Abstract
A multiresidue method was developed and optimized for the identification/quantification of organochlorine pesticides (OCPs) and pyrethroids (PYRs) in beef meat samples. Samples extraction was performed by an automated solvent extractor and the extracts were cleaned-up by a tandem-cartridge system consisting of an Extrelut NT3 combined with a Sep-Pack C18 cartridge and a florisil minicartridge. Analysis was finally carried out by gas chromatography coupled with quadrupole mass spectrometry (GC/MS). The performance of the method was investigated in terms of linearity, accuracy, precision, detection limit (LOD) and quantification limit (LOQ). Good linearity was obtained, with correlation coefficients (r2) higher than 0.98. Mean recoveries were found in the ranges 70–110 % and 84–99 % for the investigated OCPs and PYRs, respectively, with the exception of extremely volatile hexachlorobenzene (HCB). RSD% turned out to range from 2 to 15 %. LOQ values were in the range 0.005–0.1 mg/kg for either class of compounds. The method developed was successfully tested on 50 commercial beef meat samples from the market area of Rome (Italy), proving to be a useful tool in routine multiresidue analysis of OCPs and PYRs for monitoring purposes. None of the compounds of interest were observed above their respective LOQ.
Introduction
Organochlorine pesticides (OCPs) have been largely used worldwide against a variety of pests. Low volatility and high stability, together with lipophilic behaviour, are responsible for their persistence in the environment and concentration in fatty tissues. By means of the food chain, bioaccumulation can occur, particularly in products of animal origin such as meat, fat, butter and milk.
Synthetic pyrethroids (PYRs) are effective broad-spectrum insecticides with low mammalian toxicity and short-term environmental persistence. Owing to their animal metabolism, they tend to bioaccumulate, becoming a potential source of contamination through foodstuffs.
As a consequence of their physical-chemical properties and toxicological profile, both OCPs and PYRs are to be monitored in foodstuffs, in order to control food quality and thus prevent any possible risk for human health. For consumers safety's sake, European Legislation[ Citation 1 ] set the legal limits (Maximum Residue Levels or MRLs) on products of animal origin such as meat, milk and eggs down to a level ranging from 0.01 to 0.1 mg/kg for either class of compounds.
Due to their structure and physical-chemical properties, the analysis of OCPs and PYRs residues is usually carried out by gas chromatography coupled with electron capture detection (GC/ECD) or with mass spectrometry (GC/MS).[ Citation 2 , Citation 3 , Citation 4 , Citation 5 , Citation 6 , Citation 10 , Citation 11 , Citation 12 , Citation 14 , Citation 17 , Citation 18 , Citation 19 , Citation 20 , Citation 21 ]
Multiresidue methods, which allow the identification/quantification of residues of different analytes at the same time, are advantageously used for monitoring purposes. The multiresidue analysis of OCPs and PYRs in fat and meat samples involves several steps, first of all the selective extraction of the residues from the homogenized matrix. In this stage, which is the most critical in the whole analytical procedure, the co-extraction of lipidic material incompatible with the GC detection system can occur, so that the extracts need to be purified prior to analysis.
The analytical methods developed for the determination of OCPs and PYRs involve traditional extraction techniques such as extraction by Soxhlet[ Citation 2 , Citation 3 ] and sonication[ Citation 4 , Citation 5 ] as well as modern extraction techniques such as supercritical fluid extraction (SFE),[ Citation 6 , Citation 7 , Citation 8 , Citation 9 ] microwave-assisted extraction (MAE),[ Citation 10 ] accelerated solvent extraction (ASE)[ Citation 2 , Citation 11 ]and extraction by Polytron.[ Citation 2 , Citation 12 ] SFE, MAE and ASE techniques work automatically and require low quantities of solvent, with a consequent reduction of solvent waste. On the other hand, a purification step is required, so to remove co-extracted fatty acids, sterols and glycerides incompatible with those detectors usually employed for the trace analysis of OCPs and PYRs, such as the electron capture detector (ECD), the mass detector (MS), and the tandem mass detector (MS/MS). The clean-up of the extracts is accomplished by liquid-liquid partition, gel permeation chromatography (GPC)[ Citation 2 , Citation 13 ] adsorption chromatography on different adsorbents such as silica, florisil or alumina,[ Citation 14 ] solid-phase extraction (SPE) on ready-to-use cartridges[ Citation 14 , Citation 15 , Citation 16 , Citation 17 , Citation 18 , Citation 19 ] and solid-matrix dispersion partition (SMDP).[ Citation 21 ]
This work describes the development and validation of a multiresidue GC/MS method for the identification/quantification of 26 OCPs and PYRs in beef meat samples. An automated system allowed the extraction of 6 samples simultaneously with small volumes of solvent over a 24-hour period. Prior to analysis, a rapid simple clean-up procedure by SPE (florisil, C18 cartridge) was carried out. The final eluates proved to have a reduced fat residue and hence were suitable for injection in the GC/MS apparatus. The method developed was not only validated, but also tested on 50 commercial beef meat samples from the market area of Rome (Italy), in order to prove its suitability for routine analysis in monitoring programmes.
Materials and methods
Chemicals and materials
The following OCPs and PYRs standards (90 to 99.5% pure) were purchased from Dr. Ehrenerstorfer ™ (Ausberg, Germany): Tecnazene, α -HCH, HCB, β -HCH, γ -HCH, Heptachlor, Aldrin, Oxychlordane, β -HEPO, γ -Chlordane, α -Endosulfan, α -Chlordane, Dieldrin, p,p′-DDE, Endrin, β -Endosulfan, o,p′-DDT, p,p′-TDE, Endosulfan sulphate, p,p′-DDT, Bifenthrin, lambda Cyhalothrin, Permethrin, Cipermethrin, Fenvalerate, Deltamethrin, PCB 209.
Acetone, light petroleum, n-hexane, acetonitrile, toluene and isooctane (all pesticide grade) were obtained from Carlo Erba (Milan, Italy). Anhydrous sodium sulfate and sea sand (both reagent grade) were supplied by Merck (Darmstadt, Germany). Further purification from interfering substances was carried out by heating for 3 hours at 600°C and for 12 hours at 120°C in a muffle furnace and in an oven, respectively.
Ready-to-use Extrelut NT3 cartridges were purchased from Merck (Darmstadt, Germany).
Ready-to-use Sep-pack C18 cartridges were obtained from Waters (Milford, MA, USA), whereas ready-to-use Bakerbond SPE Florisil cartridges (2000 mg) were obtained from J.T. Baker (Deventer, Holland).
Cellulose extraction thimbles (33 i.d. × 80 e.l. mm) were supplied by Whatman (England).
Stock solutions of each standard were prepared at 1.0 mg/mL in toluene. Each solution was further diluted to 10 μ g/mL in isooctane.
Standards mixtures were prepared at 0.1−1.0 μ g/mL and 0.5−5.0 μ g/mL for OCPs and PYRs, respectively. PCB 209 was used as an internal standard (IS). A PCB 209 solution was prepared at 1.0 μ g/mL in isooctane.
Apparatus
An extractor unit by solvents SER148 (VELP Scientifica, USMATE-Milano) was used for the extraction of ground beef meat samples.
An Agilent System 6890 series Plus gas chromatograph (USA), equipped with an Agilent 5973 mass selective detector, an Agilent 7683 series Autosampler and a split/splitless capillary injector port, was employed. Chromatographic separation was achieved on a 30 m × 0.25 mm i.d. HP-5ms capillary column (J & W Scientific, USA) with 0.25 μ m film thickness. The following oven temperature programme was adopted: 60°C (2 min), 10°C/min to 160°C and finally 3°C/min to 260°C (20 min). The carrier gas (He) flow rate was kept in constant flow mode at 1.0 ml/min. Sample injection was carried out splitless at 240°C with 1 min purge off time and a purge flow to split vent of 51.6 mLl/min. The injection volume was 1 μ L. The mass spectrometer was used in electron ionization mode (EI) and in Selected Ion Monitoring mode (SIM) with ionization energy of 70 eV. The transfer line temperature was kept at 280°C.
Both data acquisition and processing were accomplished by software Chemstation (Agilent Technologies™).
Samples collection and storage
Fifty samples of ground beef meat (ca. 250 g each) were collected from the market area of Rome (Italy) in 2005. After sampling, all samples were stored at −20°C until analysis. Before analysis, each sample was homogenized and divided into two portions. One portion was used to detect and quantify the compounds of interest under the experimental conditions described above. The other portion was further divided into 10 g subsamples and employed to check the performance of the method.
Extraction by automatic extractor
Ten g of ground beef meat sample were accurately weighted into a 100 mL beaker and mixed with 10 g of anhydrous sodium sulfate and 10 g of sea sand to obtain a free-flowing powder. The mixture was quantitatively transferred into an extraction thimble and covered with cotton wool. Before use, the extraction thimbles had been washed with 15 mL of acetone and 15 mL of hexane. Extraction was carried out with 80 mL of light petroleum at 70°C by placing the extractor slider into the “immersion” position for 10 min and, successively, into the “washing” position for 50 min.
The final extract was quantitatively transferred into a 150 mL volumetric flask. Residual water was removed through anhydrous sodium sulfate (1 g). The extract was then concentrated to small volume by rotary evaporator (bath temperature 45°C; reduced pressure) and evaporated to dryness under gentle stream of nitrogen. Finally, the residue was re-dissolved in 1 mL n-hexane.
Clean-up procedure
One mL residue solution in n-hexane was introduced in the tandem-cartridge system, consisting of an Extrelut NT3 glass column combined with a Sep-Pack C18 cartridge and a florisil minicartridge. The solution was let to drain for 10 min, so to allow its homogeneous distribution into the filling material.
The tandem cartridge system was then eluted with 4 × 5 mL acetonitrile. The eluate was collected in a 50 mL erlenmeyer flask, concentrated to small volume by rotary evaporator (bath temperature 45°C; reduced pressure) and, finally, evaporated to dryness under a gentle stream of nitrogen. Prior to analysis, the residue was re-dissolved in 1 mL isooctane.
Calibration standard solutions and linearity
Five multi-level calibration standard solutions were prepared in isooctane at 0.005–0.1 μ g/mL, 0.01–0.2 μ g/mL, 0.02–0.5 μ g/mL, 0.03–0.75 μ g/mL and 0.5–1.0 μ g/mL. In each case the internal standard was added at 0.05 μ g/mL. The linearity of the GC/MS method was checked by plotting 5-point calibration curves. Each point of the linearity curves was obtained as average of three consecutive injections.
Recovery tests
In order to investigate the accuracy of the method, some of those meat samples which had turned out to contain no OCPs and PYRs at the level of interest were used for the determination of recovery rates. For this purpose, known volumes of standard mixtures were added to the selected samples at two spiking levels ranging from 0.02 to 0.5 mg/kg (on fat basis) and from 0.01 to 0.25 mg/kg (on fat basis), corresponding to the MRL and half MRL (on fat basis) for every pesticide, respectively. Six replicates for each fortification level were processed.
Results and discussion
A wide range of pesticides with different polarities was selected, on the basis of both use and tendency to bioaccumulate in animal tissues. The multiresidue method developed was optimized for routine analysis, meeting essential requirements such as rapidity and low solvent consumption. Six samples could be processed simultaneously by the automatic extraction system adopted, resulting in shorter analysis time compared with traditional extraction techniques. The use of small volumes of solvent allowed the extraction of the analytes of interest with no waste of volatile compounds, with the only exception of extremely volatile HCB.
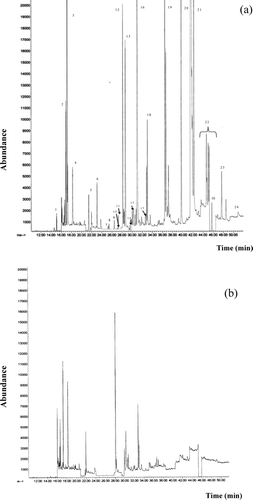
The tandem-cartridge system employed for clean-up remarkably simplified the purification step and allowed the preparation of suitable samples for GC/MS analysis, despite the complexity of the original matrix. As shown by the chromatogram of a control meat sample in , the blank of the method was satisfactory, so that the OCs and PYRs of interest could be determined at trace levels under the experimental conditions used.
Fig. 1 Gas chromatography/mass spectrometry (GC/MS) chromatograms (SIM mode) of (a) standard mixture of pesticides at MRL levels and (b) control meat sample. 1) Tecnazene, 2) -HCH, 3) HCB, 4) β -HCH + γ -HCH, 5) Heptachlor, 6) Aldrin, 7) Oxychlordane, 8) β -HEPO, 9) γ -chlordane, 10) α -Endosulfan, 11) α -chlordane, 12) Dieldrin, 13) p,p′-DDE, 14) Endrin, 15) β -Endosulfan, 16) o,p′-DDT + p,p′-DDD, 17) Endosulfan Sulfate, 18) p,p′-DDT, 19) Bifenthrin, 20) Lambda Cyhalothrin, 21) Permethrin, 22) Cypermethrin, 23) Fenvalerate, 24) Deltamethrin. HP 5 MS 30 m × 0.25 mm ID, 0.25 μ m film thickness column; oven program: 60°C (2 min.); 160°C (10°C/min); 260°C (3°C/min, 20 min); Injector temperature: 240°C; Transfer line temperature: 280°C; Ionisation energy: 70 eV.
Furthermore, the method was tested in terms of linearity, accuracy, precision, detection limit (LOD) and quantification limit (LOQ).
The linearity of the method was studied using standard solutions of the compounds of interest. Calibration curves were obtained by plotting the ratio between analyte concentration and IS concentration vs. the ratio between analyte peak area and IS peak area. In all cases, good linearity was observed, with r2 > 0.98 for all compounds, as reported in .
Table 1 Retention times (RT), selected ions, limits of determination (LOQ) and correlation coefficients (r2).
In order to verify the method accuracy, recovery rates were determined. For every compound, the response of the processed spiked matrix was considered with respect to the response of a matrix-matched standard solution. Results were expressed as per cent ratio between the mass of analyte after the clean-up step and the mass of analyte previously added to the investigated matrix. Two spiking levels ranging from 0.02 to 0.5 mg/kg (on fat basis) and from 0.01 to 0.25 mg/kg (on fat basis) were considered. As presented in , mean recoveries (n = 6) proved to fall in the range 70–110% at either spiking level, except for HCB, whose recoveries were found to be 49% and 57% at 0.1 and 0.2 mg/kg, respectively. HCB low recoveries can be ascribed to its high volatility and consequent waste under the temperature conditions adopted during the extraction step. At any rate, further experimental work is on-going, aiming to the improvement of HCB recovery rates at the spiking levels of interest.
Table 2 Spiking levels (mg/kg), average recoveries and relative standard deviations (RSD%).
The precision of the method proved to be satisfactory, too, with relative standard deviation (%RSD) below 20% in all cases, as shown in .
The identification of the target compounds was accomplished by means of their relative retention time (RRT), calculated as ratio between analyte retention time and IS retention time. The retention times of the OCs and PYRs under investigation are presented in . Each compound was further confirmed by comparison of the intensities of the response of its most abundant ion in the sample solution and in the matrix-matched standard solution at the same concentration level. Quantitative analysis was carried out by the GC/MS system operating in SIM mode. The internal standard method was adopted, working at a single calibration level with matrix-matched standard solutions. The ions selected for each pesticide are also listed in .
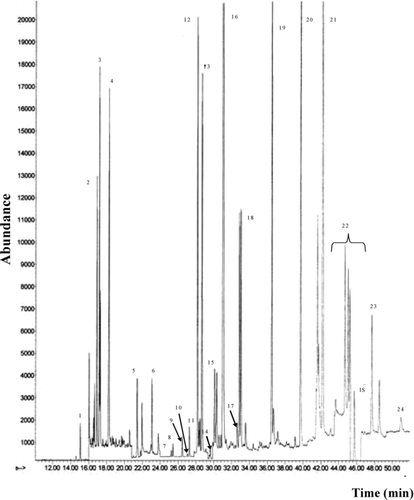
shows representative chromatograms of (a) standard solution matched in matrix at MRL levels and (b) control meat sample; shows a representative chromatogram of a meat sample fortified at MRL level with internal standard PCB 209 at 0.05 mg/kg
Fig. 2 Gas chromatography/mass spectrometry (GC/MS) chromatograms (SIM mode) of meat sample fortified at MRL levels with Internal Standard PCB 209 at 0.05 mg/kg. 1) Tecnazene, 2) -HCH, 3) HCB, 4) β -HCH + γ -HCH, 5) Heptachlor, 6) Aldrin, 7) Oxychlordane, 8) β -HEPO, 9) γ -chlordane, 10) α -Endosulfan, 11) α -chlordane, 12) Dieldrin, 13) p,p′-DDE, 14) Endrin, 15) β -Endosulfan, 16) o,p′-DDT + p,p′-DDD, 17) Endosulfan Sulfate, 18) p,p′-DDT, 19) Bifenthrin, 20) Lambda Cyhalothrin, 21) Permethrin, 22) Cypermethrin, 23) Fenvalerate, 24) Deltamethrin. HP 5 MS 30 m × 0.25 mm ID, 0.25 μ m film thickness column; oven program: 60°C (2 min.); 160°C (10°C/min); 260°C (3°C/min, 20 min); Injector temperature: 240°C; Transfer line temperature: 280°C; Ionisation energy: 70 eV.
The limit of detection (LOD) and the limit of determination (LOQ) were assessed for each compound as the lowest concentration originating a response equal to three times the noise signal and the lowest concentration producing a response equal to ten times the noise signal, respectively. For this purpose, the solution of standards prepared at 0.005–0.125 μ g/mL, corresponding for each analyte to the lowest concentration where linearity was checked, was injected and the noise value was hence computed automatically by the instrument software. LOD and LOQ were subsequently calculated according to the above definitions. LOD values were found in the range 0.001–0.008 mg/kg (on fat basis). As far as LOQ is concerned, all results are summarized in .
Finally, 50 commercial beef meat samples were analysed using the developed method, in order to prove its suitability for routine analysis in monitoring programmes. Final samples suitable for injection in the GC/MS apparatus were obtained. The average fat residue, which was observed to be 1.2831 g in the light petroleum extracts before clean-up, was drastically reduced and found to be 2.6 mg in the final eluates. Results showed that none of the compounds of interest were above their respective LOQ.
Conclusion
A GC/MS multiresidue method for the identification/ quantification of OCPs and PYRs in beef meat samples was developed. The use of an automatic extractor for sample extraction, together with a tandem-cartridge system for clean-up, makes this method an excellent candidate for routine analysis in monitoring programmes. As a result, one single operator was able to carry out the extraction of 6 samples simultaneously over a 24-hour period. Also the purification procedure proved to be simple and fast.
The analytical procedure was validated at the MRL and half MRL set by European Regulation 396/2005 for each compound of interest. In particular, satisfactory results were obtained in terms of recovery rates and %RSD, except for HCB, probably due to its high volatility and consequent waste in the extraction step.
The method developed was successfully tested on 50 commercial beef meat samples from the market area of Rome (Italy), proving to be suitable in routine multiresidue analysis of OCPs and PYRs for monitoring purposes. None of the compounds of interest were found above their respective LOQ.
Related Research Data
References
- European Commission . Commission Regulation (EC) 149/2008 of 29 January 2008 amending Regulation (EC) 396/2005 of the European Parliament and of the Council by establishing Annex II, III and IV setting maximum residue levels for products covered by Annex I thereto. Official Journal of the European Union L58/1 1.03.2008
- Garrido Frenich , A. , Martinez Vidal , J. L. , Cruz Sicilia , A. D. , Gonzalez Rodriguez , M. J. and Plaza Bonalos , P. 2006 . Multiresidue analysis of organochlorine and organophosphorous pesticides in muscle of chicken, pork amd lamb by gas chromatography-triple quadrupole mass spectrometry . Analytica Chimica Acta , 558 : 42 – 52 .
- Covaci , A. , Gheorghe , A. and Schepens , P. 2004 . Distribution of organochlorine pesticides, polychlorinated biphenyls and α -HCH enanatiomers in pork tissues . Chemosphere , 56 : 757 – 766 .
- Lino , C. M. and da Silveira , M. I. N. 1997 . Extraction and clean-up methods for the determination of organochlorine pesticide residues in medicinal plants . J. Chromatogr. A , 769 : 275 – 283 .
- Manirakiza , P. , Covaci , A. and Schepens , P. 2000 . Single step clean-up and GC-MS quantification of organochlorine pesticide residues in spice powder . Chromatographia , 52 : 787 – 790 .
- Juhler , R. K. 1998 . Supercritical fluid extraction of pesticides from meat: a systematic approach for optimisation . Analyst , 123 : 1551 – 1556 .
- Lehotay , S. J. and Eller , K. I. 1995 . Development of a method of analysis for 46 pesticides in fruit and vegetables by supercritical fluid extraction and gas chromatography/ion trap mass spectrometry . J. AOAC Int. , 78 : 821 – 830 .
- Nam , K. S. and King , W. 1994 . Supercritical fluid extraction and enzyme immunoassay for pesticide detection in meat products . J. Agric. Food Chem. , 42 : 1469 – 1474 .
- Hopper , M. L. 1999 . Automated one-step supercritical fluid extraction and clean-up system for the analysis of pesticide residues in fatty matrices . J. Chromatogr. A , 840 : 93 – 105 .
- Pastor , A. , Vazquez , E. , Ciscar , R. and de la Guardia , M. 1997 . Efficiency of the microwave-assisted extraction of hydrocarbons and pesticides from sediments . Anal. Chim. Acta , 344 : 241 – 249 .
- Adou , K. , Boutoyan , W. R. and Sweeney , P. J. 2001 . Multiresidue method for the analysis of pesticide residues in fruits and vegetables by accelerated solvent extraction and capillary gas chromatography . J. of Agric food Chem. , 49 : 4153 – 4160 .
- Garrido-Frenich , A. , Romero-Gonzalez , R. , Martinez-Vidal , J. L. , Plaza-Bolanos , P. , Cuadros-Rodriguez , L. and Herrera-Abdo , M. A. 2006 . Characterization of recovery profiles using gas chromatography-triple quadrupole mass spectrometry for the determination of pesticide residues in meat samples . J. Chromatogr. A , 1133 : 315 – 321 .
- Rimkus Gerhard , G. , Rummler , M. and Nausch , I. 1996 . Gel permeation chromatography-high performance liquid chromatography combination as an automated clean-up technique for the multiresidue analysis of fats . J. Chromatogr. A , 737 : 9 – 14 .
- Attard Barbini , D. , Vanni , F. , Girolimetti , S. and Dommarco , R. 2007 . Development of an analytical method for the determination of the residues of four pyrethroids in meat by GC-ECD and confirmation by GC-MS . Anal. Bioanal. Chem. , 389 : 1791 – 1798 .
- Doong , R. and Lee , C. 1999 . Determination of organochlorine pesticide residues in foods using solid-phase extraction clean-up cartridge . Analyst , 124 : 1287 – 1289 .
- Pagliuca , G. , Gazzotti , T. , Zironi , E. and Sticca , P. 2005 . Residue analysis of organophosphorous pesticides in animal matrices by dual column capillary gas chromatography with nitrose-phosphorous detection . J. Chromatogr. A , 1071 : 67 – 70 .
- Schenck , F. J. and Donoghue , D. J. 2000 . Determination of Organochlorine and organophosphorous pesticide residues in eggs using a solid phase extraction cleanup . J. Agric. Food Chem. , 48 ( 12 ) : 6412 – 6415 .
- Kodba , Z. C. and Voncina , D. B. 2007 . A rapid method for the determination of organochlorine, pyrethroid pesticides and polychlorobiphenyls in fatty food using GC with electron capture detection . Chromatographia , 66 : 619 – 624 .
- Di Muccio , A. , Generali , T. , Attard Barbini , D. , Pelosi , P. , Ausili , A. , Vergori , F. and Girolimetti , S. 1997 . Single-step separation of organochlorine pesticide residues from fatty materials by combined use of solid-matrix partition and C18 cartridge . J of A Chromatography , 765 : 61 – 68 .
- Park , J. W. , Abd El-Aty , A. M. , Lee , M. H. , Song , S. O. and Shim , J. H. 2006 . Residue analysis of organophosphorous and organochlorine pesticides in fatty matrices by gas chromatography coupled with electron-capture detection . J. of Biosciences C Section , 61 ( 5–6 ) : 341 – 346 .
- Di Muccio , A. , Pelosi , P. , Attard Barbini , D. , Generali , T. , Girolimetti , S. , Stefanelli , P. , Leonelli , A. , Amendola , G. , Vergori , L. and Fresquet , E. V. 1999 . Determination of pyrethroid pesticide residues in fatty materials by solid-matrix dispersion partition, followed by mini-column size exclusion chromatography . J of A Chromatography , 833 : 19 – 34 .