup demarcation, the VP2 genogroups described by the two classifications largely coincide, except for Australian strains, which are grouped into a single genogroup (and two lineages) by Michel and Jackwood (Citation2017) and into two genogroups by Islam et al. (Citation2021). While the two methods are practically interchangeable when working with only VP2 sequences, it would be preferable to also consider the VP1 gene to obtain more meaningful results.
The present work reports the identification of atypical IBDV strains in multiple broiler farms located in different Portuguese regions. The two above-mentioned classification methods were therefore applied to assess whether these strains constitute a previously unrecognized genogroup.
Materials and methods
Sample collection and analysis
An epidemiological survey was conducted in Portugal as part of the routine diagnostic activities of the laboratory of viral diseases of the Department of Animal Medicine, Production and Health (MAPS) of the University of Padua. A total of 130 flocks, two for each farm, were included in the survey and pools of 10 bursae were obtained from each one. Tissues were mechanically homogenized in 20 ml of PBS, and RNA extraction was performed with High Pure Viral Nucleic Acid kit (Roche™, Basel, Switzerland) from 200 µl of supernatant after centrifugation. A one-step RT–PCR was conducted by using SuperScript™ III One-Step RT–PCR System with Platinum™ Taq DNA Polymerase kit (Invitrogen™, Waltham, MA, USA) with 743–1 (5′-GCCCAGAGTCTACACCAT-3ʹ) and 743–2 (5′-CCCGGATTATGTCTTTGA-3ʹ) primers targeting a portion of the VP2 gene (Jackwood & Sommer-Wagner, Citation2005). Positive samples were subsequently Sanger-sequenced using both primers. The obtained chromatograms were inspected for quality evaluation, trimmed with 4peaks (Nucleobytes B.V., Aalsmer, the Netherlands) and assembled in consensus sequences using ChromasPro (Technelysium Pty Ltd, Helensvale, QLD, Australia).
A preliminary BLAST search (Altschul et al., Citation1990) revealed eight samples to be divergent from any known strains. The samples were therefore selected for further molecular and phylogenetic analyses. A portion of the VP1 gene was amplified and sequenced using B-Univ-F (5ʹ-AATGAGGAGTATGAGACCGA-3ʹ) and B-Univ-R (5ʹ-CCTTCTCTAGGTCAATTGAGTACC-3ʹ) primers published by Islam et al. (Citation2011).
Classification according to Michel and Jackwood (Citation2017).
A reference dataset was prepared by including the reference strains at genogroup (Michel & Jackwood, Citation2017) and lineage (Jackwood et al., Citation2018) level available in GenBank. Duplicate and short sequences were excluded.
The sequences were aligned with the MUSCLE method (Edgar, Citation2004) implemented in MegaX (Kumar et al., Citation2018) and trimmed to obtain a portion of the hvVP2 suitable for classification purpose. The evolutionary history was then inferred with the Maximum Likelihood method (1000 bootstrap, Tamura-Nei substitution model) implemented in MegaX (Kumar et al., Citation2018).
Classification according to Islam et al. (Citation2021)
Four different datasets were prepared: two full datasets, one for VP1 and one for VP2, which included all the sequences considered in the original article and available in GenBank, were used to estimate the evolutionary distances between different genogroups; two reduced datasets, one for VP1 and one for VP2, including a limited number of representative sequences for each genogroup, were used to reconstruct phylogenetic trees.
The Portuguese strain sequences were merged to the reference sequences and each dataset was aligned with the MUSCLE method (Edgar, Citation2004) and trimmed according to the guidelines given by the original article. For VP2, a 366 bp-long region corresponding to the hypervariable region was considered. For VP1, the considered portion was a 466 bp-long marker region largely corresponding to the B marker region described by Alfonso-Morales et al. (Citation2015).
Following the proposed classification, the evolutionary distances between the different genogroups were estimated using the Maximum Composite Likelihood model (Tamura et al., Citation2004) implemented in MegaX (Kumar et al., Citation2018). The phylogenetic trees were obtained by using the Maximum Likelihood method (1000 bootstrap, Tamura-Nei substitution model) implemented in MegaX (Kumar et al., Citation2018).
Recombination analysis
To assess whether the Portuguese strains may have originated from a recombination event, all three full datasets were analyzed with different approaches. A first analysis was performed on each dataset with RDP4 (Martin et al., Citation2015). The primary scan was performed using RDP, GENECONV, Chimaera, and 3Seq, while recombination was confirmed with the whole set of available methods. The parameters of each method were adjusted based on the dataset features following the RDP4 manual recommendations. Recombination events were accepted only if detected by more than two methods with a significance level of P < 0.001 with Bonferroni correction. Evidence of recombination occurrence was further investigated using the Single Breakpoint Recombination (SBP) method, implemented in HYPHY (Martin et al., Citation2015).
Amino acid sequence analysis
A larger dataset was prepared by including all the partial VP2 sequences available in GenBank. After a preliminary alignment with the MUSCLE method (Edgar, Citation2004), sequences that were highly divergent or too short were deleted. Sequences were then realigned and compared with Portuguese strains at amino acid level searching for unique changes. In particular, the comparison was focused on the four loop structures located within hvVP2, namely PBC (aa 219-224), PDE (aa 249-254), PFG (aa 283-286), PHI (316-324) (Jackwood, Citation2012).
Phylodynamic analysis
Portuguese IBDV clade origin (time to the most recent common ancestor; tMRCA) was estimated using the Bayesian serial coalescent-based approach implemented in BEAST 1.10 (Suchard et al., Citation2018). For this purpose, all the sequences with known collection dates from the partial VP1 and VP2 datasets proposed by Islam et al. (Citation2021) were selected. Vaccine and vaccine-like sequences were removed from the alignment and the strengths of phylogenetic and temporal signals were preliminary evaluated using Iq-Tree 2 (for likelihood mapping) (Minh et al., Citation2020) and TempEst (Rambaut et al., Citation2016), respectively.
The best substitution model was selected based on the Bayesian information criterion, calculated using Jmodeltest (Darriba et al., Citation2012), while the molecular clock and population dynamic model were selected based on marginal likelihood calculation and comparison using the Path Sampling and Stepping Stone methods (Baele et al., Citation2012). The final estimations were obtained by performing a 100 million generation Markov Chain Monte Carlo run, sampling parameters and trees every 20,000 generations. Results were visually inspected using Tracer 1.7 (Rambaut et al., Citation2018) and accepted only if mixing and convergence were adequate and the Estimated Sample Size was greater than 200 for all parameters.
Parameter estimation was summarized in terms of mean and 95% Highest Posterior Density (HPD) after the exclusion of a burn-in equal to 20% of the run length. Maximum clade credibility (MCC) trees were constructed and annotated using TreeAnnotator (BEAST package) (Drummond & Rambaut, Citation2007).
Results
Sample collection and analysis
The eight samples featured by a distinctive genetic sequence were collected in December 2020 and January 2021 from six different broiler farms belonging to two separate poultry companies. The farms were located in four different regions of Central and Northern Portugal. No signs were reported in any of the considered flocks.
The strains were named according to the nomenclature system proposed by Jackwood et al. (Citation2018). The birds were 35–38 days old and vaccinated with different IBDV vaccines. A partial VP2 sequence was obtained from all samples, while a partial VP1 sequence was obtained only from six samples. The respective names and GenBank accession numbers are listed in .
Table 1. GenBank accession numbers of the partial VP1 and VP2 sequences of the Portuguese strains.
Classification according to Michel and Jackwood (Citation2017)
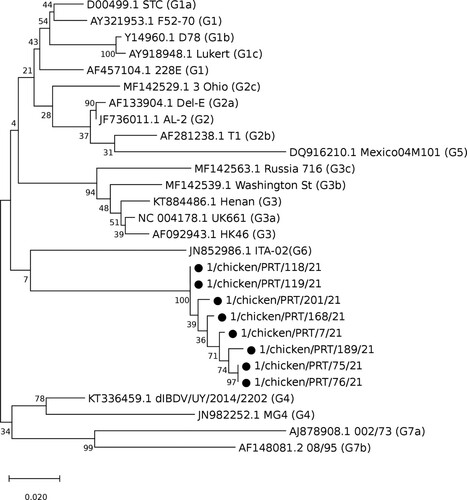
The final dataset consisted of 28 strains, including the eight Portuguese strains. The length of the considered hvVP2 portion was 541 bp. The obtained phylogenetic tree is shown in . The closest strain to the Portuguese clade is ITA-02, the reference strain for genogroup G6 (ITA), which includes strains from the Kingdom of Saudi Arabia, Russia, and most notably the ITA strains, found in Italy and associated with subclinical infections (Michel & Jackwood, Citation2017; Lupini et al., Citation2020).
Figure 1. Phylogenetic tree showing the evolutionary relationships of the Portuguese strains (marked with a black circle ●) with the reference sequences of the genogroups and lineages identified by Michel and Jackwood (Citation2017) and Jackwood et al. (Citation2018), respectively.
Classification according to Islam et al. (Citation2021)
The full VP1 dataset comprised 440 sequences, among which 143 belonged to genogroup B1, 164 to B2, 44 to B3, 20 to B4, 63 to B5, and 6 were Portuguese strains. The full VP2 dataset included 63 sequences belonging to genogroup A1, 11 to genogroup A2, 329 to genogroup A3, 20 to A4, four to A5, seven to A6, nine to A7, 10 to A8, eight to A0, and eight Portuguese strains, for a total of 469 sequences.
The reduced VP1 and VP2 datasets comprised 82 and 103 sequences, respectively. For VP2, a 366 bp-long region corresponding to the hypervariable region was considered. For VP1, the considered portion was 466 bp-long and located in a marker region identified by Alfonso-Morales et al. (Citation2015). The phylogenetic trees are shown in (a), for segment A, and 2(b), for segment B.
Figure 2. Phylogenetic tree showing the evolutionary relationships of the Portuguese strains (marked with a black circle ●) with the reference sequences of the genogroups identified by Islam et al. (Citation2021), based on partial sequences of segment A (a) and segment B (b).
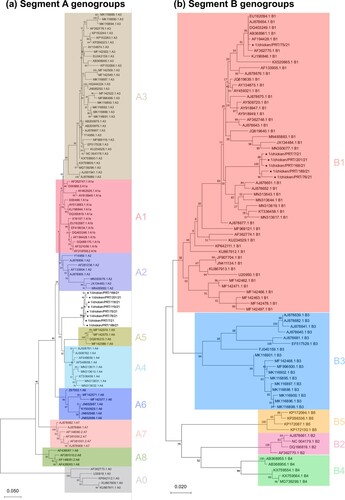
The closest segment A genogroups are A5, grouping recombinant strains steadily identified in Mexico in the last 20 years, and A4, which includes the so-called distinct IBDV (dIBDV) strains found mainly in South America. As for segment B classification, the Portuguese strains are placed within genogroup B1 (Classical-like). The Portuguese VP1 sequences form a single cluster, except for 1/chicken/PRT/75/21, which is found in a different branch.
The mean evolutionary distances between different genogroups are shown in , for VP1, and , for VP2. Since the 1/chicken/PRT/75/21 VP1 sequence did not cluster together with the other Portuguese strains, it was not included in the analysis.
Table 2. Estimated mean evolutionary distances between genogroups based on a 466 bp-long region of VP1 gene.
Table 3. Estimated evolutionary distances between genogroups based on a 366 bp-long portion of the hypervariable region of VP2 gene.
Recombination analysis
No recombination events were identified in the Portuguese strains.
Amino acid sequence analysis
The final amino acid sequence dataset comprised 1390 partial VP2 sequences. Within the PBC loop, the Portuguese strains featured a unique K221-S222 pattern. The Portuguese strains featured an N254 at the tip of the PDE loop, which also characterizes antigenic variant and Mexican recombinant strains. No relevant changes were found in the PFG loop, while a K328 flanking the PHI loop was found only in the Portuguese genogroup and in a single very virulent strain.
Portuguese clade origin
The selected VP2 dataset demonstrated an adequate phylogenetic and temporal signal and was thus processed for further analysis. The tMRCA of the Portuguese clade was estimated in 2017.13 [95HPD: 2012.45-2019.11] (Supplementary figure 1). The VP1 dataset did not demonstrate a sufficient temporal signal and further analyses were not carried out.
Discussion
The analyses performed according to both classification methods indicate that the detected strains belong to a hitherto undescribed genotype. Based on the classification by Michel and Jackwood (Citation2017), which relies on a purely phylogenetic approach, the Portuguese strains cluster outside every described genogroup. According to this classification, the Portuguese genogroup would therefore be named G8. When considering the classification proposed by Islam et al. (Citation2021), the Portuguese clade falls outside every established VP2 genogroup, too. As for VP1 genogroups, the Portuguese strains fall into the B1 genogroup.
Despite being within the same genogroup, the 1/chicken/PRT/75/21 VP1 sequence is notably different from the other Portuguese sequences. This strain was found in the same farm and at the same time as 1/chicken/PRT/76/21, which shares an identical VP2 sequence. Aside from a reassortment event, another tentative explanation for the observed VP1 diversity could involve the co-circulation within the farm of a field strain and the administered live attenuated vaccine. The amplified VP1 sequence might thus belong to the vaccine strain rather than to the field strain. Apart from genogroup B2, which is almost unique to very virulent strains, B1 is the most frequently detected VP1 genogroup. A significant number of classical, variant and very virulent strains falls into this genogroup, together with all dIBDV, Italian and serotype 2 strains. It is therefore not surprising that the Portuguese strains possess such features at VP1 level.
To establish a new genogroup, the classification by Islam et al. (Citation2021) sets that at least three strains should be retrieved from two different outbreaks, and they should have at least 12% and 9% nucleotide divergence from known genogroups at the VP1 and VP2 level, respectively. The Portuguese strains had a 6.4% average nucleotide divergence from VP1 genogroup B1, thus being comprised in it. On the other hand, their divergence from VP2 genogroups ranges from 10.7% to 18.8%, enough to consider them a new genogroup, which would be named A9. Considering both segments, the Portuguese strains would form the new genotype A9B1.
Based on recombination analyses, the Portuguese genotype does not seem to have been originated by recombination events, which were identified in other atypical IBDV strains, specifically within segment A in strains belonging to the Mexican genogroup (Jackwood, Citation2012). It is, however, worth noting that, despite their biological significance, the length of the considered genomic segments is nonetheless limited and it does not allow to definitely rule out the occurrence of recombination events.
Since several genogroups identified by both classification methods appear to circulate in geographically limited areas, it would be interesting to understand whether this is true for the newly identified group. A BLAST query found only one VP2 sequence (GenBank accession number KP878300) similar enough to the Portuguese strains to be considered as a potential member of the same genogroup, with a genetic identity ranging between 96.51–97.17%. Based on the available metadata, this VP2 sequence, named CAT278-09, was collected in 2009 in Spain and classified as a variant, although the 480-bp sequenced region was not long enough to be included in either of the two proposed classifications. This information suggests that the newly described genotype has been circulating in the Iberian Peninsula for at least a decade. tMRCA estimation largely agrees with this hypothesis, since the clade origin was estimated several years before its first detection in Portugal, even if at a later time compared to the Spanish one. An introduction from Spain can thus not be excluded. However, the sparse nature of the data prevents any robust speculation. Finally, the detection of Portuguese strains in different areas and farms belonging to different companies further corroborates this hypothesis.
Several amino acid positions within the VP2 have been recognized as pathogenicity and antigenicity determinants. The Portuguese strains were thus compared to 1390 IBDV hvVP2 sequences from GenBank at the amino acid level to assess whether the new genotype possesses any distinctive features in key positions.
In particular, the significant variability of position 222 and its location in the PBC hydrophilic loop on the surface of the VP2 projection domain makes it one of the most studied positions (Schnitzler et al., Citation1993; Coulibaly et al., Citation2005). A change from P to T at this site is thought to have played a role in the emergence of antigenic variant strains and subsequent vaccine failures (Brown et al., Citation1994; Michel & Jackwood, Citation2017). The S222 mutation found in Portuguese strains is also found in 93 (6.6%) of the considered strains, mostly belonging to the antigenic variant genogroup but also to the classical, very virulent and dIBDV genogroups.
More interesting is the residue found at position 221, which is far more conserved than 222, where 1321 of the analyzed strains (95%) show a Q221. K221 is found only in the Portuguese strains and in 52 antigenic variant strains (3.7%), of which 49 belong to a Chinese cluster of strains associated with subclinical infections (Fan et al., Citation2019). Since all these antigenic variant strains are featured by a T222, besides three strains possessing an A222, the K221-S222 pattern is unique to the Portuguese genotype.
Another well-studied amino acid feature is the one at position 254 within the PDE loop, a domain responsible for cell tropism and virulence (Qi et al., Citation2013). The N254 residue found in Portuguese strains was frequently observed in viruses that could break through maternal immunity and it is thought to contribute to antigenic drift (Jackwood & Wagner, Citation2011). Lastly, the Portuguese genotype is characterized by a K328 mutation in the proximity of the PHI loop, located in the second hvVP2 hydrophilic region. Out of all the considered sequences, K328 is observed in only one other strain, detected in Russia and belonging to the very virulent genogroup. While the attention paid to these amino acid changes is justified by their meaningful positions, it should be stressed that their actual effect on antigenicity and pathogenicity is not currently known.
In conclusion, this study allowed us to demonstrate the circulation of a previously unidentified IBDV genotype in Portugal, thus stressing the importance of steady and standardized monitoring to identify the emergence of new viral threats and evaluate the efficacy of the implemented control measures. In a similar way to the previously proposed classification methods, this work had two main limitations: the molecular characterization based on relatively short portions of VP1 and VP2, and the impossibility of assessing the pathogenicity of a given strain directly from its genetic features. The importance of the size and position of the considered region is highlighted by the fact that, even if the hvVP2 portions considered by the two classifications were largely overlapping, the topology of the respective phylogenetic trees showed significant differences. As for the need for a more precise characterization, further studies are needed to fully understand the actual impact of the newly discovered Portuguese genotype. In particular, future research efforts will be devoted to the isolation of strains belonging to this novel genotype, which would be pivotal to assess their pathogenicity with in vivo trials, determine whether the available vaccines are effective against them, and obtain full genome sequences.
Ethical statement
The considered samples were collected within the context of routine diagnostic activities and not for experimental purpose. No ethical approval was therefore required.
Supplemental Material
Download JPEG Image (1.5 MB){kind=link}
Disclosure statement
No potential conflict of interest was reported by the authors.
Related Research Data
References
- Alfonso-Morales, A., Rios, L., Martinez-Perez, O., Dolz, R., Valle, R., Perera, C.L., Bertran, K., Frías, M.T., Ganges, L., Díaz de Arce, H., Majó, N., Núñez, J.I. & Pérez, L.J. (2015). Evaluation of a phylogenetic marker based on genomic segment B of infectious bursal disease virus: facilitating a feasible incorporation of this segment to the molecular epidemiology studies for this viral agent. PLoS One, 10, e0125853.
- Alkie, T.N. & Rautenschlein, S. (2016). Infectious bursal disease virus in poultry: current status and future prospects. Veterinary Medicine (Auckland, N.Z.), 19, 9–18.
- Altschul, S.F., Gish, W., Miller, W., Myers, E.W. & Lipman, D.J. (1990). Basic local alignment search tool. Journal of Molecular Biology, 215, 403–410.
- Baele, G., Lemey, P., Bedford, T., Rambaut, A., Suchard, M.A. & Alekseyenko, A. (2012). Improving the accuracy of demographic and molecular clock model comparison while accommodating phylogenetic uncertainty. Molecular Biology and Evolution, 29, 2157–2167.
- Boot, H., Hoekman, A. & Gielkens, A. (2005). The enhanced virulence of very virulent infectious bursal disease virus is partly determined by its B-segment. Archives of Virology, 150, 137–144.
- Brandt, M., Yao, K., Liu, M., Heckert, R.A. & Vakharia, V.N. (2001). Molecular determinants of virulence, cell tropism, and pathogenic phenotype of infectious bursal disease virus. Journal of Virology, 75, 11974–11982.
- Brown, M.D., Green, P. & Skinner, M.A. (1994). VP2 sequences of recent European “very virulent” isolates of infectious bursal disease virus are closely related to each other but are distinct from those of “classical” strains. Journal of General Virology, 75, 675–680.
- Coulibaly, F., Chevalier, C., Gutsche, I., Pous, J., Navaza, J., Bressanelli, S., Delmas, B. & Rey, F.A. (2005). The birnavirus crystal structure reveals structural relationships among icosahedral viruses. Cell, 120, 761–772.
- Darriba, D., Taboada, G.L., Doallo, R. & Posada, D. (2012). Jmodeltest 2: more models, new heuristics and parallel computing. Nature Methods, 9, 772.
- Drummond, A.J. & Rambaut, A. (2007). BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evolutionary Biology, 7, 214.
- Edgar, R.C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research, 32, 1792–1797.
- Escaffre, O., Le Nouën, C., Amelot, M., Ambroggio, X., Ogden, K.M., Guionie, O., Toquin, D., Müller, H., Islam, M.R. & Eterradossi, N. (2013). Both genome segments contribute to the pathogenicity of very virulent infectious bursal disease virus. Journal of Virology, 87, 2767–2780.
- Fan, L., Wu, T., Hussain, A., Gao, Y., Zeng, X., Wang, Y., Gao, L., Li, K., Wang, Y., Liu, C., Cui, H., Pan, Q., Zhang, Y., Liu, Y., He, H., Wang, X. & Qi, X. (2019). Novel variant strains of infectious bursal disease virus isolated in China. Veterinary Microbiology, 230, 212–220.
- Islam, M.R., Rahman, S., Noor, M., Chowdhury, E.H. & Müller, H. (2011). Differentiation of infectious bursal disease virus (IBDV) genome segment B of very virulent and classical lineage by RT-PCR amplification and restriction enzyme analysis. Archives of Virology, 157, 333–336.
- Islam, M.R., Nooruzzaman, M., Rahman, T., Mumu, T.T., Rahman, M.M., Chowdhury, E.H., Eterradossi, N. & Müller, H. (2021). A unified genotypic classification of infectious bursal disease virus based on both genome segments. Avian Pathology, 50, 190–206.
- Jackwood, D.J. & Sommer-Wagner, S.E. (2005). Molecular epidemiology of infectious bursal disease viruses: distribution and genetic analysis of newly emerging viruses in the United States. Avian Diseases, 49, 220–226.
- Jackwood, D.J. & Sommer-Wagner, S.E. (2011). Amino acids contributing to antigenic drift in the infectious bursal disease birnavirus (IBDV). Virology, 409, 33–37.
- Jackwood, D.J. (2012). Molecular epidemiologic evidence of homologous recombination in infectious bursal disease viruses. Avian Diseases, 56, 574–577.
- Jackwood, D.J., Schat, K.A., Michel, L.O. & de Wit, S. (2018). A proposed nomenclature for infectious bursal disease virus isolates. Avian Pathology, 47, 576–584.
- Kumar, S., Stecher, G., Li, M., Knyaz, C. & Tamura, K. (2018). MEGA x: molecular evolutionary genetics analysis across computing platforms. Molecular Biology and Evolution, 35, 1547–1549.
- Lasher, H. & Davis, V. (1997). History of infectious bursal disease in the U.S.A.: the first two decades. Avian Diseases, 41, 11–19.
- Lee, H.J., Jang, I., Shin, S.H., Lee, H.S. & Choi, K.S. (2017). Genome sequence of a novel reassortant and very virulent strain of infectious bursal disease virus. Genome Announcements, 5, e00730–17.
- Lupini, C., Felice, V., Silveira, F., Mescolini, G., Berto, G., Listorti, V., Cecchinato, M. & Catelli, E. (2020). Comparative in vivo pathogenicity study of an ITA genotype isolate (G6) of infectious bursal disease virus. Transboundary Emerging Diseases, 67, 1025–1031.
- Maraver, A., Oña, A., Abaitua, F., González, D., Clemente, R., Ruiz-Díaz, J.A., Castón, J.R., Pazos, F. & Rodriguez, J.F. (2003). The oligomerization domain of VP3, the scaffolding protein of infectious bursal disease virus, plays a critical role in capsid assembly. Journal of Virology, 77, 6438–6449.
- Martin, D.P., Murrell, B., Golden, M., Khoosal, A. & Muhire, B. (2015). RDP4: detection and analysis of recombination patterns in virus genomes. Virus Evolution, 1, 1–5.
- Mató, T., Tatár-Kis, T., Felföldi, B., Jansson, D.S., Homonnay, Z., Bányai, K. & Palya, V. (2020). Occurrence and spread of a reassortant very virulent genotype of infectious bursal disease virus with altered VP2 amino acid profile and pathogenicity in some European countries. Veterinary Microbiology, 245, 108663.
- Michel, L.O. & Jackwood, D.J. (2017). Classification of infectious bursal disease virus into genogroups. Archives of Virology, 162, 3661–3670.
- Minh, B.Q., Schmidt, H.A., Chernomor, O., Schrempf, D., Woodhams, M.D., von Haeseler, A. & Lanfear, R. (2020). IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Molecular Biology and Evolution, 37, 1530–1534.
- OIE. (2018). Chapter 3.3.12. Infectious bursal disease virus (Gumboro disease). In OIE terrestrial manual 2018 (pp. 931–951). Paris:World Animal Health Organization.
- Pikuła, A., Śmietanka, K. & Perez, L.J. (2020). Emergence and expansion of novel pathogenic reassortant strains of infectious bursal disease virus causing acute outbreaks of the disease in Europe. Transboundary Emerging Diseases, 67, 1739–1744.
- Qi, X., Zhang, L., Chen, Y., Gao, L., Wu, G., Qin, L., Wang, Y., Ren, X., Gao, Y., Gao, H. & Wang, X. (2013). Mutations of residues 249 and 256 in VP2 are involved in the replication and virulence of infectious bursal disease virus. PLoS One, 8, e70982.
- Rambaut, A., Lam, T.T., Carvalho, L.M. & Pybus, O.G. (2016). Exploring the temporal structure of heterochronous sequences using TempEst. Virus Evolution, 2, vew007.
- Rambaut, A., Drummond, A.J., Xie, D., Baele, G. & Suchard, M.A. (2018). Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Systematic Biology, 67, 901–904.
- Schnitzler, D., Bernstein, F., Muller, H. & Becht, H. (1993). The genetic basis for the antigenicity of the VP2 protein of the IBDV. Journal of General Virology, 74, 1563–1571.
- Stoute, S.T., Jackwood, D.J., Crossley, B.M., Michel, L.O. & Blakey, J.R. (2019). Molecular epidemiology of endemic and very virulent infectious bursal disease virus genogroups in backyard chickens in California, 2009–2017. Journal of Veterinary Diagnostic Investigation, 31, 371–377.
- Suchard, M.A., Lemey, P., Baele, G., Ayres, D.L., Drummond, A.J. & Rambaut, A. (2018). Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evolution, 4, vey016.
- Tamura, K., Nei, M. & Kumar, S. (2004). Prospects for inferring very large phylogenies by using the neighbor-joining method. Proceedings of National Academy of Sciences (USA), 101, 11030–11035.