
ABSTRACT
In biology, molecular terms with the suffix “-omics” refer to disciplines aiming at the collective characterization of pools of molecules derived from different layers (DNA, RNA, proteins, metabolites) of living organisms using high-throughput technologies. Such omics analyses have been widely implemented in poultry research in recent years. This first part of a bipartite review on omics technologies in poultry health and productivity examines the use of multiple omics and multi-omics techniques in poultry research. More specific present and future applications of omics technologies, not only for the identification of specific diagnostic biomarkers, but also for potential future integration in the daily monitoring of poultry production, are discussed in part 2. Approaches based on omics technologies are particularly used in poultry research in the hunt for genetic markers of economically important phenotypical traits in the host, and in the identification of key bacterial species or functions in the intestinal microbiome. Integrative multi-omics analyses, however, are still scarce. Host physiology is investigated via genomics together with transcriptomics, proteomics and metabolomics techniques, to understand more accurately complex production traits such as disease resistance and fertility. The gut microbiota, as a key player in chicken productivity and health, is also a main subject of such studies, investigating the association between its composition (16S rRNA gene sequencing) or function (metagenomics, metatranscriptomics, metaproteomics, metabolomics) and host phenotypes. Applications of these technologies in the study of other host-associated microbiota and other host characteristics are still in their infancy.
Introduction
Feeding the growing world population is one of the big challenges of the twenty-first century. In this context, poultry meat and eggs play an ever-increasing role, as they constitute valuable and affordable sources of high-quality protein. In order to meet the increasing demand for safe and high-quality meat and eggs, while at the same time reducing the ecological footprint, the poultry industry is continuously increasing the production efficiency. This is done by implementing new management tools, new feeding strategies and formulations, and the genetic selection of high-performing birds. Traditional empirical approaches to improving management, feeding, and selection no longer suffice. Therefore, the poultry industry is increasingly relying on scientific research to further improve productivity and health of the birds. Genetic traits for disease resistance have been studied, but mostly on individual gene levels, while a large number of data are available on microbe- (or pathogen-)host interactions for improving knowledge on disease pathogenesis and resistance (Oakley & Kogut, Citation2016; Deblais et al., Citation2020; Mon et al., Citation2020). Also, studies on the effects of dietary interventions on the microbiota and host response have exponentially increased in the last decade (Borda-Molina et al., Citation2016; Eeckhaut et al., Citation2016; Huang et al., Citation2018; Zou et al., Citation2019) (for reviews on this topic see: prebiotics (Pourabedin & Zhao, Citation2015; Roto et al., Citation2015), plant-derived polysaccharides (Zhang et al., Citation2022), dietary fibres (Singh & Kim, Citation2021)). Tools that provide a complete quantitative overview of biological processes in the microbiota and the host are of major importance in understanding disease resistance and production efficiency, and can be used to identify biomarkers for health, disease, and performance.
Over the last decades, advances in high-throughput technologies, such as next-generation sequencing and mass spectrometry, have led to a revolution in biomedical research, thereby changing the research strategy from a traditional reductionist approach that focuses on a few individual molecules, genes or pathways of interest, to a more holistic approach where a complex biological system is characterized in great detail. These technological advances have allowed the creation of various new research fields, commonly referred to as “omics”. Molecular terms with the suffix “-omics” encompass a number of disciplines in biology aiming at the collective characterization and quantification of pools of biological molecules that translate into the structure, function and dynamics of an organism or organisms. The ultimate aim of such a holistic approach is to better understand biological events or processes in the environment, plants, animals and humans. Omics technologies indeed are research tools in the first place. In poultry, omics technologies have been applied mostly to study either host genetics (reviewed by Zampiga et al., Citation2018) or the intestinal microbiota in an attempt to gain insight in the role of the microbiota in gut health and performance (for review see Upadhyaya et al., Citation2019). This two-part review focuses on the current use and future applications of omics technologies in poultry health and productivity. Part 1 discusses the different omics technologies currently used in poultry research. Part 2 focuses more specifically on the practical applications of omics research, not only for the identification of next-generation diagnostic biomarkers, but also looking at the potential future implementation of the omics tools in the daily routine of the poultry industry.
The purpose of this first part is to critically review the current contributions of multiple omics to research targeting poultry health and productivity, taking into account the possibilities and limitations of the different omics technologies. The potential for the integration of multiple omics, referred to as multi-omics, to advance our understanding of complex biological processes, such as feed efficiency, disease resistance and microbiota-host interactions, is also discussed.
Omics and multi-omics: dissecting the different layers of the living organism
Organisms are biologically complex, because they comprise many interacting parts, including molecules, cells, tissues, organs, and organ systems (Kane & Higham, Citation2015). A biological organism consists of different biological layers (e.g. genes, proteins, etc.) (). Genomics studies the complete genome sequence of an organism. The focus of genomics studies is to find genes or genetic variants that are associated with specific phenotypes, such as disease resistance or animal performance (Tuite & Gros, Citation2006). Transcriptomics studies the expression of all genes in a cell or organism, thereby measuring the direct activity of the genome, or any change thereof, under different conditions at a specific timepoint (Khodadadian et al., Citation2020). Using proteomics, the entire set of proteins produced or modified by an organism is studied. This approach connects the genes with their functionally diverse protein products, which are the predominant mediators of cellular functions (Deblais et al., Citation2020; Almeida et al., Citation2021). Metabolomics provides a snapshot of the metabolic state of an organism or tissue. Metabolites can be seen as the intermediates and products of cellular processes. Therefore, changes in metabolite concentration and composition can reflect changes in functions of the mediating enzymes and proteins (Putri et al, Citation2013; Stanberry et al., Citation2013; Wörheide et al., Citation2021).
Figure 1. The different omics layers of the host and the microbiota and the interactions between them: holo-omics. Biomolecular interactions between host and microbiota triggered by environmental factors yield different phenotypes. Green-blue arrows indicate host-microbiota holo-omics interactions. Blue and purple arrows, respectively representing host and microbiome domain, indicate omics levels influencing host phenotype. Green arrows indicate omics levels influenced by environmental factors. Colour online.
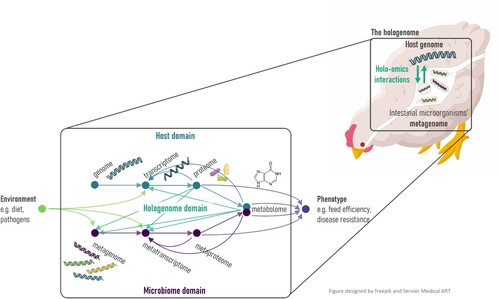
The biological processes of complex organisms are highly influenced by bidirectional interactions between the host organism and its associated microbiome (Nyholm et al., Citation2020; Wen et al., Citation2021). Moreover, the microbiome is essential for many biological processes including, amongst others, nutrient acquisition, immune development and pathogen exclusion (Shang et al., Citation2018). Therefore, information on both the host as well as its microbiomes is needed to characterize the interplay between the two and elucidate how this influences animal performance and disease resistance. The most studied microbial ecosystem in poultry is the gut microbiome, but other microbiomes, such as the respiratory microbiome or less straightforward microbiomes such as blood and bone microbiomes, have been increasingly investigated (Mandal et al., Citation2016; Johnson et al., Citation2018; Michael et al., Citation2020; Zhou et al., Citation2021). In accordance with the omics approaches used to study different biological layers of the avian organism or tissue, similar omics approaches are used to study different layers of the microbiome (Jansson & Baker, Citation2016). Microbial community genomics can be studied using two different approaches. The most common and cost-effective approach is by focusing on the 16S rRNA gene as a phylogenetic marker. 16S rRNA gene sequencing studies allow the exploration of the bacterial community composition, but no information on the functional genes and pathways present in the microbial community is available. Metagenomics studies the genomes from all microorganisms in the community, thereby obtaining information from the entire gene complement, including phylogenetic and functional genes. Metagenomics thus provides information on the metabolic potential of a microbial community, but no information on the actual metabolic activity is provided. In order to elucidate which genes are expressed and translated into proteins, metatranscriptomics and metaproteomics approaches are used, respectively, whereas metabolomics provides information on the actual metabolites present (Jansson & Baker, Citation2016). The integration of data across multiple omics levels from both host and microbiota domains has recently been introduced as holo-omics (Nyholm et al., Citation2020). Such an approach would represent a useful tool to improve our understanding of the biology of host-microbiome interactions and inform microbial approaches to improve host health (Nyholm et al., Citation2020) ().
Currently, the use of omics approaches is well integrated into poultry research. However, the majority of the studies focus only on a single omics type. Single omics approaches have resulted in increased knowledge on disease resistance (Cheng et al., Citation2013), productivity (Zampiga et al., Citation2018) and biomarker discovery (de Meyer et al., Citation2019), and understanding of the pathogenesis and host response to infectious (Deblais et al., Citation2020) and non-infectious challenges (Zampiga et al., Citation2021). Although these studies are highly valuable, single omics studies do not take into account the complex interplay between biomolecules from different molecular layers, and the analysis of only one omics subset might provide an incomplete picture of the underlying biology. For example, as protein function is mediated and altered by post-translational modifications (e.g. phosphorylation, complex formation, post-translational processing of pre-proteins), correlations between gene expression levels and protein expression levels of cellular proteomes are relatively low (Washburn et al., Citation2003; Olivier et al., Citation2019). Additionally, changes in the metabolome or proteome can modulate gene expression levels, thereby creating complex inter-omics interactions (Wörheide et al., Citation2021). As such, each single omics has its own features that could compensate for limitations of other omics techniques, and an integrated analysis of multiple omics datasets is needed to obtain a full picture of the underlying biological processes that steer animal performance and disease resistance.
Current use of omics in poultry research
Single omics and the need for multiple omics to study host parameters associated with production traits
Single omics technologies have been used to understand biological processes behind production traits, such as feed efficiency, nutrition, meat quality, disease resistance and fertility. The use of different single omics approaches in poultry research has recently been reviewed (Zampiga et al., Citation2018; Long, Citation2020). In the current review, we highlight the use of omics approaches to study host disease resistance, and host fertility, with special emphasis on how multi-omics approaches might further contribute to these research fields. Disease resistance is primarily investigated using host genomics. The release of the chicken genome sequence in 2004 greatly enhanced the ability to select for improved disease resistance via genetic markers and to understand more deeply the biological basis of host resistance. Genome-wide associations studies (GWAS) are commonly used, aiming to identify genetic variants such as Quantitative Trait Loci (QTL) associated with diseases such as Marek’s disease and susceptibility to Salmonella and Campylobacter (Cheng et al., Citation2013). When looking at Salmonella resistance in chickens, a major QTL controlling spleen bacterial load was identified on chromosome 5 and named SAL1 (Mariani et al., Citation2001). This QTL was shown to be involved in bacterial clearance by macrophages. Another QTL linked to Salmonella contamination in the caecum has raised interest due to its proximity with immune-response genes such as the Major Histocompatibility Complex (MHC) (Tilquin et al., Citation2005). These genomic regions, which often contain several hundred genes, are interesting candidates for more in-depth studies. However, some of those genes might not be expressed in response to infection and are unlikely to explain phenotypic variations. Therefore, genomic information alone is not enough to unravel the biological host–pathogen interaction, and future research could benefit from the use of multiple omics. Functional omics, such as transcriptomics, proteomics and metabolomics, connect genome to gene functions and are needed to identify factors responsible for the successful or unsuccessful infection by a pathogen (Deblais et al., Citation2020). These findings might help to develop resistant chicken lines as well as novel antibacterial strategies. For instance, by investigating the microRNAome response in chicken spleen to avian pathogenic Escherichia coli (APEC) infection, Jia et al. (Citation2017) identified gga-miR-429 miRNA, which is associated with the enhanced susceptibility of APEC in chickens. Therefore, suppression of gga-miR-429 expression could increase the chicken’s resistance to APEC infection. As a second example, omics have also allowed the identification and the validation of biomarkers, such as genes, transcripts, proteins, and metabolites, that are associated with fertility phenotypes, which holds great potential to improve the reproductive efficiency of poultry (Da Silva et al., Citation2019; Long, Citation2020). The identification of molecular markers for abnormal embryonic development is of importance for poultry production. Ninety-one nonredundant proteins have been identified through GeLC-MS/MS and shotgun strategies, delineating the chicken amniotic fluid proteome at day 11 of development, before egg white transfer. These proteins are essentially associated with the metabolism of nutrients, immune response, and developmental processes. These results constitute a reference starting point for analyses of pathological conditions due to infection or impaired development of the chicken embryo. This may lead to the identification of valuable biomarkers, where differences in concentration in certain proteins could help with detecting hazardous situations such as heat stress, inflammation, or infection, that may negatively affect the development of the chicken embryo (Da Silva et al., Citation2019).
Selection of broilers for rapid growth has been accompanied by an increase in abdominal fat, which has low commercial value and decreases feed efficiency. Adipose tissue is well known to be involved in the regulation of appetite and body growth in mammals, due to the expression of adipokines such as leptin (Friedman & Halaas, Citation1998). The latter is hardly expressed in avian adipose tissue (Friedman-Einat & Seroussi, Citation2019), therefore questioning its function. Some suggest that adipose tissue might play a role in the higher predisposition of broilers to metabolic disorders compared to layers (Bornelöv et al., Citation2018). Multiple omics studies have been employed to investigate the adipose tissue and its role in chicken health further (Bornelöv et al., Citation2018; Zhang et al., Citation2020). Transcriptome and proteome data from chicken abdominal fat have revealed the expression of new reproduction-related proteins and suggest a direct crosstalk of the chicken visceral fat with the reproductive system and a lower involvement in the regulation of appetite, inflammation and insulin resistance as compared to mammals (Bornelöv et al., Citation2018).
Multi-omics or integrated omics to better understand host physiology
While many papers characterize themselves as multi-omics studies, they are reporting the simultaneous use of different omics without performing a real effort to integrate the different data layers, which is what multi-omics or integrative omics should aim to do. This approach allows us to understand complex biological processes more accurately and has only been used in poultry research since 2017. At the time of writing this paper, only one study has integrated transcriptomic and metabolomic data to understand how the liver responds under chronic heat stress and has highlighted multiple pathways which are affected under these conditions (Jastrebski et al., Citation2017). The integration of transcriptomic and proteomic data seems to be the most employed multi-omics approach in poultry studies. Fatty acid metabolism/fat storage in birds is a major subject of such studies, highlighting the importance of this trait for poultry production. Transcriptomics and proteomics have been combined in a few studies, and differentially abundant genes, proteins or pathways have been investigated in fat and lean chicken lines to identify key genes involved in the regulation of abdominal fat deposition (Na et al., Citation2018; Wang et al., Citation2021). Genes related to fatty acid metabolism, fatty acid biosynthesis and PPAR signaling are notably down-regulated in lean lines (Na et al., Citation2018; Wang et al., Citation2021). In order to further investigate lipid metabolism in two groups of broiler chickens with high and low abdominal fat, gene expression data from RNAseq have been integrated with microRNAs expression data (Ghafouri et al., Citation2021). An interactive gene–microRNA bipartite network has been created and has revealed that a gene set involved in the PPAR pathway, among others, plays a role in abdominal fat storage of animals, consistent with previous findings which identified PPAR as a key pathway (Wang, et al., Citation2021). In addition, multi-omics have been employed to understand the host response when subject to disease, especially in the case of Newcastle disease (Saelao et al., Citation2018; Chanthavixay et al., Citation2020), reticulo-endotheliosis (Zhai et al., Citation2018; Gao et al., Citation2019) and Mycoplasma synoviae infection (Liu et al., Citation2020).
16S rRNA gene sequencing: the composition of the gut microbiota plays a role in chicken productivity and health
The gastrointestinal (GI) tract of chickens harbours a diverse and complex microbiota that greatly influences overall poultry health and performance, by playing a vital role in digestion and absorption of nutrients, immune system development and pathogen exclusion (Shang et al., Citation2018). The diversity of the chicken GI microbiota is largely influenced by the age of the birds, the location in the digestive tract, the environment, and the diet (Oakley et al., Citation2014; Shang et al., Citation2018; Rychlik, Citation2020). Understanding and optimizing the gut microbiota composition and functionality towards health and productivity has therefore become a major objective for the poultry industry and a main topic in poultry research, where omics technologies can play a key role (for review see Upadhyaya et al., Citation2019).
16S rRNA gene amplicon sequencing has been the most widely used omics approach in microbiome research and has allowed researchers to gain significant knowledge on which microbes are present in the various segments of the poultry gut. As early as 2013, Wei and colleagues used phylogenetic profiling of 16S rRNA gene-based sequences to compare the global diversity of intestinal microbiome between chickens and turkeys, which served as the working framework for describing bacterial diversity in the poultry gut (Wei et al., Citation2013). Since then, studies using 16S rRNA gene amplicon sequencing consistently aimed to identify microbiome characteristics associated with bird health and to develop modulation strategies (e.g. optimal diet formulation or probiotics) to enhance the abundance of beneficial bacteria. Caecal bacterial taxa such as Faecalibacterium prausnitzii, Ruminococcus genus, Lachnospiraceae family and Bacteroides genus, are associated with good bird performance or low feed conversion ratio (FCR) (Stanley et al., Citation2012, Citation2013, Citation2014, Citation2016; Singh et al., Citation2012; Johnson et al., Citation2018). These bacteria can degrade complex indigestible carbohydrates present in chicken feed, such as cellulose and hemicellulose, into end metabolites that can be utilized by the bird, thereby providing additional energy to the bird. Microbial carbohydrate fermentation is accompanied by the production of short chain fatty acids (SCFA), such as butyrate, which is beneficial for gut health as it provides energy to epithelial cells, stimulates cell proliferation, reinforces the gut barrier and may inhibit certain pathogens (Guilloteau et al., Citation2010). In contrast to the above-mentioned taxa, which are consistently associated with good performance, contradictory findings are reported for the genus Lactobacillus, even if strains belonging to this genus are commonly used as probiotics in poultry. This might suggest that different strains within this same genus have different effects on feed efficiency and performance (Stanley et al., Citation2016; Yan et al., Citation2017; Johnson et al., Citation2018). 16S rRNA gene amplicon sequencing has also provided insight into the early development of the intestinal microbiome. When the caecal development in newly hatched chickens with or without contact with an adult hen is investigated, it appears that hens are efficient donors of Bacteroidetes, Actinobacteria, Selenomonadales and Faecalibacterium, but not of most important Gram-positive bacteria inhabiting the gut such as Clostridiales or Lactobacilli, which appear to come mostly from the environment (Kubasova et al., Citation2019a). In a follow-up study, these authors confirmed that these vertically inherited bacteria can efficiently colonize the caecum of newly hatched chicks after a single dose, while bacteria commonly used as probiotics such as Bacillus, Lactobacillus or Enterococcus do not (Kubasova et al., Citation2019b). These observations may have consequences for the design of probiotics for poultry.
In addition to the gut microbiome, there is increasing scientific interest in the impact of other microbiomes on poultry productivity. Johnson et al. (Citation2018) found that potential respiratory pathogens in the trachea detected by 16S rRNA gene sequencing are negatively correlated with performance. Studies based on 16S rRNA gene sequencing also have found that bacterial chondronecrosis with osteomyelitis (BCO), an important cause of lameness in commercial broiler chickens, might be related to microbial communities present in bones (Jiang et al., Citation2015) or blood (Mandal et al., Citation2016). When caecum and extra-intestinal microbiota (blood, femur, tibia) of chickens raised under stress conditions were investigated, it was hypothesized that bacteria translocated across the impaired gut barrier due to stress. These extra-intestinal sites display a high abundance of novel taxa that need to be further explored for their role in health and disease of chickens (Mandal et al., Citation2020).
Omics techniques and the functional analysis of the gut microbiota: metagenomics
The modulation of the microbiome to improve poultry health and production requires knowledge of how the intervention will impact the host. However, because commercially hatched chicks have no contact with the parent hens, the initial gut microbiota composition is greatly affected by the environment, and the microbiome composition of young birds is highly variable between studies (Rychlik, Citation2020). The outcome of an intervention might depend on this initial microbiota, which questions its reproducibility and biological relevance. In contrast to the high variability in microbial populations, the functional pathways of the microbiome seem more stable and conserved across studies (Qi et al., Citation2019). Therefore, investigating the functional profile of the gut microbiota appears of major importance. Consequently, researchers tend to shift towards metagenomic approaches, which provide a measure of the metabolic capabilities of the microbiome. One of the first examples combined both 16S rRNA gene and metagenomic sequencing to assess the effect of anticoccidial and antibiotic treatments on the caecal microbiome (Danzeisen et al., Citation2011). Shortly afterwards, Sergeant et al. (Citation2014) performed an in-depth metagenomic analysis on a single sample, to gain insight into the function of the caecal microbiota in chickens. Pathways for non-starch polysaccharides (NSPs) utilization, SCFA production and hydrogen consumption were identified (Sergeant et al., Citation2014). The first gene catalogue of the chicken gut microbiome was created in 2018, covering all intestinal compartments, and generated from metagenomic data (Huang et al., Citation2018). Since then, many research groups have used metagenomics to further elucidate the functionality of the chicken intestinal microbiome, leading to a better characterization of acquired antimicrobial resistance genes in poultry farms (Luiken et al., Citation2020; Feng et al., Citation2021), the identification of the genes involved in SCFA production, and detailed annotation of carbohydrate-active enzymes (CAZymes) (Glendinning et al., Citation2020; Segura-Wang et al., Citation2021). These CAZymes could be useful biomarkers of the functional capacity of the gut microbiota to digest complex carbohydrates without focusing on single isolated bacteria that are not representative of the community (Segura-Wang et al., Citation2021).
As an alternative to full shotgun metagenome sequencing, metagenome functional predictions from 16S rRNA gene libraries can be performed using computational approaches (e.g. PICRUST or Tax4Fun) (Langille et al., Citation2013; Aßhauer et al., Citation2015; Douglas et al., Citation2020; Wemheuer et al., Citation2020). High correlations between inferred and metagenomically measured gene content have been reported for the human, mammalian gut and soil microbiome (Douglas et al., Citation2020; Wemheuer et al., Citation2020). However, the accuracy and reliability of these tools depend on the availability of whole genome sequences in public databases. A large number of these sequences is available for the human gut microbiome, whereas the poultry microbiome is less studied. Therefore, functional predictions are mainly based on whole genome sequences from human microbiota. As the same genera are present in the avian gut and human gut, the major metabolic characteristics of those bacterial species will be comparable, and overall functional predictions of the poultry microbiome are believed to be reliable (Rychlik, Citation2020). However, host-specific differences in microbial strains enable them to colonize and interact with their respective host (Chung et al., Citation2012). This indicates that strains belonging to the same species, but colonizing different hosts (e.g. poultry versus human strains) should have some metabolic differences linked to host adaptation, which will not be reflected in the predicted functional metagenomes (Ma et al., Citation2018; Rychlik, Citation2020). Indeed, recent research revealed an extensive uncharacterized microbial diversity in the chicken gut microbiome (Glendinning et al., Citation2020; Feng et al., Citation2021; Gilroy et al., Citation2021; Segura-Wang et al., Citation2021). This clearly indicates how underrepresented chicken microbial strains are in public database. Therefore, the inferred metagenomic functions should be interpreted with caution.
Multiple omics and multi-omics to study the gut microbiota: getting closer to the actual functions and roles of bacteria in chicken health
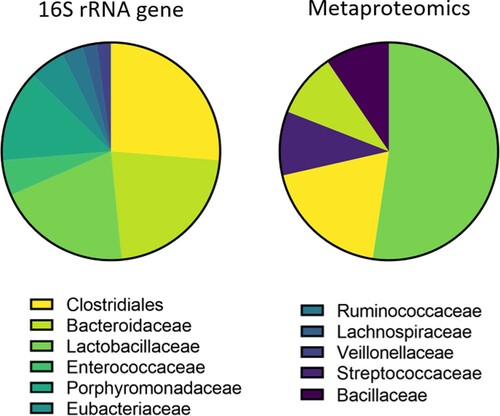
So far, studies of the chicken microbiota have mainly been performed using 16S rRNA gene amplicon sequencing or metagenomics. However, these DNA-based approaches also detect dormant or even dead microorganisms that do not contribute to the activity of the microbial community (Jansson & Baker, Citation2016). Therefore, other approaches are needed to elucidate the true functionality of the microbiome. Indeed, multiple studies have observed large discrepancies between the active microbiome derived from metaproteomics, and the total microbiome obtained through 16S rRNA gene sequencing (Tang et al., Citation2014; Borda-Molina et al., Citation2016, Citation2018) (). This shows that metaproteomics brings another layer of information that is otherwise not accessible by purely compositional studies. The caecal metaproteome of chickens has also been investigated to identify promising probiotic strains. It was found that Anaerostipes, Anaerotruncus and Subdoligranulum might be good candidates as they express both spore-forming proteins and enzymes required for butyrate production (Polansky et al., Citation2016). In addition to metaproteomics, metabolomics is a frequently used omics approach. In one example, metabolomics was employed to understand the caecum microbiome-host interaction during Salmonella infection in young layers (Mon et al., Citation2020). Microbial metabolite pathway enrichment analysis revealed that arginine and proline metabolism were enriched after Salmonella Enteritidis infection, which was consistent with host caecal tonsil RNA-seq data showing the upregulation of arginine-associated pathways at 3 days post-infection. The upregulation of this pathway might be a way for the host to regulate the intestinal inflammation during Salmonella infection. This kind of study might lead to the development of novel preventive and control strategies for Salmonella infections in poultry production. In addition, novel omics such as kinomics (the study of protein phosphorylation) show potential to uncover the complex of signals among the microbiome, intestinal lumen metabolites, and the host (Lee et al., Citation2022).
Figure 2. Phylogenetic differences in faecal microbiome composition obtained by 16S rRNA gene sequencing or metaproteomics. Only families (or order level for Clostridialesa) with more than one count in either the 16S rRNA gene data or metaproteomics data were plotted. Large differences are observed between the microbiome composition obtained by 16S rRNA gene sequencing or inferred by metaproteomics. The Clostridiales, Bacteroidaceae and Lactobacillaceae were detected by both 16S rRNA gene sequencing and metaproteomics, whereas the families Streptococcaceae and Bacillaceae were only present in the metaproteome data. For the other families, the 16S rRNA gene was detected, without the identification of proteins produced by these families, indicating that these bacterial families might be less metabolically active in the broiler faeces. Differences between the 16S rRNA gene sequencing and metaproteomics approach might be due to a combination of true biological variation (differences in metabolic activity between the bacterial families), as well as methodological differences (e.g. extraction methods, differences in databases, etc.). Data were obtained from Tang et al. (Citation2014). aDue to the resolution of the 16S rRNA gene sequencing, no family information is available for the order Clostridiales.
While most previous microbiome studies did not integrate the different omics layers studied, a few studies have done so. Bacterial relative abundances have been correlated with metabolite levels in the caecum to investigate the growth-promoting mechanism of feed additives (Chen et al., Citation2020). Another integrative study combined metagenomics and metatranscriptomics to reveal the abundance, diversity, and expression of antibiotic-resistance genes (ARGs) in chickens and other species (Wang et al., Citation2020). Multi-omics studies on the gut microbiota have the potential to move from predictive analyses to more accurate descriptions of the actual microbial activities and the gut microbiome-host interactions. Metaproteomics and metabolomics are intended to gain more precise insights into the actual functions carried out by bacteria of a microbiome.
Combining microbiome and host omics: towards understanding the crosstalk in healthy and diseased conditions
The co-extraction of host RNA, proteins or metabolites may be beneficial to gain concomitant information about the microbiome and host status. Few studies have combined omics targeting both the host and the gut microbiome. Correlations between the kidney metabolome and the caecal microbial community showed contributions of gut microbiota in the progression of nephropathogenic infectious bronchitis virus (NIBV) infection. Indeed, the relative abundance in the caecal microbiota of Bacteroides vulgatus and Lactobacillus, which were decreased by NIBV, negatively correlated with the uric acid concentration in serum, which was increased by NIBV (Xu et al., Citation2019). Some studies have combined 16S rRNA gene sequencing, performed on gut content, with transcriptomics or proteomics on host gut tissue, to investigate associations between microbiota and the intestinal immune function (Oakley & Kogut, Citation2016; Willson et al., Citation2018; Rodrigues et al., Citation2020). Faecalibacterium was found to be negatively correlated with the expression of the pro-inflammatory cytokines IL-1β and IL-18, and a relative increase in Bacteroidetes was associated with better tight junctions and Toll-like receptor expression levels, once again highlighting a potential beneficial effect of these bacteria for chicken health.
Conclusion
The use of omics in poultry research has started with host genomics, more particularly with association studies identifying correlations between genetic variants and phenotypes, to further focus on functional omics such as transcriptomics, proteomics, and metabolomics in the host. The combination of different omics indeed shows great potential to understand chicken metabolic pathways affected by an intervention or a disease. The genomics era also has shown gut microbes to play a major role in chicken health and to interact with host biological response. While earlier studies primarily tried to link gut microbiota composition (16S rRNA gene sequencing) with host phenotype, research since has focused more on the functional profile of the gut and the role of bacteria in health and disease, via metagenomics, metatranscriptomics, metaproteomics and metabolomics. Phenotypes are shaped by the bidirectional interactions between the host and its associated microbiota. The integration of multiple omics layers or multi-omics shows the potential to create more accurate insight in the biological mechanisms at play, but such studies are still scarce. Furthermore, the incorporation of data from multiple omics levels from both host and microbiota, referred to as holo-omics, would allow full understanding of the gut microbiome-host interaction that governs animal health status. However, while the techniques to generate these data are available and understood, a challenge remains in the analysis and interpretation of these integrative approaches.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Almeida, A.M., Ali, S.A., Ceciliani, F., Eckersall, P.D., Hernández-Castellano, L.E., Han, R., Hodnik, J.J., Jaswal, S., Lippolis, J.D., McLaughlin, M., Miller, I., Mohanty, A.K., Mrljak, V., Nally, J.E., Nanni, P., Plowman, J.E., Poleti, M.D., Ribeiro, D.M., Rodrigues, P., Roschitzki, B., Schlapbach, R., Starič, J., Yang, Y. & Zachut, M. (2021). Domestic animal proteomics in the 21st century: a global retrospective and viewpoint analysis. Journal of Proteomics, 241, 104220.
- Aßhauer, K.P., Wemheuer, B., Daniel, R. & Meinicke, P. (2015). Tax4Fun: predicting functional profiles from metagenomic 16S rRNA data. Bioinformatics, 31, 2882–2884.
- Borda-Molina, D., Seifert, J. & Camarinha-Silva, A. (2018). Current perspectives of the chicken gastrointestinal tract and its microbiome. Computational and Structural Biotechnology Journal, 16, 131–139.
- Borda-Molina, D., Vital, M., Sommerfeld, V., Rodehutscord, M. & Camarinha-Silva, A. (2016). Insights into broilers’ gut microbiota fed with phosphorus, calcium, and phytase supplemented diets. Frontiers in Microbiology, 7, 2033.
- Bornelöv, S., Seroussi, E., Yosefi, S., Benjamini, S., Miyara, S., Ruzal, M., Grabherr, M., Rafati, N., Molin, A.M., Pendavis, K., Burgess, S.C., Andersson, L. & Friedman-Einat, M. (2018). Comparative omics and feeding manipulations in chicken indicate a shift of the endocrine role of visceral fat towards reproduction. BMC Genomics, 19, 1–15.
- Chanthavixay, G., Kern, C., Wang, Y., Saelao, P., Lamont, S.J., Gallardo, R.A., Rincon, G. & Zhou, H. (2020). Integrated transcriptome and histone modification analysis reveals NDV infection under heat stress affects bursa development and proliferation in susceptible chicken line. Frontiers in Genetics, 11, 1176.
- Chen, Y., Wang, J., Yu, L., Xu, T. & Zhu, N. (2020). Microbiota and metabolome responses in the cecum and serum of broiler chickens fed with plant essential oils or virginiamycin. Scientific Reports, 10, 1–14.
- Cheng, H.H., Kaiser, P. & Lamont, S.J. (2013). Integrated genomic approaches to enhance genetic resistance in chickens. Annual Review of Animal Biosciences, 1, 239–260.
- Chung, H., Pamp, S.J., Hill, J.A., Surana, N.K., Edelman, S.M., Troy, E.B., Reading, N.C., Villablanca, E.J., Wang, S., Mora, J.R., Umesaki, Y., Mathis, D., Benoist, C., Relman, D.A. & Kasper, D.L. (2012). Gut immune maturation depends on colonization with a host-specific microbiota. Cell, 149, 1578–1593.
- Da Silva, M., Dombre, C., Brionne, A., Monget, P., Chessé, M., De Pauw, M., Mills, M., Combes-Soia, L., Labas, V., Guyot, N., Nys, Y. & Réhault-Godbert, S. (2019). The unique features of proteins depicting the chicken amniotic fluid. Molecular & Cellular Proteomics, 18, S174–S190.
- Danzeisen, J.L., Kim, H.B., Isaacson, R.E., Tu, Z.J. & Johnson, T.J. (2011). Modulations of the chicken cecal microbiome and metagenome in response to anticoccidial and growth promoter treatment. PLoS One, 6, e27949.
- de Meyer, F., Eeckhaut, V., Ducatelle, R., Dhaenens, M., Daled, S., Dedeurwaerder, A., de Gussem, M., Haesebrouck, F., Deforce, D. & van Immerseel, F. (2019). Host intestinal biomarker identification in a gut leakage model in broilers. Veterinary Research, 50, 46.
- Deblais, L., Kathayat, D., Helmy, Y.A., Closs, G. & Rajashekara, G. (2020). Translating ‘big data’: better understanding of host-pathogen interactions to control bacterial foodborne pathogens in poultry. Animal Health Research Reviews, 21, 15–35.
- Douglas, G.M., Maffei, V.J., Zaneveld, J.R., Yurgel, S.N., Brown, J.R., Taylor, C.M., Huttenhower, C. & Langille, M.G.I. (2020). PICRUSt2 for prediction of metagenome functions. Nature Biotechnology, 38, 685–688.
- Eeckhaut, V., Wang, J., van Parys, A., Haesebrouck, F., Joossens, M., Falony, G., Raes, J., Ducatelle, R. & van Immerseel, F. (2016). The probiotic Butyricicoccus pullicaecorum reduces feed conversion and protects from potentially harmful intestinal microorganisms and necrotic enteritis in broilers. Frontiers in Microbiology, 7, 1416.
- Feng, Y., Wang, Y., Zhu, B., Gao, G.F., Guo, Y. & Hu, Y. (2021). Metagenome-assembled genomes and gene catalog from the chicken gut microbiome aid in deciphering antibiotic resistomes. Communications Biology, 4, 1–9.
- Friedman, J.M. & Halaas, J.L. (1998). Leptin and the regulation of body weight in mammals. Nature, 395, 763–770.
- Friedman-Einat, M. & Seroussi, E. (2019). Avian leptin: bird’s-eye view of the evolution of vertebrate energy-balance control. Trends in Endocrinology & Metabolism, 30, 819–832.
- Gao, S., Jiang, H., Sun, J., Diao, Y., Tang, Y. & Hu, J. (2019). Integrated analysis of miRNA and mRNA expression profiles in spleen of specific pathogen-free chicken infected with avian reticuloendotheliosis virus strain SNV. International Journal of Molecular Sciences, 20, 1041.
- Ghafouri, F., Bahrami, A., Sadeghi, M., Miraei-Ashtiani, S.R., Bakherad, M., Barkema, H.W. & Larose, S. (2021). Omics multi-layers networks provide novel mechanistic and functional insights into fat storage and lipid metabolism in poultry. Frontiers in Genetics, 12, 1127.
- Gilroy, R., Ravi, A., Getino, M., Pursley, I., Horton, D.L., Alikhan, N.F., Baker, D., Gharbi, K., Hall, N., Watson, M., Adriaenssens, E.M., Foster-Nyarko, E., Jarju, S., Secka, A., Antonio, M., Oren, A., Chaudhuri, R.R., la Ragione, R., Hildebrand, F. & Pallen, M.J. (2021). Extensive microbial diversity within the chicken gut microbiome revealed by metagenomics and culture. PeerJ, 9, e10941.
- Glendinning, L., Stewart, R.D., Pallen, M.J., Watson, K.A. & Watson, M. (2020). Assembly of hundreds of novel bacterial genomes from the chicken caecum. Genome Biology, 21, 1–16.
- Guilloteau, P., Martin, L., Eeckhaut, V., Ducatelle, R., Zabielski, R. & Van Immerseel, F. (2010). From the gut to the peripheral tissues: the multiple effects of butyrate. Nutrition Research Reviews, 23, 366–384.
- Huang, P., Zhang, Y., Xiao, K., Jiang, F., Wang, H., Tang, D., Liu, D., Liu, B., Liu, Y., He, X., Liu, H., Liu, X., Qing, Z., Liu, C., Huang, J., Ren, Y., Yun, L., Yin, L., Lin, Q., Zeng, C., Su, X., Yuan, J., Lin, L., Hu, N., Cao, H., Huang, S., Guo, Y., Fan, W. & Zeng, J. (2018). The chicken gut metagenome and the modulatory effects of plant-derived benzylisoquinoline alkaloids. Microbiome, 6, 211.
- Jansson, J.K. & Baker, E.S. (2016). A multi-omic future for microbiome studies. Nature Microbiology, 1, 16049.
- Jastrebski, S.F., Lamont, S.J. & Schmidt, C.J. (2017). Chicken hepatic response to chronic heat stress using integrated transcriptome and metabolome analysis. PLoS One, 12, e0181900.
- Jia, X., Nie, Q., Zhang, X., Nolan, L.K. & Lamont, S.J. (2017). Novel microRNA involved in host response to avian pathogenic Escherichia coli identified by deep sequencing and integration analysis. Infection and Immunity, 85, e00688-16.
- Jiang, T., Mandal, R.K., Wideman, R.F., Khatiwara, A., Pevzner, I. & Kwon, Y.M. (2015). Molecular survey of bacterial communities associated with bacterial chondronecrosis with osteomyelitis (BCO) in broilers. PLoS One, 10, e0124403.
- Johnson, T.J., Youmans, B.P., Noll, S., Cardona, C., Evans, N.P., Peter Karnezos, T., Ngunjiri, J.M., Abundo, M.C. & Lee, C.W. (2018). A consistent and predictable commercial broiler chicken bacterial microbiota in antibiotic-free production displays strong correlations with performance. Applied and Environmental Microbiology, 84, e00362-18.
- Kane, E.A. & Higham, T.E. (2015). Complex systems are more than the sum of their parts: using integration to understand performance, biomechanics, and diversity. Integrative and Comparative Biology, 55, 146–165.
- Khodadadian, A., Darzi, S., Haghi-Daredeh, S., Eshaghi, F.S., Babakhanzadeh, E., Mirabutalebi, S.H. & Nazari, M. (2020). Genomics and transcriptomics: the powerful technologies in precision medicine. International Journal of General Medicine, 13, 627–640.
- Kubasova, T., Kollarcikova, M., Crhanova, M., Karasova, D., Cejkova, D., Sebkova, A., Matiasovicova, J., Faldynova, M., Pokorna, A., Cizek, A. & Rychlik, I. (2019a). Contact with adult hen affects development of caecal microbiota in newly hatched chicks. PLoS One, 14, e0212446.
- Kubasova, T., Kollarcikova, M., Crhanova, M., Karasova, D., Cejkova, D., Sebkova, A., Matiasovicova, J., Faldynova, M., Sisak, F., Babak, V., Pokorna, A., Cizek, A. & Rychlik, I. (2019b). Gut anaerobes capable of chicken caecum colonisation. Microorganisms, 7, 597.
- Langille, M.G., Zaneveld, J., Caporaso, J.G., McDonald, D., Knights, D., Reyes, J.A., Clemente, J.C., Burkepile, D.E., Vega Thurber, R.L., Knight, R., Beiko, R.G. & Huttenhower, C. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nature Biotechnology, 31, 814–821.
- Lee, M.D., Ipharraguerre, I.R., Arsenault, R.J., Lyte, M., Lyte, J.M., Humphrey, B., Angel, R. & Korver, D.R. (2022). Informal nutrition symposium: leveraging the microbiome (and the metabolome) for poultry production. Poultry Science, 101, 101588.
- Liu, R., Xu, B., Yu, S., Zhang, J., Sun, H., Liu, C., Lu, F., Pan, Q. & Zhang, X. (2020). Integrated transcriptomic and proteomic analyses of the interaction between chicken synovial fibroblasts and Mycoplasma synoviae. Frontiers in Microbiology, 11, 576.
- Long, J.A. (2020). The ‘omics’ revolution: use of genomic, transcriptomic, proteomic and metabolomic tools to predict male reproductive traits that impact fertility in livestock and poultry. Animal Reproduction Science, 220, 106354.
- Luiken, R.E.C., Van Gompel, L., Bossers, A., Munk, P., Joosten, P., Hansen, R.B., Knudsen, B.E., García-Cobos, S., Dewulf, J., Aarestrup, F.M., Wagenaar, J.A., Smit, L.A.M., Mevius, D.J., Heederik, D.J.J. & Schmitt, H. (2020). Farm dust resistomes and bacterial microbiomes in European poultry and pig farms. Environment International, 143, 105971.
- Ma, T., Suzuki, Y. & Guan, L.L. (2018). Dissect the mode of action of probiotics in affecting host-microbial interactions and immunity in food producing animals. Veterinary Immunology and Immunopathology, 205, 35–48.
- Mandal, R.K., Jiang, T., Al-Rubaye, A.A., Rhoads, D.D., Wideman, R.F., Zhao, J., Pevzner, I. & Kwon, Y.M. (2016). An investigation into blood microbiota and its potential association with bacterial chondronecrosis with osteomyelitis (BCO) in broilers. Scientific Reports, 6, 1–11.
- Mandal, R.K., Jiang, T., Wideman, R.F., Lohrmann, T. & Kwon, Y.M. (2020). Microbiota analysis of chickens raised under stressed conditions. Frontiers in Veterinary Science, 7, 696.
- Mariani, P., Barrow, P.A., Cheng, H.H., Groenen, M.A., Negrini, R. & Bumstead, N. (2001). Localization to chicken chromosome 5 of a novel locus determining salmonellosis resistance. Immunogenetics, 53, 786–791.
- Michael, M.E., Ngunjiri, J.M., Taylor, K.J.M., Ji, H., Ghorbani, A., Kc, M., Elaish, M., Jang, H., Weber, B., Johnson, T.J. & Lee, C.W. (2020). Evaluation of sampling methods for the study of avian respiratory microbiota. Avian Diseases, 64, 277–285.
- Mon, K.K.Z., Zhu, Y., Chanthavixay, G., Kern, C. & Zhou, H. (2020). Integrative analysis of gut microbiome and metabolites revealed novel mechanisms of intestinal Salmonella carriage in chicken. Scientific Reports, 10, 1–14.
- Na, W., Wu, Y.Y., Gong, P.F., Wu, C.Y., Cheng, B.H., Wang, Y.X., Wang, N., Du, Z.Q. & Li, H. (2018). Embryonic transcriptome and proteome analyses on hepatic lipid metabolism in chickens divergently selected for abdominal fat content. BMC Genomics, 19, 1–17.
- Nyholm, L., Koziol, A., Marcos, S., Botnen, A.B., Aizpurua, O., Gopalakrishnan, S., Limborg, M.T., Gilbert, M.T.P. & Alberdi, A. (2020). Holo-omics: integrated host-microbiota multi-omics for basic and applied biological research. IScience, 23, 101414.
- Oakley, B.B. & Kogut, M.H. (2016). Spatial and temporal changes in the broiler chicken cecal and fecal microbiomes and correlations of bacterial taxa with cytokine gene expression. Frontiers in Veterinary Science, 3, 11.
- Oakley, B.B., Lillehoj, H.S., Kogut, M.H., Kim, W.K., Maurer, J.J., Pedroso, A., Lee, M.D., Collett, S.R., Johnson, T.J. & Cox, N.A. (2014). The chicken gastrointestinal microbiome. FEMS Microbiology Letters, 360, 100–112.
- Olivier, M., Asmis, R., Hawkins, G.A., Howard, T.D. & Cox, L.A. (2019). The need for multi-omics biomarker signatures in precision medicine. International Journal of Molecular Sciences, 20, 4781.
- Polansky, O., Sekelova, Z., Faldynova, M., Sebkova, A., Sisak, F. & Rychlik, I. (2016). Important metabolic pathways and biological processes expressed by chicken cecal microbiota. Applied and Environmental Microbiology, 82, 1569–1576.
- Pourabedin, M. & Zhao, X. (2015). Prebiotics and gut microbiota in chickens. FEMS Microbiology Letters, 362, 122.
- Putri, S.P., Yamamoto, S., Tsugawa, H. & Fukusaki, E. (2013). Current metabolomics: technological advances. Journal of Bioscience and Bioengineering, 116, 9–16.
- Qi, Z., Shi, S., Tu, J. & Li, S. (2019). Comparative metagenomic sequencing analysis of cecum microbiotal diversity and function in broilers and layers. 3 Biotech, 9, 316.
- Rodrigues, D.R., Wilson, K.M., Trombetta, M., Briggs, W.N., Duff, A.F., Chasser, K.M., Bottje, W.G. & Bielke, L. (2020). A proteomic view of the cross-talk between early intestinal microbiota and poultry immune system. Frontiers in Physiology, 11, 20.
- Roto, S.M., Rubinelli, P.M. & Ricke, S.C. (2015). An introduction to the avian gut microbiota and the effects of yeast-based prebiotic-type compounds as potential feed additives. Frontiers in Veterinary Science, 2, 28.
- Rychlik, I. (2020). Composition and function of chicken gut microbiota. Animals, 10, 103.
- Saelao, P., Wang, Y., Chanthavixay, G., Yu, V., Gallardo, R.A., Dekkers, J.C.M., Lamont, S.J., Kelly, T. & Zhou, H. (2018). Integrated proteomic and transcriptomic analysis of differential expression of chicken lung tissue in response to NDV infection during heat stress. Genes, 9, 579.
- Segura-Wang, M., Grabner, N., Koestelbauer, A., Klose, V. & Ghanbari, M. (2021). Genome-resolved metagenomics of the chicken gut microbiome. Frontiers in Microbiology, 12, 2390.
- Sergeant, M.J., Constantinidou, C., Cogan, T.A., Bedford, M.R., Penn, C.W. & Pallen, M.J. (2014). Extensive microbial and functional diversity within the chicken cecal microbiome. PLoS One, 9, e91941.
- Shang, Y., Kumar, S., Oakley, B. & Kim, W.K. (2018). Chicken gut microbiota: importance and detection technology. Frontiers in Veterinary Science, 5, 254.
- Singh, A.K. & Kim, W.K. (2021). Effects of dietary fiber on nutrients utilization and gut health of poultry: a review of challenges and opportunities. Animals, 11, 1–18.
- Singh, K.M., Shah, T., Deshpande, S., Jakhesara, S.J., Koringa, P.G., Rank, D.N. & Joshi, C.G. (2012). High through put 16S rRNA gene-based pyrosequencing analysis of the fecal microbiota of high FCR and low FCR broiler growers. Molecular Biology Reports, 39, 10595–10602.
- Stanberry, L., Mias, G., Haynes, W., Higdon, R., Snyder, M. & Kolker, E. (2013). Integrative analysis of longitudinal metabolomics data from a personal multi-omics profile. Metabolites, 3, 741–760.
- Stanley, D., Denman, S.E., Hughes, R.J., Geier, M.S., Crowley, T.M., Chen, H., Haring, V.R. & Moore, R.J. (2012). Intestinal microbiota associated with differential feed conversion efficiency in chickens. Applied Microbiology and Biotechnology, 96, 1361–1369.
- Stanley, D., Geier, M.S., Denman, S.E., Haring, V.R., Crowley, T.M., Hughes, R.J. & Moore, R.J. (2013). Identification of chicken intestinal microbiota correlated with the efficiency of energy extraction from feed. Veterinary Microbiology, 164, 85–92.
- Stanley, D., Hughes, R.J. & Moore, R.J. (2014). Microbiota of the chicken gastrointestinal tract: influence on health, productivity and disease. Applied Microbiology and Biotechnology, 98, 4301–4310.
- Stanley, D., Hughes, R.J., Geier, M.S. & Moore, R.J. (2016). Bacteria within the gastrointestinal tract microbiota correlated with improved growth and feed conversion: challenges presented for the identification of performance enhancing probiotic bacteria. Frontiers in Microbiology, 7, 187.
- Tang, Y., Underwood, A., Gielbert, A., Woodward, M.J. & Petrovska, L. (2014). Metaproteomics analysis reveals the adaptation process for the chicken gut microbiota. Applied and Environmental Microbiology, 80, 478–485.
- Tilquin, P., Barrow, P.A., Marly, J., Pitel, F., Plisson-Petit, F., Velge, P., Vignal, A., Baret, P.V., Bumstead, N. & Beaumont, C. (2005). A genome scan for quantitative trait loci affecting the Salmonella carrier-state in the chicken. Genetics, Selection, Evolution, 37, 539.
- Tuite, A. & Gros, P. (2006). The impact of genomics on the analysis of host resistance to infectious disease. Microbes and Infection, 8, 1647–1653.
- Upadhyaya, I., Upadhyay, A. & Venkitanarayanan, K. (2019). Applications of “omics” technologies to study gut health in poultry. K. Venkitanarayanan, S. Thakur, & S.C. Ricke (Eds.). Food Safety in Poultry Meat Production, book series: Food Microbiology and Food Safety (pp. 211–234). Cham: Springer Nature Switzerland AG
- Wang, L., Leng, L., Ding, R., Gong, P., Liu, C., Wang, N., Li, H., Du, Z.Q. & Cheng, B. (2021). Integrated transcriptome and proteome analysis reveals potential mechanisms for differential abdominal fat deposition between divergently selected chicken lines. Journal of Proteomics, 241, 104242.
- Wang, Y., Hu, Y., Liu, F., Cao, J., Lv, N., Zhu, B., Zhang, G. & Gao, G.F. (2020). Integrated metagenomic and metatranscriptomic profiling reveals differentially expressed resistomes in human, chicken, and pig gut microbiomes. Environment International, 138, 105649.
- Washburn, M.P., Koller, A., Oshiro, G., Ulaszek, R.R., Plouffe, D., Deciu, C., Winzeler, E. & Yates, J.R. (2003). Protein pathway and complex clustering of correlated mRNA and protein expression analyses in Saccharomyces cerevisiae. Proceedings of the National Academy of Sciences of the United States of America, 100, 3107–3112.
- Wei, S., Morrison, M. & Yu, Z. (2013). Bacterial census of poultry intestinal microbiome. Poultry Science, 92, 671–683.
- Wemheuer, F., Taylor, J.A., Daniel, R., Johnston, E., Meinicke, P., Thomas, T. & Wemheuer, B. (2020). Tax4Fun2: prediction of habitat-specific functional profiles and functional redundancy based on 16S rRNA gene sequences. Environmental Microbiomes, 15, 1–12.
- Wen, C., Yan, W., Mai, C., Duan, Z., Zheng, J., Sun, C. & Yang, N. (2021). Joint contributions of the gut microbiota and host genetics to feed efficiency in chickens. Microbiome, 9, 1–23.
- Willson, N.L., Nattrass, G.S., Hughes, R.J., Moore, R.J., Stanley, D., Hynd, P.I. & Forder, R.E.A. (2018). Correlations between intestinal innate immune genes and cecal microbiota highlight potential for probiotic development for immune modulation in poultry. Applied Microbiology and Biotechnology, 102, 9317–9329.
- Wörheide, M.A., Krumsiek, J., Kastenmüller, G. & Arnold, M. (2021). Multi-omics integration in biomedical research – a metabolomics-centric review. Analytica Chimica Acta, 1141, 144–162.
- Xu, P., Liu, P., Zhou, C., Shi, Y., Wu, Q., Yang, Y., Li, G., Hu, G. & Guo, X. (2019). A multi-omics study of chicken infected by nephropathogenic infectious bronchitis virus. Viruses, 11, 1070.
- Yan, W., Sun, C., Yuan, J. & Yang, N. (2017). Gut metagenomic analysis reveals prominent roles of Lactobacillus and cecal microbiota in chicken feed efficiency. Scientific Reports, 7, 1–11.
- Zampiga, M., Flees, J., Meluzzi, A., Dridi, S. & Sirri, F. (2018). Application of omics technologies for a deeper insight into quali-quantitative production traits in broiler chickens: a review. Journal of Animal Science and Biotechnology, 9, 1–18.
- Zampiga, M., Laghi, L., Zhu, C., Cartoni Mancinelli, A., Mattioli, S. & Sirri, F. (2021). Breast muscle and plasma metabolomics profile of broiler chickens exposed to chronic heat stress conditions. Animal, 15, 100275.
- Zhai, J., Ga, C., Fu, L., Jing, L., Dang, S. & Zheng, S. (2018). Integrative analyses of transcriptome sequencing identify functional miRNAs in the chicken embryo fibroblasts cells infected with reticuloendotheliosis virus. Frontiers in Genetics, 9, 340.
- Zhang, B., Liu, N., Hao, M., Zhou, J., Xie, Y. & He, Z. (2022). Plant-derived polysaccharides regulated immune status, gut health and microbiota of broilers: a review. Frontiers in Veterinary Science, 8, 1576.
- Zhang, H., Liang, Q., Wang, N., Wang, Q., Leng, L., Mao, J., Wang, Y., Wang, S., Zhang, J., Liang, H., Zhou, X., Li, Y., Cao, Z., Luan, P., Wang, Z., Yuan, H., Wang, Z., Zhou, X., Lamont, S.J., Da, Y., Li, R., Tian, S., Du, Z. & Li, H. (2020). Microevolutionary dynamics of chicken genomes under divergent selection for adiposity. IScience, 23, 101193.
- Zhou, Y., Zhang, M., Liu, Q. & Feng, J. (2021). The alterations of tracheal microbiota and inflammation caused by different levels of ammonia exposure in broiler chickens. Poultry Science, 100, 685–696.
- Zou, X., Ji, J., Qu, H., Wang, J., Shu, D.M., Wang, Y., Liu, T.F., Li, Y. & Luo, C.L. (2019). Effects of sodium butyrate on intestinal health and gut microbiota composition during intestinal inflammation progression in broilers. Poultry Science, 98, 4449–4456.