
Abstract
This annual review is the sixth of its kind since 2016 (see references). Our objective is to explore and share articles which we deem influential and significant in the field of biotransformation and bioactivation. These fields are constantly evolving with new molecular structures and discoveries of corresponding pathways for metabolism that impact relevant drug development with respect to efficacy and safety. Based on the selected articles, we created three sections: (1) drug design, (2) metabolites and drug metabolizing enzymes, and (3) bioactivation and safety (). Unlike in years past, more biotransformation experts have joined and contributed to this effort while striving to maintain a balance of authors from academic and industry settings.
Table 1. Articles covered in this review.
Beyond biotransformation
Since the last review, significant events occurred that made 2020 a unique year; from environmental disasters (the COVID-19 pandemic and catastrophic wildfires) to cultural reactions about basic human rights (the Black Lives Matter movement and stop Asian hate movement). These events highlighted that we all have an important role to play in contributing to stronger diverse and inclusive communities. With all the privileges given to us, we need to give back in any way we can. As Dr. Martin Luther King, Jr. said, ‘The time is always right to do what is right.’
We would be pleased to hear your opinions, and we extend an invitation to anyone who would like to contribute to a future edition of this review.
Cyrus Khojasteh, on behalf of the authors.
Metabolic and pharmaceutical aspects of fluorinated compounds
Benjamin M. JohnsonYue-Zhong ShuXiaoliang ZhuoNicholas A. MeanwellSource: J. Med. Chem. 2020;63:6315–6386
SYNOPSIS
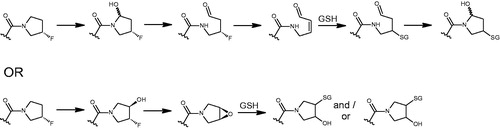
This perspective article on the metabolic and pharmaceutical aspects of fluorinated compounds is extensive and thoroughly researched. The incorporation of fluorine in drugs, drug candidates, and agricultural chemicals is increasing, and with this development comes an improved understanding of the metabolic pathways of fluorinated molecules. At the same time, this improved understanding is bringing to light the ‘cryptic liabilities’ of these compounds. The authors describe the chemistry of fluorine and why it can be considered a useful tool in drug design. For example, fluorination can improve the physicochemical properties of a molecule, particularly with ameliorating the metabolic profile of a new chemical entity (NCE). Strategies for incorporating fluorine into NCEs are illustrated with numerous examples across the structural classes, from simple fluoroalkanes and ethers to more complex heterocyclic systems. In cases where metabolic defluorination is observed, the pathways and enzymes responsible are used to depict the benefits and liabilities of these biotransformation pathways (e.g. mechanism-based inhibition and/or defluorination resulting in the formation of electrophilic reactive metabolites) (). In sum, the perspective recognizes the benefits of fluorination and its continued utilization in drug discovery but advocates judicious use of this approach whilst incorporating the increased understanding of its metabolic characteristics into this strategy.
Commentary
In 2018 alone, 18 of the 38 FDA approved small molecule drugs contained fluorine (Pan Citation2019), and this level of representation follows decades of fluorinated drugs being approved for use from fluorocortisone in the 1950’s to the blockbuster atorvastatin (Mei et al. Citation2020). Approved fluorine-containing drugs cover a wide range of therapeutic areas including oncology, cardiology, and neurology as well as agrochemicals and veterinary drugs (Wang et al. Citation2013). This breadth of application clearly highlights our improved understanding of the impact of fluorination on the physical, (bio)chemical and metabolic characteristics of compounds, leading to fewer unpredicted deleterious issues whilst benefitting from improved physicochemical properties.
Fluorine is employed in drug design because of its potentially beneficial impact on various physicochemical and biochemical parameters; however, another well-known application of fluorine use is for 18F-based positron emission tomography (PET). This inclusion is not to improve the DMPK characteristics of the molecule but to serve as an effective PET tracer. In this case, the 18F needs to remain part of the molecule, and therefore, the molecule’s DMPK properties will need to be fully profiled as it would be for a drug candidate.
An additional beneficial property of fluorine is the fact that its nuclear spin number of ½ allows it to be detectable by NMR. Within the caveats of NMR performance in terms of sensitivity, fluorine NMR can provide a limited but useful amount of structural information related to biotransformation, particularly if the metabolism results in defluorination to a fluoride ion. Where NMR really comes into its own is the absolute quantitative information it can provide, and as fluorine is not naturally occurring, the specificity of the 19F signal means that the limit of quantitation can be valuable in determining absolute levels of parent and its metabolites. Examples exist of 19F NMR being used as an alternative to the classic radiolabeling strategy for mass balance studies (Hu et al. Citation2017). and its use in quantifying covalent binding has been recently reported (Kakutani et al. Citation2021).
Based on the continued increase in the use of fluorine in NCEs, all these points highlight the reason why this perspective is a timely one. Importantly, the review emphasizes that even with a clear understanding of the risks and rewards of incorporating fluorine into a molecule, unpredictable outcomes can still occur, be it from complex metabolic pathways resulting in defluorination and bioactivation or conformational changes based on a steric interaction. Perspectives such as this one, in addition to focused standalone articles, will help drug design programs appreciate that simply adding fluorine as a replacement for hydrogen by virtue of its small atomic size, greater electronegativity and greater bond dissociation energy (Gillis et al. Citation2015) will not always be a quick fix.
Figure 1. Metabolic pathway elucidated for a fluorinated pyrrolidine in rat liver microsomes, and an alternative epoxide-based metabolic pathway that could be contemplated.
References
- Gillis EP, Eastman KJ, Hill MD, Donnelly DJ, Meanwell NA. 2015. Applications of fluorine in medicinal chemistry. J Med Chem. 58:8315–8359.
- Hu H, Katyayan KK, Czeskis BA, Perkins EJ, Kulanthaivel P. 2017. Comparison between radioanalysis and 19F nuclear magnetic resonance spectroscopy in the determination of mass balance, metabolism, and distribution of pefloxacin. Drug Metab Dispos. 45(4):399–408.
- Kakutani N, Iwai T, Ohno Y, Kobayashi S, Nomura Y. 2021. Evaluation of covalent binding of flutamide and its risk assessment using 19F-NMR. Xenobiotica. 51(1):88–94.
- Mei H, Remete AM, Zou Y, Moriwaki H, Fustero S, Kiss L, Soloshonok VA, Han J. 2020. Fluorine-containing drugs approved by the FDA in 2019. Chin Chem Lett. 31(9):2401–2413.
- Pan Y. 2019. The dark side of fluorine. ACS Med Chem Lett. 10(7):1016–1019.
- Wang J, Sanchez-Rosello M, Acena JL, Pozo C, Sorochinsky AE, Fustero S, Soloshonok VA, Liu H. 2013. Fluorine in pharmaceutical industry: fluorine-containing drugs introduced to the market in the last decade (2001 − 2011). Chem Rev. 114:2432–2506.
Effective application of metabolite profiling in drug design and discovery
Matthew A. CernyAmit S. KalgutkarR. Scott ObachRaman SharmaDouglas K. SpracklinGregory S. WalkerSource: J Med Chem. 2020;63(12):6387–6406
SYNOPSIS
This perspective paper describes how a strategic approach has evolved at Pfizer that utilizes the increasingly pivotal role of biotransformation studies in drug design and development. This approach is founded on the authors’ combined extensive experiences over a period of time when biotransformation developed from being a late-stage descriptive activity to one that plays a key role in assisting in the design of better molecules, guiding clinical development and underwriting patient safety. The perspective begins with an introduction into how the drug research industry has evolved over the century or so, especially the ADME function. The mindset of drug research began with the theory of chemoreceptors and specific interactions with chemicals based on their specific designs to where we are now in applying the increasing wealth of knowledge and technology to assist therapeutic drug design. A key part of this evolution has been the advances in structural elucidation capabilities in the area of biotransformation and how one can use this key data to aid chemistry design and subsequent structure–activity relationship (SAR) development. The strategic approach described () is based on five key areas: 1. Understanding clearance mechanisms 2. Modulating metabolism to reduce metabolic liability 3. Does bioactivation result in reactive metabolite formation? 4. Presence of active/inactive metabolites 5. Metabolite safety & Metabolites In Safety Testing (MIST) For each of these areas, the authors provide case examples to support their observations and assertions as to how design can assist discovery programs. Examples include identification of the species disconnect in the metabolism of myeloperoxidase inactivators, the potential for deuteration to be used to modulate metabolism and the characterization of intrinsically electrophilic groups that can benefit the design of molecules less likely to undergo bioactivation. The section on metabolite safety comes last as the need to support drug design decreases after a candidate is nominated into development, but biotransformation input is still required to provide cross-species metabolite profiling comparisons. The authors assert that the advance in analytical technology was the critical development in allowing the area of biotransformation to play this increasingly pivotal role in the modern-day collaborative drug discovery and development environment.
Commentary
This perspective should resonate with many in the field of biotransformation and will help support their assertions that the role they play in drug design and the wider aspects of drug discovery and development is a key one. With this in mind, the role should not be considered as simply a data generator working in isolation, but a collaborative one where biotransformation scientists can share their knowledge and experience with wider discovery/development teams. This would not only be input into the chemistry, but also the design of the studies used to underpin the many aspects of discovery programs. For example, when in vitro assays that have a metabolic component are to be employed, the biotransformation scientist can add value to these efforts by providing suggestions for the design of the assay based on an understanding of the metabolic competency of the assay platform. This input is important to help select the right assay platform and address the issue at hand while balancing the conclusions derived from said assays based on the assays’ possible limitations.
The advances in analytical technologies that are used to support metabolite profiling activities are highlighted, both from a qualitative and quantitative perspective and it is certainly true that without them, metabolite profiling would have less of an impact than it does today. In the labs of many pharma companies and academia, there are a plethora of different models of high resolution mass spectrometers (HRMS) and nuclear magnetic resonance (NMR) spectrometers, but these achievements come at a high cost and require experts to get the most out of them. For those wishing to carry out analytical studies but do not have the resource to support them, there is the option to utilize the services of contract research organizations (CROs). CROs are playing an increasing role in providing not just a bench-extension to many companies by making efficient use of state-of-the-art technologies, but also providing the expert assistance that some companies or academia may be missing.
Quantitative metabolite profiling typically comes after qualitative profiling because the understanding of ‘how much’ is often as important as ‘what is it’, especially as the drug makes its way along the discovery and development path. NMR is described as a valid technique for providing early absolute quantitation, well before any authentic or radiolabel synthesis. A branch of mass spectrometry not mentioned is accelerator mass spectrometry (Young et al. Citation2020), and this technique may play an increasing and, specifically, earlier role due technological advances that reduce the size and cost of the instrument. Finally, the ‘characterize’ part of the strategy () will undoubtedly be supported by both biotransformation scientists and, increasingly, software programs offered by both MS and NMR vendors and independent companies alike.
As the authors have rightly said, the science of biotransformation has ‘transformed’ itself, and will continue to do so through increased understanding of the field, greater cross-functional and company collaboration, supported by advances in analytical and computational technology.
Figure 2. Strategic approach using structural elucidation to enable chemistry design/SAR development.
References
- Young GC, Croft M. 2020. AMS in drug development: exploring the current utility of AMS and future opportunities for absolute bioavailability and ADME investigations. In: Ma S and Chowdhury SK, editors. Identification and quantification of drugs, metabolites, drug metabolizing enzymes, and transporters. 2nd ed. Elsevier Science; p. 185–210.
Understanding the metabolism of proteolysis targeting chimeras (PROTACs): the next step toward pharmaceutical applications
Laura GoracciJenny DesantisAurora ValeriBeatrice CastellaniMichela EleuteriGabriele CrucianiSource: J Med Chem. 2020;63(20):11615–11638
SYNOPSIS
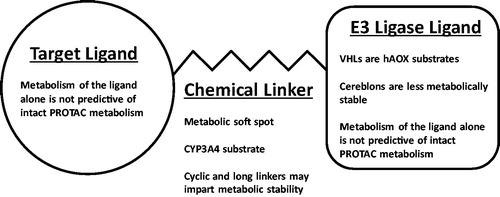
The work by Goracci et al. (Goracci et al. Citation2020) represents the first published comparison of the in vitro human hepatocyte metabolism of multiple proteolysis targeting chimeras (PROTACS). The authors employed a systematic approach to profile and compare the metabolism in cryopreserved hepatocytes across a total of 40 structurally diverse PROTAC molecules; reporting both the half-life and structures of biotransformation products formed for each compound. PROTACs consist of three distinct building blocks: a target protein ligand, an E3 ligase ligand, and a chemical linker that joins them (). Through this work, it was established that the linker region of PROTACs are metabolic soft spots, and that this metabolism is catalyzed by human cytochrome P450 3A4 (CYP3A4). From here, the impact of linker length and makeup (aliphatic chains, polyethylene glycol-based, or cyclic linkers) on metabolism was investigated, and the authors concluded that both increased linker length and the introduction of a cyclic linker generally increased PROTAC metabolic stability (though not in all cases). Interestingly, through the comparison of the metabolism of PROTAC molecules and that of their individual components, the authors found that the metabolism of each PROTAC building block was not predictive of the metabolism of the intact PROTAC, with respect to both half-life and the structure of metabolites formed. Finally, the authors also reported that von Hippel-Lindau (VHL) PROTACs are substrates for metabolism by human aldehyde oxidase (hAOX).
Commentary
PROTACs are large molecular weight (600–1400 Da) compounds with two protein binding ligands attached by a chemical linker: one ligand binds a target protein and the other an E3 ligase. Through simultaneous PROTAC binding of the target protein and E3 ligase, the target protein is polyubiquitinated and then subject to proteosomal degradation (Maple et al. Citation2019; Wang et al. Citation2020). The target protein ligands can vary widely, while there is a much smaller selection of commonly employed classes of E3 ligase binding ligands, two of which were studied in this work: cereblons and VHLs. PROTACs are a relatively new class of drug molecules, with their mechanism first demonstrated in 2001 (Sakamoto et al. Citation2001) and the first human trials of PROTAC drug molecules began in 2019 (Wang et al. Citation2020).
This study included five target protein binding ligands, five E3 ligase binding ligands (two cereblons and three VHLs), and 19 chemical linkers in the library of 40 PROTAC molecules. The identification of the PROTAC linker as a metabolic soft spot through this work gives medicinal chemists a point of focus for optimizing the metabolic stability of these complex molecules, as does the investigation of linker length and makeup, with increased length and cyclic linkers tending to impart metabolic stability. In addition, of the two classes of E3 ligase investigated, the authors noted that the cereblons were less metabolically stable than VHLs, attributing some of this property to proposed non-enzymatic degradation of cereblons. This observation is not surprising as the cereblon ligands are very similar to thalidomide, both containing multiple lactam moieties, and the non-enzymatic hydrolysis of thalidomide has been long understood (Schumacher et al. Citation1965; Lepper et al. Citation2006).
When making their comparisons of structural differences on PROTAC half-lives, Goracci et al. hypothesized how the changes would impact the interactions of these molecules with their drug metabolizing enzymes (Goracci et al. Citation2020). Another possible factor not addressed by this work would be changes in compound membrane permeability, which would limit substrate availability to intracellular drug metabolizing enzymes in cryopreserved hepatocytes. PROTAC molecules have demonstrated poor cellular permeability (Klein et al. Citation2020; Atilaw et al. Citation2021), and, therefore, a comparison of compound permeability or perhaps a metabolic screen in a more ‘open’ system, such as human liver microsomes, would prove insightful.
The extensive metabolism of PROTACs by CYP3A4 demonstrated in this work, while perhaps unsurprising given the enzyme’s flexible substrate pocket, nevertheless presents a drug-drug interaction risk (Zhou Citation2008). This finding may prompt a search for other linker options more resistant to CYP3A metabolism. To this point, the author’s observed hAOX metabolism of VHL PROTACs could help to relieve some of the risk associated with CYP3A-only metabolism. This strategy may be somewhat of a double-edged sword, as hAOX metabolism can cause poor in vitro-in vivo extrapolation for drug candidates (Zientek et al. Citation2010; Argikar et al. Citation2016). It is worth noting that while this hAOX metabolism was observed in this study, the half-lives of the compounds screened in human liver cytosol were high (≫90 minutes), and therefore further work is needed to establish the relevance of hAOX metabolism in the overall clearance of PROTAC molecules.
The above highlighted findings from Goracci et al. are summarized in . Given the novelty of PROTAC drugs, a thorough understanding of their metabolism remains to be established. Goracci et al. cites a prior study that investigated the metabolism of a single PROTAC molecule (Zhou et al. Citation2018), and there is additional study on screening PROTAC metabolic clearance that was published in 2020 (Pike et al. Citation2020). This publication by Goracci et al. is unique in that it provides a thorough first-look at PROTAC metabolism across multiple, structurally diverse compounds and includes metabolite structural characterization, giving many jumping-off points for future work.
Figure 3. The building blocks of PROTAC drug molecules and conclusions on their metabolism from Goracci et al. Citation2020.
References
- Argikar UA, Potter PM, Hutzler JM, Marathe PH. 2016. Challenges and opportunities with non-CYP enzymes aldehyde oxidase, carboxylesterase, and UDP-glucuronosyltransferase: focus on reaction phenotyping and prediction of human clearance. AAPS J. 18(6):1391–1405.
- Atilaw Y, Poongavanam V, Svensson Nilsson C, Nguyen D, Giese A, Meibom D, Erdelyi M, Kihlberg J. 2021. Solution conformations shed light on PROTAC cell permeability. ACS Med Chem Lett. 12(1):107–114.
- Goracci L, Desantis J, Valeri A, Castellani B, Eleuteri M, Cruciani G. 2020. Understanding the metabolism of proteolysis targeting chimeras (PROTACs): the next step toward pharmaceutical applications. J Med Chem. 63(20):11615–11638.
- Klein VG, Townsend CE, Testa A, Zengerle M, Maniaci C, Hughes SJ, Chan KH, Ciulli A, Lokey RS. 2020. Understanding and improving the membrane permeability of VH032-based PROTACs. ACS Med Chem Lett. 11 (9):1732–1738.
- Lepper ER, Smith NF, Cox MC, Scripture CD, Figg WD. 2006. Thalidomide metabolism and hydrolysis: mechanisms and implications. Curr Drug Metab. 7(6):677–685.
- Maple HJ, Clayden N, Baron A, Stacey C, Felix R. 2019. Developing degraders: principles and perspectives on design and chemical space. Medchemcomm. 10(10):1755–1764.
- Pike A, Williamson B, Harlfinger S, Martin S, McGinnity DF. 2020. Optimising proteolysis-targeting chimeras (PROTACs) for oral drug delivery: a drug metabolism and pharmacokinetics perspective. Drug Discov Today. 25(10):1793–1800.
- Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ. 2001. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc Natl Acad Sci USA. 98(15):8554–8559.
- Schumacher H, Smith RL, Williams RT. 1965. The metabolism of thalidomide: the fate of thalidomide and some of its hydrolysis products in various species. Br J Pharmacol Chemother. 25(2):338–351.
- Wang Y, Jiang X, Feng F, Liu W, Sun H. 2020. Degradation of proteins by PROTACs and other strategies. Acta Pharm Sin B. 10(2):207–238.
- Zhou B, Hu J, Xu F, Chen Z, Bai L, Fernandez-Salas E, Lin M, Liu L, Yang CY, Zhao Y, et al. 2018. Discovery of a small-molecule degrader of bromodomain and extra-terminal (BET) proteins with picomolar cellular potencies and capable of achieving tumor regression. J Med Chem. 61(2):462–481.
- Zhou SF. 2008. Drugs behave as substrates, inhibitors and inducers of human cytochrome P450 3A4. Curr Drug Metab. 9(4):310–322.
- Zientek M, Jiang Y, Youdim K, Obach RS. 2010. In vitro-in vivo correlation for intrinsic clearance for drugs metabolized by human aldehyde oxidase. Drug Metab Dispos. 38(8):1322–1327.
Late-stage lead diversification coupled with quantitative nuclear magnetic resonance spectroscopy to identify new structure-activity relationship vectors at nanomole-scale synthesis: application to loratidine, a human histamine H1 receptor inverse agonist
Manjinder S. LallAsser BassyouniJames BradowMaria BrownMark BundesmannJinshan ChenGregory CiszewskiAnne E. HagenDennis HyekStephen JenkinsonBo LiuR. Scott ObachSenliang PanUsa ReillyNeal SachDaniel J. SmaltzDouglas K. SpracklinJeremy StarrMelissa WagenaarGregory S. WalkerSource: J Med Chem. 2020;63:7268–7292
SYNOPSIS
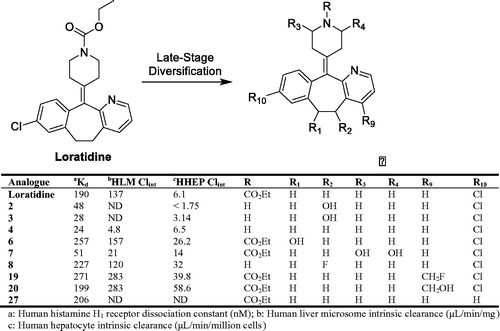
Late-stage lead diversification (LSLD) of small molecules is an encouraging and paradigm shifting methodology that can be utilized in parallel with the canonical approaches to drug discovery. Traditionally, the LSLD process takes advantage of the enzymatic reactions (mainly from mammalian and/or bacterial P450s) to efficiently and directly oxidize unreactive C(sp3)-H bonds in a lead molecule, a process which poses significant challenges to the synthetic medicinal chemist (Fessner Citation2019). Additionally, lead molecules can be subjected to direct chemical oxidation procedures via electrochemistry and biomimetic metalloporphyrin catalysts, as well as direct fluorination, Minisci chemistry, alkyl sulfonate radical precursor chemistry, and photoredox catalysis to generate additional analogues. LSLD requires low amounts (1–2 mg) of lead compound that are subjected to the aforementioned oxidative and/or chemical conditions, and the selected hits are scaled and purified using liquid chromatography with automated fraction collection. The purified fractions are characterized and quantified by high-resolution mass spectrometry and NMR spectroscopy, and are subsequently tested in pharmacological potency and metabolic stability assays in a high-throughput manner (Walker et al. Citation2014). To illustrate the LSLD strategy, loratidine was utilized as a model substrate due to the various intrinsic functional group handles on the respective ring systems. From the LSLD approaches employed, forty analogues were identified, twenty-six of which were chosen for further scaling, purification, and structural characterization. The results indicated that nine analogues (2, 3, 4, 6, 7, 8, 19, 20, and 27) were comparable and up to eight-fold more potent than the loratidine (). Additionally, analogues 2, 3, 4, and 7 had improved metabolic stability compared with loratidine. Overall, the comprehensive LSLD approach described by Lall et al. (Lall et al. Citation2020) provides a framework for an additional strategy to identify lead molecules within drug discovery programs.
Commentary
Much akin to diversifying portfolio investments within the stock market to maximize returns and dividends, Lall et al. provides a highly comprehensive and efficient diversification strategy to generate several analogues of lead compounds for drug discovery programs. This strategy was conducted mainly by exploiting the promiscuity of mammalian and bacterial cytochrome P450 enzymes, chemical oxidation approaches with electro- and/or metalloporphyrin chemistry, and direct modifications with derivatizing reagents. Essentially, the LSLD strategy capitalizes on areas of chemical space that are otherwise nearly inaccessible by conventional synthetic chemistry endeavors (i.e. C–H bond activation). Diversification of a lead molecule by a ‘single step’ biological or chemical reaction is a highly attractive and effective option to produce additional analogues for discovery programs (Cerny et al. Citation2020; Shanu-Wilson et al. Citation2020). However, implementation of the LSLD paradigm may be challenging due to the specific set of technical expertise, skills, and knowledge required from multiple scientific disciplines. Therefore, inter-departmental collaborative efforts are essential and critical to maximize the highest return on investment from an LSLD strategy.
Figure 4. Loratidine analogues with improved potency and/or metabolic stability.
References
- Cerny MA, Kalgutkar AS, Obach RS, Sharma R, Spracklin DK, Walker GS. 2020. Effective application of metabolite profiling in drug design and discovery. J Med Chem. 63(12):6387–6406.
- Fessner ND. 2019. P450 monooxygenases enable rapid late-stage diversification of natural products via C–H bond activation. ChemCatChem. 11(9):2226–2242.
- Lall MS, Bassyouni A, Bradow J, Brown M, Bundesmann M, Chen J, Ciszewski G, Hagen AE, Hyek D, Jenkinson S, et al. 2020. Late-stage lead diversification coupled with quantitative nuclear magnetic resonance spectroscopy to identify new structure-activity relationship vectors at nanomole –scale synthesis: application to loratidine, a human histamine H1 receptor inverse agonist. J Med Chem. 63(13):7268–7292.
- Shanu-Wilson J, Evans L, Wrigley S, Steele J, Atherton J, Boer J. 2020. Biotransformation: impact and application of metabolism in drug discovery. ACS Med Chem Lett. 11(11):2087–2107.
- Walker GS, Bauman JN, Ryder TF, Smith EB, Spracklin DK, Obach RS. 2014. Biosynthesis of drug metabolites and quantitation using NMR spectroscopy for use in pharmacologic and drug metabolism studies. Drug Metab Dispos. 42(1):1627–1639.
GLORYx: prediction of the metabolites resulting from phase 1 and phase 2 biotransformations of xenobiotics
Christina de Bruyn KopsMartin ŠíchoAngelica MazzolariJohannes KirchmairSources: Chem Res Toxicol. 2021;34:286–299
SYNOPSIS
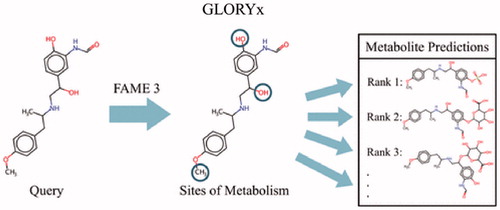
de Bruyn Kops et al. (de Bruyn Kops et al. Citation2021) reported a new tool called GLORYx for predicting metabolite structures formed by both Phase 1 and Phase 2 reactions in humans. The approach used the FAst MEtabolizer (FAME) 3 to predict the likelihood for metabolism at every atom in a molecule using extremely randomized trees. Based on the prediction values, GLORYx then applied reaction rules for generating and ranking possible metabolites. Overall performance evaluation relied on predicting metabolism and corresponding metabolite structures for a reference set of molecules that yielded insights on how to optimize the design of GLORYx. Both Phase 1 and 2 predictions improved with the inclusion of a binary common/uncommon reaction classifier and reaction type rules. Further improvements for Phase 2 reactions were achieved through individual reaction type models rather than a single model that predicted all reaction types. After optimization, GLORYx was then tested against a manually curated data set for the top-selling drugs in 2018. The sensitivity (recall) was 77%, reflecting the ability to correctly identify experimentally observed metabolites and the correcting rank of metabolites demonstrated in a receiver operator curve based on the area under the curve (AUC) of 0.79. Taken together, GLORYx possibly provided the most comprehensive array of predictions for xenobiotic metabolic pathways during drug development and yielded insights on improving model design.
Commentary
Xenobiotic metabolism plays a critical role in exposure and, hence, health outcomes associated with drugs, natural products and pollutants. Identification and assessment of metabolic pathways traditionally relies on experimental approaches, yet this strategy incurs high costs in time, effort and resources that can hamper decision-making. As attractive alternatives, computational tools provide more accessible, rapid ways to acquire potential information on metabolism. In this article, the investigators present a robust, comprehensive ensemble model GLORYx that predicts a broad array of Phase 1 and 2 reactions and the corresponding metabolites (). The most dominant enzymes are cytochromes P450 that account for almost 75% of drugs that undergo metabolism (Wienkers and Heath Citation2005), and thus, early models focused on identifying P450 sites of metabolism and the likelihood of those events. Models for Phase 2 reactions are lagging even though glucuronidation within that class is the second most important pathway for drugs (Wienkers and Heath Citation2005). Importantly, there are many other Phase 1 and 2 reactions that may be minor in overall contribution to metabolism, yet they are critical for certain drugs and thus warrant consideration. Those reactions have been mostly ignored in the modeling field. For GLORYx, the third generation FAst MEtabolizer (FAME) 3 predicts those major and minor Phase 1 and 2 pathways and generates information for predicting the corresponding metabolite structures that are readily interpretable and testable. Other metabolite models exist but generally do not provide as much coverage of reaction space (Liu et al. Citation2012; Tomberg et al. Citation2015). Taken together, these qualities make GLORYx a very practical ensemble model that reflects important elements and evolutions in design, notably, the data, reaction rules and scaled predictions.
The development of GLORYx relied on the accessibility and diversity of data sets. First, the initial FAst MEtabolizer was trained on the MDL Metabolite database that proved problematic. As a privately held resource, the data could not be independently verified to ensure data quality, and access to the data eventually was no longer possible due to its discontinuation. The investigators circumvented those shortcomings by building subsequent FAME iterations (Šícho et al. Citation2019) on the MetaQSAR database (Pedretti et al. Citation2018). This resource is publicly available and amenable to readily incorporating new reaction data for more molecules, and thus, strengthen model training and scientific rigor. Moreover, the GLORYx study included the public release of compiled data including a large reference set and smaller test set of molecules and associated reactions to expand on freely available training and testing resources. Second, the accessibility domain defines the chemical space predicted by a model to build confidence in predictions. Such an analysis of GLORYx was not possible due to the limited representative examples of certain reaction types, mostly the minor ones. Nevertheless, FAME 3 guides metabolite predictions and seems to model the effects of diverse environments on atoms targeted for reactions (Šícho et al. Citation2019). The investigators demonstrated this quality using an atom-based distance measure called FAMEscore during FAME 3 development to improve model performance. If FAME 3 adequately modeled the environmental effects that impact reactions, then there would be higher confidence in the quality of metabolite predictions based on FAME 3 predictions. In fact, GLORYx performance with a test data set yielded a sensitivity (recall) of 77% reflecting the ability to correctly identify experimentally observed metabolites.
Reaction type rules were essential for translating the site of metabolism from FAME 3 predictions into actual metabolite structures but also improved GLORYx performance. The investigators compiled reaction rules based on biochemical principles and those from modeling efforts by others (Ridder and Wagener Citation2008) that described specific chemical biotransformations. During model development, they explored which ones were best suited for predicting Phase 1 and 2 reactions and corresponding metabolites. This approach was not dependent on data sets and so avoided possible limitations from poorly represented reactions. In fact, the combination of site of metabolism scores and reaction rules improved the positive identification of known metabolites for reference data based on sensitivity increasing from 72 to 84%. Not all reactions in the data set fell under predicted families, so there is the possibility of missing some metabolic pathways through this approach.
Like other metabolite models (Judson Citation2014), GLORYx generates possibilities that often exceed those observed experimentally such that the ranking of molecules is necessary for interpretability. Rather than categorical values, FAME 3 scales predictions continuously from 0 to 1.0 resembling a statistical probability so that it is possible to rank the likelihood for reactions and subsequently metabolites. While the approach is not novel, the development of GLORYx explored different sets of rules to optimize predictions for a broad array of reactions. As mentioned previously, the effort involved trying different sets of reaction type rules eventually finding the final set. Further improvements in model performance were made possible by considering the relative frequency of reactions based on expert opinion. Reactions were divided into common and uncommon classes and the binary classifier weighed with the site of metabolism predictions. While subjective, the simple descriptor improved metabolite ranking from 75 to 80% (AUC). Those beneficial effects suggest that more information on the relative importance of reactions may be necessary to improve model performance than just relying on the reaction representation in a data set.
Figure 5. Representative workflow for metabolite prediction. Inputted molecule is subjected to modelling with FAME 3 to predict the sites of metabolism for a variety of Phase 1 and 2 reactions. Results then serve as a basis for predicting and ranking possible metabolite structures.
References
- de Bruyn Kops C, Šícho M, Mazzolari A, Kirchmair J. 2021. GLORYx: prediction of the metabolites resulting from phase 1 and phase 2 biotransformations of xenobiotics. Chem Res Toxicol. 34:286–299.
- Judson PN. 2014. Knowledge-based approaches for predicting sites of and products metabolism, in drug metabolism prediction. p. 293–318. Weinheim, Germany: WIley-VCH.
- Liu R, Liu J, Tawa G, Wallqvist A. 2012. 2D SMARTCyp reactivity-based site of metabolism prediction for major drug-metabolizing cytochrome P450 enzymes. Chem Inf Model. 52:1698–1712.
- Pedretti A, Mazzolari A, Vistoli G, Testa B. 2018. MetaQSAR: an integrated database engine to manage and analyze metabolic data. J Med Chem. 61:1019–1030.
- Ridder L, Wagener M. 2008. SyGMa: combining expert knowledge and empirical scoring in the prediction of metabolites. ChemMedChem. 3:821–832.
- Šícho M, Stork C, Mazzolari A, de Bruyn Kops C, Pedretti A, Testa B, Vistoli G, Svozil D, Kirchmair J. 2019. FAME 3: predicting the sites of metabolism in synthetic compounds and natural products for phase 1 and phase 2 metabolic enzymes. J Chem Inf Model. 59:3400–3412.
- Tomberg A, Pottel J, Liu Z, Labute P, Moitessier N. 2015. Understanding P450-mediated bio-transformations into epoxide and phenolic metabolites. Angew Chem Int Ed Engl. 54:13743–13747.
- Wienkers LC, Heath TG. 2005. Predicting in vivo drug interactions from in vitro drug discovery data. Nat Rev Drug Discov. 4:825–833.
Inhibition of human UDP-glucuronosyltransferase (UGT) enzymes by kinase inhibitors: effects of dabrafenib, ibrutinib, nintedanib, trametinib and BIBF 1202
Porntipa KorprasertthawornNuy ChauPramod C. NairAndrew RowlandJohn O. MinersSource: Biochem Pharmacol. 2019;169:113616
SYNOPSIS
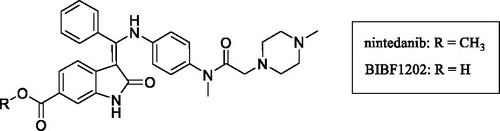
Korprasertthaworn et al. (Citation2019) demonstrate inhibition of human liver microsomal uridine diphosphoglucuronosyltransferases (UGTs) by kinase inhibitors. Dabrafenib, ibrutinib, nintedanib, and trametinib were identified as potent inhibitors of UGT1A1, with unbound inhibition constants (Ki,u) of 7.5, 3, 4.9, and 1.1 µM, respectively. In spite of the potent in vitro inhibition, the potential for in vivo inhibition of UGT1A1 is thought to be low after taking the free circulating concentrations of these kinase inhibitors into account. Studies with BIBF1202, a major hydrolytic metabolite of nintedanib did not demonstrate considerable inhibition of UGTs in in vitro experiments. Structural overlay of chemically diverse kinase inhibitors led the authors to conclude that lack of UGT inhibition was linked to the terminal electronegative carboxyl group in BIBF1202.
Commentary
UGTs are responsible for the metabolism of many endogenous molecules, drugs and exogenous chemicals (Foti and Argikar Citation2019). Inhibition of UGTs is responsible for a number of clinically observed drug interactions (Remmel et al. Citation2008; Miners et al. Citation2010). Kinase inhibitors have been investigated for their potential UGT1A1 inhibition, with an aim to predict or de-risk hyperbilirubinemia. Kinase inhibitors such as lapatinib, pazopanib, regorafenib, and sorafenib were previously demonstrated to be potent inhibitors of human liver microsomal UGT1A1, with Ki,u values of 0.6, 2.3, 0.02, and 0.03 µM, respectively (Miners et al. Citation2017). The potent inhibition by regorafenib and sorafenib is thought to contribute to the observed hyperbilirubinemia in vivo. In this article, dabrafenib, ibrutinib, nintedanib, and trametinib were identified by the authors as potent inhibitors of UGT1A1, with Ki,u values of 7.5, 3, 4.9, and 1.1 µM, respectively (Korprasertthaworn et al. Citation2019). However, after taking into account the systemic concentrations of these inhibitors and accounting for in vitro and in vivo free fractions, the anticipated possibility of drug interactions is thought be low. The article also documents computational modelling and comparison of in silico physico-chemical characteristics of approximately thirty structurally diverse kinase inhibitors. Interestingly, the overlaid structures of kinase inhibitors indicated that almost all marketed kinase inhibitors are likely to inhibit UGTs 1A1, 1A7, 1A8, 1A9, and 1A10 to some extent. However, the prediction of in vivo UGT1A1 inhibition and association with hyperbilirubinemia will need to be investigated further after considering factors such as unbound systemic and microsomal concentrations as noted above. Furthermore, the shape of these molecules is an important factor for binding within the active site of UGT1A1. BIBF1202, a major metabolite of nintedanib formed via hydrolysis of the methyl ester (), did not demonstrate considerable inhibition of UGTs in in vitro experiments. Computational modelling revealed that the unmasked carboxylic acid, was a key structural feature present in BIBF1202 in contrast to the other kinase inhibitors. Therefore, the authors concluded that this functionality and presence of a terminal electronegative group in general prevents tight binding of BIBF1202 in UGT1A1 active site, resulting in no UGT1A1 inhibition in in vitro experiments. This is a crucial observation and is likely to become a key element of medicinal chemistry strategies in the design of potent, selective, and safe kinase inhibitors.
Figure 6. Representative structures of nintedanib and its hydrolytic metabolite BIBF1202.
References
- Foti RS, Argikar UA. 2019. Uridine diphospho glucuronosyltransferases. In: Pearson PG, Weinkers LC, editors. Handbook of drug metabolism. 3rd ed. Boca Raton, FL: CRC Press; p. 109–159.
- Korprasertthaworn P, Chau N, Nair PC, Rowland A, Miners JO. 2019. Inhibition of human UDP-glucuronosyltransferase (UGT) enzymes by kinase inhibitors: effects of dabrafenib, ibrutinib, nintedanib, trametinib and BIBF 1202. Biochem Pharmacol. 169:113616.
- Miners JO, Plasek TM, Mackenzie PI, Knights KM. 2010. The in vitro characterization of inhibitory drug-drug interactions involving UDP-glucuronosyltransferase. In: Pang KS, Rodrigues AD, Peter R, editors. Enzyme and transporter based drug-drug interactions. New York: Springer; p. 217–236.
- Miners JO, Chau N, Rowland A, Burns K, McKinnon RA, Mackenzie PI, Tucker GT, Knights KM, Kichenadasse G. 2017. Inhibition of human UDP-glucuronosyltransferase enzymes by lapatinib, pazopanib, regorafenib and sorafenib: implications for hyperbilirubinemia. Biochem Pharmacol. 129:85–95.
- Remmel RP, Zhou J, Argikar UA. 2008. UDP-glucuronosyltransferases. In: Rodrigues AD, editor. Drug-drug interactions. Drugs and the pharmaceutical sciences. Handbook of drug metabolism. Vol. 186. 2nd ed. Informa healthcare; p. 87–134.
Highly selective inhibition of tyrosine kinase (TYK2) for the treatment of autoimmune diseases: discovery of the allosteric inhibitor BMS-986165
Stephen T. WrobleskiRyan MoslinShuqun LinYanlei ZhangSteven SpergelJames KempsonJohn S. TokarskiJoann StrnadAdriana Zupa-FernandezLihong ChengDavid ShusterKathleen GilloolyXiaoxia YangElizabeth HeimrichKim W. McIntyreCharu ChaudhryJaved KhanMax RuzanovJeffrey TredupDawn MulliganDianlin XieHuadong SunChristine HuangCelia D'ArienzoNelly AranibarManoj ChineyAnjaneya ChimalakondaWilliam J PittsLouis LombardoPercy H. CarterJames R. BurkeDavid S. WeinsteinSource: J Med Chem. 2019;62(20):8973–8995
SYNOPSIS
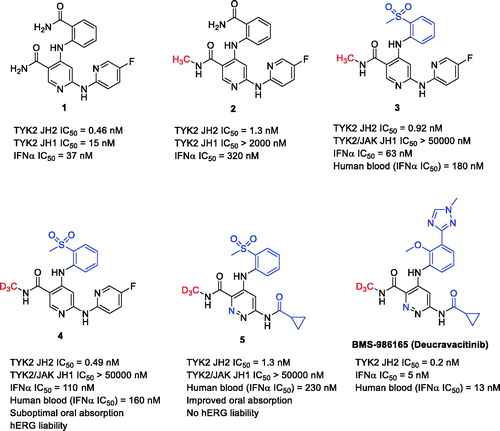
Tyrosine kinase 2 (TYK2) is a member of the Janus family of kinases (JAK1–3). The JAK family of nonreceptor TYKs mediates signaling of numerous proinflammatory cytokines, and therefore, small molecule inhibitors offer promise as effective treatments for a variety of serious inflammatory and autoimmune disorders. To date, all known JAK inhibitors are active site-directed inhibitors, which bind to the adenosine triphosphate (ATP) site within the catalytic domain and prevent the catalytic activity of the kinase by blocking ATP, downstream phosphorylation, and resulting pathway signal transduction. Achieving high selectivity towards individual JAK isoforms has, therefore, been a formidable challenge. In recent companion publications by Moslin et al. (Citation2019) and Wrobleski et al. (Citation2019), the optimization of a series of N-methyl-nicotinamide and N-methyl-pyridazine-3-carboxamide derivatives as highly selective allosteric inhibitors of TYK2 is described, ultimately culminating in the discovery of BMS-986165 (deucravacitinib, ). Deucravacitinib is the first orally active and highly selective TYK2 inhibitor in clinical trials across several immune-mediated inflammatory diseases, with positive phase 3 data for the treatment of moderate to severe plaque psoriasis. The high selectivity of BMS-986165 towards TYK2 (relative to JAK1–3 isoforms) was achieved due to its unique ability to selectively bind to the pseudokinase JH2 domain of TYK2 and inhibit its function through an allosteric mechanism, rather than interactions at the ATP binding site.
Commentary
A chemogenomics approach proved fruitful in identifying the nicotinamide derivative 1 () that allosterically inhibited TYK2-dependent interleukin (IL)-23 signaling (Moslin et al. Citation2019). However, nicotinamide 1 was fairly promiscuous, inhibiting the catalytic JH1 domains of the four JAK family members as well as numerous other kinases. A simple N-methylation of the primary carboxamide substituent in 1 led to 2, which retained the TYK2 JH2 affinity (including the sub-micromolar potency in the TYK2-dependent IL23 and interferon α (IFNα) cellular assays) of 1 and proved to be inactive against the JAK (including TYK2) JH1 domains. Moreover, N-methylation of 1 resulted in a dramatic improvement in selectivity across the kinome. A cocrystal structure of 2 with the TYK2 JH2 domain indicated that the N-methyl group accessed an atypical pocket created by a rare alanine residue in the ligand binding domain of TYK2 JH2 leading to high selectivity. Lead optimization work on 2 led to the identification of the N-methylnicotinamide 3 with considerable gains in TYK2 JH2 potency and selectivity. The TYK2 potency and selectivity gains with 3, however, were offset by the finding that 3 was subject to an appreciable degree of CYP catalyzed N-demethylation in mouse pharmacokinetics studies and that the N-demethylated metabolite lost the TYK2 JH2 selectivity (similar to observations of the unsubstituted carboxamide 1).
This dilemma was resolved via the synthesis of a trideuteromethyl amide variant of 3 (i.e. compound 4), which retained TYK2 JH2 potency and selectivity of 3 while demonstrating a dramatic reduction in N-demethylation in in vitro metabolism and pharmacokinetic assessments in mice. The observations are consistent with the fact that the C-D bond is more stable than a C–H bond. This quality leads to decreased kinetic rates of metabolism by approximately 6–10-fold when bond breakage is the rate limiting step (Katsnelson Citation2013). The heavier isotope has a lower vibrational frequency, and thus, a larger amount of energy is required to break the bond (Gant Citation2014). The metabolic stability (half-lives) of 3 (CH3) and 4 (CD3) in liver microsomal incubation across animals and human were comparable, suggesting that the deuterium incorporation was simply serving to shunt metabolism away from the amide as opposed to increasing the overall stability of the molecule towards oxidative metabolism. Having solved the TYK2 selectivity issue arising from an undesired metabolic process, additional medicinal chemistry efforts involving improvements in oral absorption profile and reduction in hERG ion channel liability associated with the N-methylnicotinamides were achieved by replacement of the central pyridine ring with the more lipophilic pyridazine ring and by substituting the aminopyridine group with the smaller cyclopropylamide functionality to yield 5. Finally, an innovative structure-based drug design strategy aimed at displacing a water molecule observed within the TYK2 JH2 binding site led to observations around enhanced TYK2 JH2 inhibitory potency with C3’ substituted analogs, and ultimately led to the identification of BMS-986165 (deucravacitinib), which contains an N-methyltriazole group at C3’ instead of the aromatic methyl sulfone.
The incorporation of deuterium into drug molecules is not an uncommon practice. However, the vast majority of examples typically involve deuterium incorporation into existing drug candidates and/or marketed drugs in an attempt to improve metabolic stability, and more importantly, improve on ADME liabilities such as drug–drug interactions (e.g. deuterated paroxetine) (Uttamsingh et al. Citation2015). Against this backdrop, the approach utilized by Moslin et al. (Citation2019) and Wrobleski et al. (Citation2019) is unique in that deuterium was incorporated during the medicinal chemistry design and optimization efforts to block and shunt an undesired metabolic pathway in vivo. It is important to note that replacement of the N–CH3 group (compound 3) with the N–CD3 group (compound 4) effectively prevented N-demethylation without any improvement in the metabolic stability of 3. This finding is consistent with previous reports (Miwa and Lu Citation1987) where CYP enzymes have been versatile enough such that metabolism can be re-routed to a different site within a molecule when deuterium is strategically placed at the site where hydrogen atom abstraction normally occurs in the nondeuterated analog. This ‘metabolic shunting’ requires the substrate to reorient itself within the CYP binding site to offer an alternative position to the heme center for metabolism. Thus, deuteration can result in alteration of the metabolite profile without a decrease in metabolic rate.
Figure 7. SAR studies leading to the discovery of the orally active, selective TYK2 inhibitor deucravacitinib. Utilization of the deuterium isotope effect prevents N-demethylation to a non-selective TYK2 inhibitor.
References
- Gant TG. 2014. Using deuterium in drug discovery; leaving the label in the drug. J Med Chem. 57(9):3595–3611.
- Katsnelson A. 2013. Heavy drugs draw heavy interest from pharma backers. Nat Med. 19(6):656.
- Miwa GT, Lu AYH. 1987. Kinetic isotope effects and ‘metabolic switching’ in cytochrome P450-catalyed reactions. BioEssays. 7(5):215–219.
- Moslin R, Zhang Y, Wrobleski ST, Lin S, Mertzman M, Spergel S, Tokarski JS, Strnad J, Gillooly K, McIntyre KW, et al. 2019. Identification on N-methyl nicotinamide and N-methyl pyridazine-3-carboxamide pseudokinase domain ligands as highly selective allosteric inhibitors of tyrosine kinase 2 (TYK2). J Med Chem. 62(20):8953–8972.
- Wrobleski ST, Moslin R, Lin S, Zhang Y, Spergel S, Kempson J, Tokarski JS, Strnad J, Zupa-Fernandez A, Cheng L, et al. 2019. Highly selective inhibition of tyrosine kinase (TYK2) for the treatment of autoimmune diseases: discovery of the allosteric inhibitor BMS-986165. J Med Chem. 62(20):8973–8995.
- Uttamsingh V, Gallegos R, Liu JF, Harbeson SL, Bridson GW, Cheng C, Wells DS, Graham PB, Zelle R, Tung R. 2015. Altering metabolic profiles of drugs by precision deuteration: reducing mechanism-based inhibition of CYP2D6 by paroxetine. J Pharmacol Exp Ther. 354(1):43–54.
Discovery of a novel deaminated metabolite of a single-stranded oligonucleotide in vivo by mass spectrometry
Jing LiJu LiuJennifer EndersMichael ArcipreteChris TranKrishna AluriLi-Hua GuanJonathan O’SheaAnna BisbeKlaus CharisséIvan ZlatevDiana NajarianYuanxin XuSource: Bioanalysis. 2019;11(21):1955–1966
SYNOPSIS
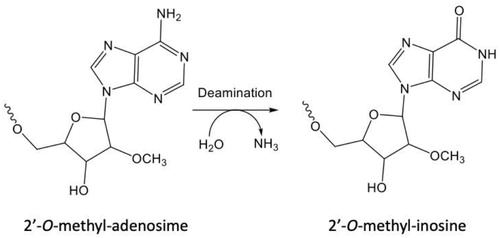
Li et al. (Citation2019) describe the first observation of deamination from adenosine to inosine (A-to-I editing) in a therapeutic oligonucleotide (). In this study, metabolite identification was conducted for a trivalent GalNAc modified REVERSIR™ oligonucleotide in vivo in monkeys. Generation of shortmers is typically expected for oligonucleotides due to nuclease activities. Nevertheless, the authors identified a novel deamination metabolite that resulted from conversion of 3’-terminal 2’-O-methyl-adenosine to 2’-O-methyl-inosine and demonstrated its presence at significant amounts in monkey liver. A-to-I editing has been reported for internal adenosine for endogenous RNAs, but this observation is a first for terminal adenosine of an exogenously dosed oligonucleotide.
Commentary
siRNAs are double-stranded RNA molecules that suppress expression of the target genes through incorporation into the RNA-induced silencing complex, which mediates degradation of the targeted mRNA (Shen and Corey Citation2018, Sutton et al. Citation2020). In recent years, siRNAs have emerged as a significant therapeutic modality due to their capability to modulate drug targets that are not reachable by traditional small molecules or protein drugs. Due to their pharmacological mechanism, siRNAs typically exhibit potent gene knockdown with prolonged duration from weeks to months. In some cases, siRNA therapeutics may benefit from a platform that can reverse the knockdown effect, thus providing an opportunity to fine-tune the drug’s pharmacology. REVERSIR molecules are short, single stranded oligonucleotides designed to serve that purpose (Zlatev et al. Citation2018). They bind to the guide strand of siRNA to block its reorganization and cleavage of target mRNA, leading to reversal of the gene silencing effect.
In this work (Li et al. Citation2019), an N-acetylgalactosamine (GalNAc)-conjugated REVERSIR molecule was studied for its biotransformation in vivo in monkeys. GalNAc is a high affinity ligand for asialoglycoprotein receptors (ASGPR) that are highly and specifically expressed on the surface of hepatocytes and, therefore, mediate rapid and efficient uptake of GalNAc-conjugated molecules into liver (Nair et al. Citation2014). In this study, the RESERSIR-A molecule conjugated with GalNAc at the 3′ end was administrated subcutaneously to monkeys, and liver, plasma, and urine samples were collected for metabolite identification. The samples were extracted using solid phase extraction, followed by ion-pair reversed phase liquid chromatography and high-resolution mass spectrometry (Q Exactive) analysis under negative mode. Metabolites resulting from cleavage of internucleotide phosphodiester bonds by nuclease activities were observed in all three matrices, with Rev-N1-N9 (loss of GalNAc and one nucleotide from 3′ end) and Rev-N1-N7 (loss of GalNAc and three nucleotides from the 3′ end) being the major metabolites. Surprisingly, another metabolite with +0.984 Da mass shift compared to Rev-N1-N9 that eluted slightly earlier was observed in monkey liver. Because the mass difference was only 0.984 Da, the spectra of multiply charged isotope envelop overlapped for the most part between the new metabolite and Rev-N1-N9, except for the first peak that is unique to Rev-N1-N9. Tandem mass analysis was conducted along with a synthetic reference standard of Rev-N1-N9 and confirmed that the mass increase occurred on the 3′ terminal 2′-O-methyl-adenosine nucleotide. Furthermore, Rev-N1-N9-inosine, with the 2′-O-methyl-ionsine replacing the 3′ terminal 2′-O-adenosine Rev-N1-N9 was synthesized (). The LC condition was developed for baseline separation of Rev-N1-N9 and Rev-N1-N9-inosine, and further confirmed that the new metabolite observed in monkey liver was Rev-N1-N9-inosine. Concentrations of the different metabolites were determined in monkey liver and the time profile indicated that REVERSIR-A was rapidly metabolized to Rev-N1-N7 and Rev-N1-N9, the latter of which then was converted to Rev-N1-N9-inosine, the most abundant metabolite in monkey liver from 24 hours to 28 days post dose. Rev-N1-N9-inosine was not detected in monkey plasma or urine.
Deamination of adenosine to inosine (A-to-I) editing has been well documented on internal adenosine of endogenous RNAs, but Li et al. are the first to report this deamination for a terminal adenosine of an exogenously dosed oligonucleotide. The mechanism of deamination was not described, but the authors proposed that if the reaction happens at a substantial level, the impact on the pharmacological activity of the oligonucleotide drug should be investigated. This work also showed the importance of surveying metabolites for GalNAc-conjugated siRNA in liver as the major organ of accumulation and action for better understanding the pharmacological relevance of the metabolism.
Figure 8. Transformation of 2′-O-methyl-adenosine to 2′-O-methyl-inosine on the 3′ terminus of an oligonucleotide.
References
- Li J, Liu J, Enders J, Arciprete M, Tran C, Aluri K, Guan LH, O’Shea J, Bisbe A, Charisse K, et al. 2019. Discovery of a novel deaminated metabolite of a single-stranded oligonucleotide in vivo by mass spectrometry. Bioanalysis. 11(21):1955–1965.
- Nair JK, Willoughby JLS, Chan A, Charisse K, Alam MR, Wang Q, Hoekstra M, Kandasamy P, Kel’in AV, Milstein S, et al. 2014. Multivalent N-acetylgalactosamine-conjugated siRNA localizes in hepatocytes and elicits robust RNAi-mediated gene silencing. J Am Chem Soc. 136(49):16958–16961.
- Shen X, Corey DR. 2018. Chemistry, mechanism and clinical status of antisense oligonucleotides and duplex RNAs. Nucleic Acids Res. 46(4):1584–1600.
- Sutton JM, Kim J, El Zahar NM, Bartlett MG. 2020. Bioanalysis and biotransformation of oligonucleotide therapeutics by liquid chromatography-mass spectrometry. Mass Spectrom Rev. DOI:https://doi.org/10.1002/mas.21641.
- Zlatev I, Castoreno A, Brown CR, Qin J, Waldron S, Schlegel MK, Degaonkar R, Shulga-Morskaya S, Xu H, Gupta S, et al. 2018. Reversal of siRNA-mediated gene silencing in vivo. Nat Biotechnol. 36(6):509–511.
In vitro metabolism of 2′‐ribose unmodified and modified phosphorothioate oligonucleotide therapeutics using liquid chromatography mass spectrometry
Jaeah KimNoha M. El ZaharMichael G. BartlettSource: Biomed Chromat. 2020;34:e4839
SYNOPSIS
Kim et al. (Citation2020) studied antisense oligonucleotides (ASOs) to understand the impact of chemical modifications on their metabolic stability in different in vitro systems, including endonuclease/exonucleases, mouse liver homogenate and human liver microsomes, by using model molecules of unmodified (phosphodiester backbone), first-generation (phosphorothioate backbone), and second-generation (phosphorothioate backbone/2′-O-methoxyethyl gapmer) ASOs. The work highlights the utilization of an optimized ion-pair liquid chromatographic condition that enables high-resolution separation of metabolites for effective identification. Phosphodiester and phosphorothioate ASOs showed different extents of instability in all the tested systems, and typical patterns of metabolism included chain truncation mediated mainly by 3′-exonucleases. In comparison, the phosphorothioate/2′-O-methoxyethyl gapmer exhibited only significant degradation in mouse liver homogenate, and the metabolism was initiated in the regions where 2′-ribose was not modified by endonucleases, followed by chain shortening by both 3′ and 5′-exonuclease activities. Overall, the results showed that modifications on backbone and/or 2′-ribose on ASOs improved their metabolic stability in in vitro systems.
Commentary
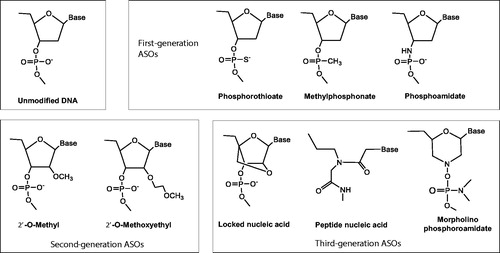
ASOs mediate the knockdown of specific genes by binding to the complementary sequence of targeted mRNA and, therefore, blocking mRNA translation or inducing degradation by RNase H (Shen and Corey Citation2018; Sutton et al. Citation2020). Unmodified ASOs with phosphodiester (PO) backbones are highly susceptible to nuclease degradation in biological matrices, so that chemical modifications are introduced to improve stability, as well as cellular delivery and target binding (). The first generation ASOs are those modified in the phosphate linkage, replacing the non-bridging oxygen with sulfur and resulting in a phosphorothioate (PS) backbone. Second generation ASOs contain additional modifications on the 2′-position of the ribose to incorporate alkyl groups such as 2′-O-methyl (2′-OMe) and 2′-methoxyethyl (2′-MOE). Modified 2′-OMe and 2′-MOE may interfere with target mRNA degradation by RNase H. Consequently, the design of gapmers was developed in which internal unmodified nucleotides are flanked by 2′-O modified bases at the 5′- and 3′- ends. The third generation of ASOs involve locked nucleic acid, peptide nucleic acid and morpholino phosphoroamidate substitutions for improved binding affinity.
In the work by Kim et al. (Citation2020), unmodified (PO backbone), first-generation (PS backbone), and second-generation (PS backbone/2′-MOE gapmer) ASOs were investigated for their metabolic stability in in vitro systems including endo/exonucleases, mouse liver homogenate and human liver microsomes. After incubation, the samples were prepared by solid phase extraction and analyzed using ion pair reversed phase liquid chromatography and high-resolution time of flight mass spectrometry (SYNAPT G2) under negative mode. The method featured an optimized LC condition with final mobile phase consisting of 30 mM N,N‐dimethylcyclohexylamine (DMCHA) as ion pairing reagent and 100 mm 1,1,1,3,3,3‐hexafluoro‐2‐propanol (HFIP) as counter anion and pH modifier. This condition was shown to improve chromatographic resolving power and provide at least 0.3 min difference in retention time between metabolites that differed in length by a single nucleotide. With the high-quality separation, the metabolites were readily assigned based on predicted molecular formulas without complications due to overlapping of the m/z series from co-eluting metabolites. Diagnostic tandem mass fragments of PO3- at m/z 79 and PO2S- at m/z 95 that originate from PO and PS linkers, respectively, as reported by Husser et al. were also observed (Husser et al. Citation2017).
The results showed that unmodified ASOs with PO backbones had poor metabolic stability and were completely degraded after an hour-long incubation with endo/exonuclease (DNase I/Exonuclease I) or mouse liver homogenate and after 12 hours with human liver microsomes. An interesting observation is that metabolism of ASOs in human liver microsomes requires NADPH, implying the possibility of the presence of an unknown metabolic pathway by membrane proteins requiring NADPH as cofactor. ASOs with PS backbones exhibited improved stability, with 45, 27, 58, and 62% degradation by exonucleases, endonucleases, mouse liver homogenate, and human liver microsomes, respectively, after a 24-hour incubation. In all the above cases, the major metabolites were 3′-end shortmers, indicating that 3′-exonucleases were the major metabolizing enzymes mediating the degradation of the tested ASOs in the studied systems. The PS/2′-MOE gapmer did not show any metabolism after a 24-hour incubation with endo/exonuclease. Degradation of 16 and 53% were observed at 24 hours and 7 days, respectively, with mouse liver homogenate but no metabolism with human liver microsomes up to 7 days. In the presence of mouse liver homogenates, the detected metabolites were those originating from endonucleases cleavage of the unmodified regions of 2′-ribose followed by exonuclease degradation, and both 3′ and 5′ activities were observed.
Overall, the results show that modifications on backbone and/or 2′-ribose on ASOs improved their metabolic stability. It would be interesting to see how well the metabolic stability and metabolite profile observed for ASOs in these in vitro system correlate to in vivo and predict in vivo metabolism and pharmacokinetics behavior. Some insight was provided by another publication from the same first author on metabolism of Eluforsen, a 33-mer PS/2′-OMe modified PS ASO (Kim et al. Citation2019). The study revealed a difference between in vitro and in vivo metabolism as the in vitro system did not generate 5′ metabolites while in vivo samples contained both 5′ and 3′ shortmers.
Figure 9. Structure of unmodified oligonucleotides and first-, second-, and third-generation antisense oligonucleotides (ASOs).
References
- Husser C, Brink A, Zell M, Muller MB, Koller E, Schadt S. 2017. Identification of GalNAc-conjugated antisense oligonucleotide metabolites using an untargeted and generic approach based on high resolution mass spectrometry. Anal Chem. 89(12):6821–6826.
- Kim J, Basiri B, Hassan C, Punt C, van der Hage E, den Besten C, Bartlett MG. 2019. Metabolite profiling of the antisense oligonucleotide eluforsen using liquid chromatography-mass spectrometry. Mol Ther Nucleic Acids. 17:714–725.
- Kim J, El Zahar NM, Bartlett MG. 2020. In vitro metabolism of 2’-ribose unmodified and modified phosphorothioate oligonucleotide therapeutics using liquid chromatography mass spectrometry. Biomed Chromatogr. 34(7):e4839.
- Shen X, Corey DR. 2018. Chemistry, mechanism and clinical status of antisense oligonucleotides and duplex RNAs. Nucleic Acids Res. 46(4):1584–1600.
- Sutton JM, Kim J, El Zahar NM, Bartlett MG. 2020. Bioanalysis and biotransformation of oligonucleotide therapeutics by liquid chromatography-mass spectrometry. Mass Spectrom Rev. :1–25.
Excretion, mass balance, and metabolism of [14C]LY3202626 in humans: an interplay of microbial reduction, reabsorption, and aldehyde oxidase oxidation that leads to an extended excretion profile
Kishore KatyayanPing YiScott MonkKenneth CassidySource: Drug Metab Dispos. 2020;48(8):698–707
SYNOPSIS
The results of excretion, mass balance, and metabolism of [14C]LY3202626, a β-site amyloid precursor protein-cleaving enzyme 1 (BACE1) inhibitor, in humans after a single 10 mg oral dose were reported. In comparison to greater than 84% recoveries of radioactivity in rats and dogs during the first 72 hours postdosing, excretion of radioactivity in humans was slow and incomplete, with approximately 75% of the total radioactivity recovered after 504 hours from the feces (31%) and urine (44%). Metabolism was the major clearance pathway in human and primarily through O-demethylation and amide hydrolysis. The unexpected slow clearance of metabolites M1, M2, and M16 in human was investigated in vitro and the experimental results suggested that an interplay between microbial reduction, reabsorption, and aldehyde oxidase (AO) oxidation (M2 → M16 → M2) could be a reason for an extended excretion profile. In addition, unextractable radioactivity observed in human plasma protein pellet and the detection of an LY3202626-dimedone adduct in human liver microsomal incubations supplemented with NADPH and dimedone suggested that a reactive sulfenic acid intermediate might be formed through bioactivation and subsequently react with sulfhydryl moiety of plasma proteins to form covalent adducts.
Commentary
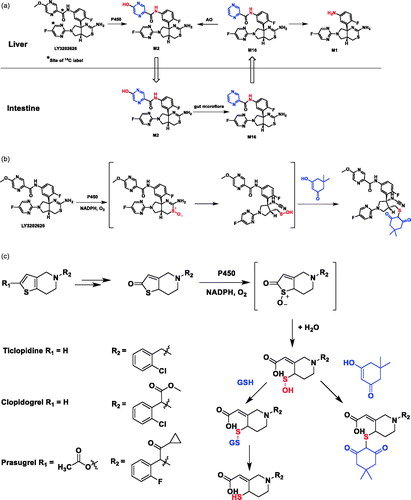
The work of Katyayan et al. demonstrates that M2, the O-demethylation metabolite of BACE1 inhibitor [14C]LY3202626, can undergo pyrazinone reduction by gut microflora under anaerobic conditions in vitro to form pyrazine M16. In addition to forming the amide hydrolysis metabolite M1 in human hepatocytes, which is metabolized the by aldehyde oxidase (AO) at the pyrazine to re-generate M2, the authors hypothesized that enterohepatic re-circulation of M2/M16 observed in human might contribute to the protraction of M1, M2, and M16 (). The hypothesis is supported by the fact that M2 is a P-glycoprotein (P-gp) substrate with low passive permeability, which can undergo intestinal secretion by P-gp to allow for gut microflora reduction to M16. The enhanced permeability of M16 may then permit for its reabsorption and subsequent AO-mediated oxidation to M2 in the liver, thereby completing its reduction/oxidation cycle.
The metabolite excretion profile in human was not anticipated preclinically perhaps for two main reasons. Firstly, pyrazinone reduction is not a common and well-recognized microbial biotransformation involved in enterohepatic circulation. Azo, nitro, alkene, ketone, N-oxide, and sulfoxide moieties are considered more susceptible to reductions in the gut (Guo et al. Citation2020). Hydrolysis of glucuronides, glycosides, sulfates, and other reactions (Roberts et al. Citation2002) mediated by gut microbial enzymes are more commonly involved in enterohepatic circulation (Wilson and Nicholson Citation2016). Secondly, the low and deficient AO activity in rats and dogs, respectively, limited the potential for M2 regeneration. As a future investigation, it would be interesting to examine if a mass balance study conducted in monkey, given its higher AO activity, would exhibit a similar protraction of M2 and M16 as humans provided monkey gut microflora could produce M16.
A small percentage of unextracted radioactivity remaining in human plasma protein pellets is likely due to LY3202626-related species forming protein adducts presumably through a reactive sulfenic intermediate, which is supported by the detection of a LY3202626-dimedone adduct in a human liver microsomal incubation in the presence of NADPH and dimedone (). The hypothesis would be strengthened if an experiment was conducted to demonstrate that the remaining radioactivity could be extracted after addition of a thiol reducing reagent dithiothreitol (DTT) to human plasma protein pellet by reducing the cysteine residue from disulfide to a free thiol group. It is known that the anti-thrombotic prodrugs ticlopidine, clopidogrel, and prasugrel require metabolic bioactivation in vivo through sulfenic acid intermediates to their active thiol metabolites (), which could covalently bind to the cysteine residue of platelet P2Y12 receptor, resulting in a disulfide bond for the intended pharmacology (Mansuy and Dansette Citation2011).
Figure 10. (a) Hypothesis illustrating enterohepatic recirculation of radioactive material and possible cause of slow excretion of radioactivity, involving microbial reduction of metabolite M2–M16 in the gut and reabsorption of M16, followed by hepatic oxidation of M16 to re-generate M2. (b) Formation of LY3202626-dimedone adduct in human liver microsome incubations supplemented with NADPH and dimedone. (c). Formation of sulfenic acid intermediates during metabolic activation of ticlopidine, and clopidogrel, and prasugrel.
References
- Guo Y, Lee H, Jeong H. 2020. Gut microbiota in reductive drug metabolism. Prog Mol Biol Transl Sci. 171:61–93.
- Mansuy D, Dansette PM. 2011. Sulfenic acids as reactive intermediates in xenobiotic metabolism. Arch Biochem Biophys. 507(1):174–185.
- Roberts MS, Magnusson BM, Burczynski FJ, Weiss M. 2002. Enterohepatic circulation: physiological, pharmacokinetic and clinical implications. Clin Pharmacokinet. 41(10):751–790.
- Wilson ID, Nicholson JK. 2016. Gut microbiome interactions with drug metabolism, efficacy, and toxicity. Transl Res. 179:204–222.
Novel homodimer metabolites of GDC-0994 via cytochrome P450-catalyed radical coupling
Ryan H. TakahashiJessica M. GrandnerSudheer BobbaYanzhou LiuPaul BerozaDonglu ZhangShuguang MaSource: Drug Metab Dispos. 2020;48:521–527
SYNOPSIS
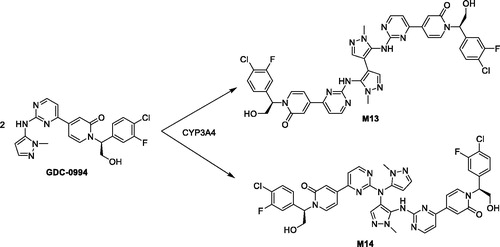
GDC-0994 is small molecule inhibitor of the extracellular signal-regulated kinases (ERK1 and ERK2) and was in clinical development for the treatment of a variety of cancer indications, including advanced or metastatic solid tumors. Metabolite profiling and structure identification studies were conducted in plasma, urine, bile, and fecal samples following a single oral 50 mg/kg (100 µCi/kg) dose of [14C]GDC-0994 to male and female Sprague–Dawley rats. Metabolites were characterized and identified by LC-MS, including accurate mass determination in conjunction with radioactive detection. Overall, fourteen metabolites were tentatively identified, twelve of which consisted of traditional phase one and phase two biotransformations. In the bile and feces, two metabolites (M13 and M14) chromatographically eluted after GDC-0994 and had identical protonated molecular ion masses [2M–2H]+, suggesting the formation of two distinct dimers. To unequivocally determine the structure of the proposed dimers, a metabolite biosynthesis approach was initiated to scale up the amount of M13 and M14 required for NMR analysis. Initial attempts with mammalian liver microsome preparations and recombinant enzymes did not result in appreciable yield. Alternatively, biosynthesis and scale up of M13 and M14 was conducted with an organometallic metalloporphyrin catalysis kit. Following isolation and purification by semi-preparative HPLC, the metabolites were characterized by NMR as the symmetric and asymmetric homodimer structures shown in . Further in vitro investigations with human liver microsomes and recombinant enzymes confirmed CYP3A4/5 as the isoforms responsible for generating the homodimer metabolites. Additionally, a radical addition mechanism and a dual radical formation mechanism were proposed for the generation of the dimers. Computational experiments with density functional theory were employed to evaluate mechanistic probability for each proposal, and ultimately supported the dual radical formation mechanism: (1) (P.+)FeIV=O (compound I) mediated hydrogen atom abstraction from the N–H linking the pyrazole and pyrimidine of one GDC-0994 molecule, (2) (P)FeIV–OH (compound II) mediated hydrogen atom abstraction from the N–H of a second GDC-0994 molecule, (3) radical coupling of the two molecules and subsequent re-aromatization to form the corresponding homodimers. In summary, this work provides an elegant mechanistic investigation to support the formation of two homodimer metabolites identified in the bile and feces from a 14C rat ADME study, and further exemplifies the diversity of P450 enzymes in producing unexpected and novel metabolites (Guengerich and Munro Citation2013).
Commentary
In drug discovery and development, metabolite identification studies are routinely conducted during in vitro, preclinical, and clinical studies to determine clearance pathways, metabolic stability, MIST, and DDI liabilities, etc. Although, the discovery of new types of biotransformation reactions are unlikely (Obach et al. Citation2012), cases routinely arise when uncommon and unexpected biotransformations occur, thus providing exciting mechanistic challenges for the biotransformation scientist. In this article, Takahashi et al. (Takahashi et al. Citation2020) conducted several mechanistic investigations to elucidate the structure of distinct homodimer metabolites and substantiate their formation, which involved a collaborative effort amongst colleagues within different scientific disciplines. Of note, proposed biochemical mechanisms were evaluated by non-routine computational probability experiments, and custom computational models were built for docking M13 and M14 into the active site of CYP3A4. Overall, this study confirms and supports that further mechanistic investigations and publications of unexpected biotransformation reactions are certainly warranted and will continually add to the richness of mechanisms seen within the biotransformation field.
Figure 11. Structures of the homodimer metabolites, M13 and M14, of GDC-0994.
References
- Guengerich FP, Munro AW. 2013. Unusual cytochrome P450 enzymes and reactions. J Biol Chem. 288(24):17065–17073.
- Obach RS, Nedderman AN, Smith DA. 2012. Radiolabelled mass-balance excretion and metabolism studies in laboratory animals: are they still necessary? Xenobiotica. 42(1):46–56.
- Takahashi RH, Grander JM, Bobba S, Liu Y, Beroza P, Zhang D, Ma S. 2020. Novel homodimer metabolites of GDC-0994 via cytochrome P450-catalyzed radical coupling. Drug Metab Dispos. 48(6):521–527.
Metabolism and disposition of volanesorsen, a 2’- O-(2 methoxyethyl) antisense oligonucleotide, across species
Noah PostRosie YuSarah GreenleeHans GausEunju HurhJohn MatsonYanfeng WangSource: Drug Metab Dispos. 2019;47:1164–1173
SYNOPSIS
Post et al. (Citation2019) report on the metabolism and disposition of volanesorsen, a partially 2′-O-(2-methoxyethyl) (2′-MOE)-modified antisense oligonucleotide across species, from mouse to human, utilizing novel extraction and quantitation techniques in samples collected from preclinical studies (mice, rats and monkeys), a 3H rat ADME study, and a phase 1 clinical trial. Volanesorsen (previously known as ISIS 304801) is a 20-nucleotide partially 2′-MOE-modified antisense oligonucleotide (ASO) gapmer, which was recently approved in the European Union as a novel, first-in-class treatment in the reduction of triglyceride levels in patients with familial chylomicronemia syndrome. The plasma metabolite profiles of volanesorsen are similar across species, with volanesorsen as the major component. Various shortened oligonucleotide metabolites (5–19 nucleotides long) were identified in tissues in the multiple-dose mouse and monkey studies, but fewer in the [3H]-volanesorsen rat study, likely due to a lower accumulation of metabolites following a single dose in rats. In urine, all metabolites identified in tissues were observed, consistent with both endo- and exonuclease-mediated metabolism and urinary excretion being the major elimination pathway for volanesorsen and its metabolites. The main mode of volanesorsen metabolism was identified as being initial endonuclease-mediated hydrolysis at various positions within the central gap of the parent compound, followed by subsequent exonuclease (3′ and 5′)-mediated hydrolysis of the deoxynucleoside ends of the formed metabolites.
Commentary
Volanesorsen, a 20-nucleotide partially 2′-MOE–modified ASO gapmer, was developed to inhibit apolipoprotein C-III (APOC3), a key player in the metabolism of triglycerides and triglyceride-rich lipoproteins. Treatment with volanesorsen leads to a robust decrease in both APOC3 production and triglyceride concentrations (Pechlaner et al. Citation2017) and has recently been approved by the European Union as a novel, first-in-class treatment of reduction of triglyceride levels in patients with familial chylomicronemia syndrome. The 2′-MOE modifications are commonly known as second generation ASOs. The 2-methoxyethyl modification led to the development of potent, pharmacologically active, specific ASOs such as mipomersen (marketed as Kynamro), an inhibitor of apolipoprotein B-100 synthesis used to reduce low-density lipoprotein cholesterol, apolipoprotein B, total cholesterol, and non-high-density lipoprotein cholesterol in patients with homozygous familial hypercholesterolemia (Kastelein et al. Citation2006), and inotersen (marketed as Tegsedi®), an inhibitor of hepatic production of transthyretin protein used to reduce serum transthyretin protein and risk of transthyretin protein deposits in tissues in patients with polyneuropathy caused by hereditary transthyretin-mediated amyloidosis (Ackermann et al. Citation2016; Benson et al. Citation2018; Shen and Corey Citation2018).
The pharmacokinetic and metabolism properties of volanesorsen have been thoroughly characterized across species from mice to human by the authors. Particularly for metabolism, volanesorsen was the most abundant oligonucleotide and accounted for >70% of the total oligonucleotides in mouse liver and kidney tissue samples taken 48 hours after 13 weeks of treatment (day 93). Metabolites consistent with both initial exonuclease-mediated cleavage (N-1 to N-3 metabolites, 17–19 nucleotides in length) and endonuclease-mediated cleavage (N-5 to N-14, 6–15 nucleotides in length) were evident in both liver and kidney samples, with the N-1 (or 19-mer) as the most abundant. Initial profiling using the UV chromatograms indicated that the N-15 (5-mer) metabolites were not present in the samples; however, when samples were run with a more sensitive method (via LC-MS/MS), both 5-mers were detected, although at a low abundance. Metabolites from both exo- and endo-nuclease-mediated metabolism increased from the time treatment ended after 13 weeks (day 93) to the recovery time point (day 182). After 13 weeks of recovery the amount of volanesorsen had decreased to <50% of the total oligonucleotides. Monkey liver and kidney tissue samples taken 48 hours after 13 weeks of treatment (day 93) exhibited a similar metabolism profile to mice, including volanesorsen being the most abundant oligonucleotide detected (>82% of total oligonucleotides). Metabolites consistent with exonuclease-mediated cleavage were evident in all liver and kidney cortex samples as well as low levels of putative endonuclease metabolism products. After 13 weeks following the last day of treatment, the amount of volanesorsen decreased to between 48 and 52% of total oligonucleotides. Mouse, monkey, and human urine samples all had metabolites consistent with both exo- and endo-nuclease-mediated metabolism. The extent of metabolites compared with intact volanesorsen was much greater in all urine samples, except for mouse urine at the 0–24-hour time point. The most abundant individual metabolite in most of the samples were the 7-mers (generated either from 39- to 59-deletions). Two metabolites were found in human urine that were not found in mouse or monkey urine, the 18-mer from a 59-deletion and the 16-mer from a 59-deletion; however, both were present at a very low level (<0.1% of total oligonucleotide excreted over 24 hours).
The authors showed consistent results with previous reports indicating that 2′-MOE partially modified ASOs are slowly metabolized by nucleases in tissues, via predominantly endonuclease hydrolysis at various positions within the deoxyphosphorothioate gap, followed by subsequent 3′- and 5′-exonuclease hydrolysis of the exposed deoxynucleoside ends of the formed metabolites (). It is worth noting that the extraction method used in this report (liquid-liquid extraction with phenol/chloroform/isoamyl alcohol 25/24/1, followed by a solid-phase extraction using a 96-well Strata-X packed plate and additional protein precipitation) has a higher absolute recovery for ASOs ranging from 5 to 20 bases in length and can capture the entire metabolic profile much better than previous methods (Geary et al. Citation2003; Yu et al. Citation2007). It is likely that the previously reported total ASO recovery in urine for ASOs utilizing the older extraction method (two-step solid-phase extraction, a strong anion exchange followed by a C18) led to an underestimate of the actual value. With these improved methods the authors were able to generate mass balance data showing approximately 16.5% recovery of the dose administered over a 24 hour post-dose interval, which would lead to complete recovery of the administered dose assuming the same excretion on each of the other 6 days of a weekly dose interval (a reasonable assumption considering the long half-life of the ASO in tissues).
Figure 12. Metabolic biotransformation and excretion pathways of second generation ASOs (e.g. volanesorsen).
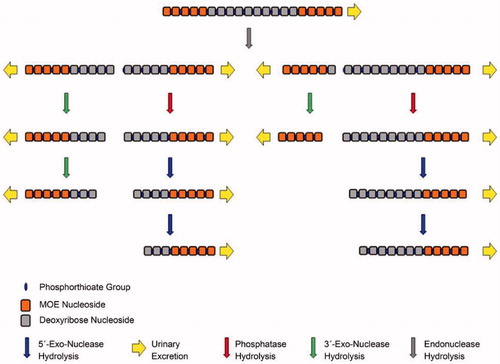
References
- Ackermann EJ, Guo S, Benson MD, Booten S, Freier S, Hughes SG, Kim TW, Jesse Kwoh T, Matson J, Norris D, et al. 2016. Suppressing transhyretin production in mice, monkeys and humans using 2nd-generation antisense oligonucleotides. Amyloid. 23:148–157.
- Benson MD, Waddington-Cruz M, Berk JL, Polydefkis M, Dyck PJ, Wang AK, Planté-Bordeneuve V, Barroso FA, Merlini G, Obici L, et al. 2018. Inotersen treatment for patients with hereditary transthyretin amyloidosis. N Engl J Med. 389:22–31.
- Geary RS, Yu RZ, Watanabe T, Henry SP, Hardee GE, Chappell A, Matson J, Sasmor H, Cummins L, Levin AA. 2003. Pharmacokinetics of a tumor necrosis factor-α phosphorothionate 2’-O-(2-methoxyethyl) modified antisense oligonucleotide: comparison across species. Drug Metab Dispos. 31(11):1419–1428.
- Kastelein JJ, Wedel MK, Baker BF, Su J, Bradley JD, Yu RZ, Chuang E, Graham MJ, Crooke RM. 2006. Protein reduction of apolipoprotein B and low-density lipoprotein cholesterol by short-term administration of an antisense inhibitor of apolipoprotein B. Circulation. 114(16):1729–1735.
- Pechlaner R, Tsimikas S, Yin X, Willeit P, Baig F, Santer P, Oberhollenzer F, Egger G, Witztum JL, Alexander VJ, et al. 2017. Very-low-density lipoprotein-associated apolipoproteins predict cardiovascular events and are lowered by inhibition of APOC-III. J Am Coll Cardiol. 69(7):789–800.
- Post N, Yu R, Greenlee S, Gaus H, Hurh E, Matson J, Wang Y. 2019. Metabolism and disposition of Volanesorsen, a 2'-O-(2 methoxyethyl) antisense oligonucleotide, across species. Drug Metab Dispos. 47:1164–1173.
- Shen X, Corey DR. 2018. Chemistry, mechanism and clinical status of antisense oligonucleotides and duplex RNAs. Nucleic Acids Res. 46:1584–1600.
- Yu RZ, Kim TW, Hong A, Watanabe TA, Gaus HJ, Geary RS. 2007. Cross-species pharmacokinetic comparison from mouse to man of a second-generation antisense oligonucleotides, ISIS 301012, targeting human apolipoprotein B-100. Drug Metab Dispos. 35:460–468.
Novel o-toluidine metabolite in rat urine associated with urinary bladder carcinogenesis
Yuya TajimaTakeshi ToyodaYuichiro HirayamaKohei MatsushitaTakanori YamadaKumiko OgawaKenji WatanabeTakeji Takamura-EnyaYukari TotsukaKeiji WakabayashiNoriyuki MiyoshiSource: Chem Res Toxicol. 2020;33:1907–1914
SYNOPSIS
Tajima et al. (Citation2020a) report on a novel metabolite of o-toluidine that may be involved in the mode of action underlying its bladder carcinogenicity. o-Toluidine is a monocyclic aromatic amine known to cause bladder cancer in experimental animals and in humans. The underlying mode of action has long been assumed to proceed via metabolic activation via cytochrome P450 catalyzed N-hydroxylation in the liver followed by acetylation in the bladder (). N-Acetoxy-o-toluidine is unstable and results via deacetylation in a DNA reactive electrophilic nitrenium ion which forms covalent adducts at the C8 position of guanine. The study by Tajima et al. (Citation2020a) shows a novel metabolite of o-toluidine, 2-methyl-N4-(2-methylphenyl)benzene-1,4-diamine (MMBD) (Tajima et al. Citation2020b) (). MMBD formation was detected in in vitro incubations of o-toluidine with S9 from phenobarbital/5,6-benzoflavone induced rat liver. MMBD was also detected in the urine of o-toluidine exposed rats. In a genotoxicity test in a Salmonella typhimurium strain that overexpressed N-acetyltransferases MMBD showed a higher genotoxic potency than o-toluidine itself. In an in vitro incubation of MMBD containing rat urine with calf thymus DNA in the presence of S9, a guanine DNA adduct of MMBD (dG-MMBD) was detected as the major adduct formed. The same dG-MMBD adducts could also be detected in the liver DNA of rats treated with o-toluidine. The authors conclude that the bladder carcinogenesis of o-toluidine can be at least in part ascribed to his newly identified metabolite MMBD.
Commentary
o-Toluidine is listed by the international Agency for Research on Cancer (IARC) as a Group 1 carcinogen (‘carcinogenic to humans’). Exposure to o-toluidine is known to cause bladder cancer in both experimental animals and humans by a genotoxic mode of action. Although the pathway leading to o-toluidine DNA adducts has been suggested to proceed via formation of a nitrenium ion intermediate (), direct detection of the potentially resulting o-toluidine-dG DNA adducts in relevant biological samples has not been reported (Tajima et al. Citation2020a). As a result, the exact mode of action underlying the bladder carcinogenicity of o-toluidine remained to be investigated to a further extent. The formation of MMBD as a novel o-toluidine metabolite represents a novel bioactivation pathway for o-toluidine. MMBD can be formed by head-to-tail dimerization of two o-toluidine units, and its formation was shown to be dependent on active S9 in the presence of NADPH as a cofactor. The authors do not comment on the mechanism underlying the dimerization, but the radical type intermediates formed upon hydrogen or electron abstraction from the substrate by the high valency iron-oxo intermediate in the active site of cytochromes P450 may result in o-toluidine radicals that could dimerize to give the respective dimer. The dimerisation was observed in vivo and in S9 incubations in the presence of NADPH, but whether it is indeed cytochrome P450 mediated remains to be established. Interestingly and also open to further research is that the levels of the dimers resulting from tail-to-tail and head-to-head dimerization, 2,2′-dimethylhydrazobenzene (DHB) and 3,3′-dimethylbenzidine (DBZ), respectively, () in the urinary samples of o-toluidine treated rats were under the detection limit. DBZ (also called o-tolidine) is already known to be a possible human carcinogen, being classified as a Group 2 carcinogen by IARC. MMBD is structurally related to 4-aminobiphenyl, a well-known bladder carcinogen, classified as a Group1 carcinogen by IARC so it is conceivable that o-toluidine is a carcinogen. Based on the fact that another aromatic amine, o-anisidine appeared able to form dimers, made the authors to speculate that dimerization might be a critical step in the carcinogenicity of monocyclic aromatic amines in general.
The authors did not identify the position of the dG adduct formation by MMBD and indicate if it could have occurred at the C8, N2, or the N7 position of dG. Exact identification of the nature of the MMBD-dG adduct must await synthesis of the authentic reference compound. Formation of the MMBD-dG adducts in the target organ for tumor formation, the bladder, also remains to be established. The authors could not complete the detection of the MMBD-dG adducts in bladder DNA of the o-toluidine exposed rats, because they could not isolate sufficient DNA from this tissue for their LC-MS/MS based analyses.
In spite of these limitations, the results of the study provide a potential novel pathway of importance for the mode of action underlying o-toluidine induced bladder carcinogenicity that may even prove relevant for other monocyclic aromatic amines that could dimerize to potentially genotoxic carcinogens.
Figure 13. Conversion of o-toluidine to its reactive nitrenium ion via N-hydroxylation and subsequent acetylation, and to MMBD via head-to-tail dimerization. For comparison, the structures of the dimers resulting from tail-to-tail dimerization (DBZ) and head-to-head dimerization (DHB) are also shown.
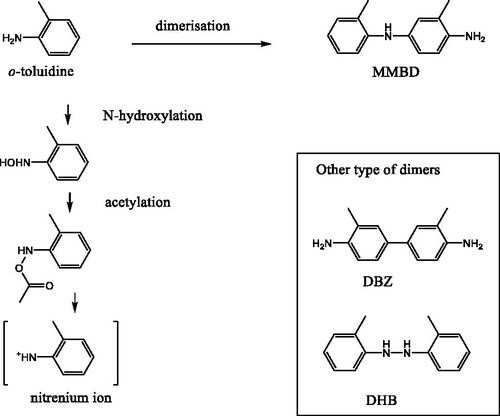
References
- Tajima Y, Toyoda T, Hirayama Y, Matsushita K, Yamada T, Ogawa K, Watanabe K, Takamura-Enya T, Totsuka Y, Wakabayashi K, et al. 2020a. Novel o-toluidine metabolite in rat urine associated with urinary bladder carcinogenesis. Chem Res Toxicol. 33(7):1907–1914.
- Tajima Y, Toyoda T, Hirayama Y, Matsushita K, Yamada T, Ogawa K, Watanabe K, Takamura-Enya T, Totsuka Y, Wakabayashi K, et al. 2020b. Correction to a novel o-toluidine metabolite in rat urine associated with urinary bladder carcinogenesis. Chem Res Toxicol. 33(12):3065–3065.
Identifying cysteine, N-acetylcysteine, and glutathione conjugates as novel metabolites of aristolochic acid I: emergence of a new detoxifying pathway
Jiayin ZhangChi-Kong ChanYat-Hing HamWan ChanSource: Chem Res Toxicol. 2020;33:1374–1381.
Synopsis
This paper by Zhang et al. (Citation2020) describes a series of novel metabolites formed from aristolochic acid I (AAI) that point at a novel detoxification pathway for this potent kidney carcinogen. Formation of the aristolactam-nitrenium ion upon metabolism of AAI () is considered to lead to a DNA reactive intermediate responsible for tumor formation. In the present study it is shown, both in in vitro studies and in rats exposed to AAI in vivo, that the aristolactam-nitreniun ion intermediate can also react with endogenous thiols including cysteine, N-acetylcysteine and glutathione. This reaction represents a novel, previously unreported detoxification pathway for the DNA reactive aristolactam-nitrenium intermediate of AAI.
Commentary
Aristolochic acids, including AAI, are natural constituents of all Aristolochia species. These botanicals are used as ingredients for traditional medicines including herbal Chinese medicines (Arlt et al. Citation2002; Zhang et al. Citation2020) or may end up in the food chain because of contamination of soil or crops via Aristolochia type weeds (Zhang et al. Citation2020). In many countries, products containing AAs were prohibited after discovery of the carcinogenic potential of AAs (Mengs et al. Citation1982). The risks of exposure to AAs became even more evident in 1993 when more than 1800 Belgian women were accidentally exposed to AAs via slimming pills. Many of these young women developed chronic kidney failure and/or cancer of the kidneys and the urinary tract (Vanherweghem et al. Citation1993; Vanhaelen et al. Citation1994). A large body of evidence suggests that AA-induced DNA adduct formation, followed by cellular proliferation and fixation of mutations, is responsible for development of the kidney cancers in AA-treated animals and humans (Schmeiser et al. Citation1996; Arlt et al. Citation2002; Nortier and Vanherweghem Citation2002). AAs have also been suggested to be involved in the etiology of Balkan endemic nephropathy (BEN). This renal fibrotic disease in the Balkan Peninsula is due to AA contamination of cultivation soil and food crops by AA from Aristolochic clematis I. growing as an abundant weed in this area (Arlt et al. Citation2007; Zhang et al. Citation2020).
Formation of the aristolactam-nitrenium ion upon nitroreduction of AAI () is considered to represent the formation of a DNA reactive intermediate responsible for covalent adduct formation with the exocyclic amino group of purine bases and subsequent tumor formation. To date, cytochrome P450 mediated O-demethylation to aristolochic acid Ia (AAIa) and subsequent glucuronidation and/or sulfation and excretion have been considered as a detoxification pathway for AAI (). The study by Zhang et al. (Zhang et al. Citation2020) provides evidence for an additional detoxification pathway, following formation of the aristolactam-nitrenium ion intermediate. Zhang et al. generated the adducts of AAI with cysteine (AAI-Cys), N-acetylcysteine (AAI-NAC) and glutathione (AAI-SG) in in vitro incubations of AAI with these thiols in the presence of preactivated Zinc dust (Zinc/H+). The exact structure of the adducts was not characterized by NMR due to the low amount of reference material available, but Zhang et al. (Citation2020) provided three lines of indirect evidence for the fact that the adducts were carbon-sulfur linked conjugates, instead of sulfonamide conjugates formed at the amide nitrogen atom of the aristolactam moiety. Evidence included: (i) the conjugates were stable in acidic conditions (pH 2.0) while sulfonamide adducts are not, (ii) theoretical calculations revealed the electron density on C7 to be lower than that on the N in the aristolactam-nitrenium ion intermediate, and (iii) there is abundant evidence that the aristolactam-nitrenium ion binds covalently via its C7 to the exocyclic amine groups of purine bases in DNA. The adducts thus prepared were used as reference materials, to be detected by LC-MS/MS, the respective adducts in serum samples of rats orally exposed to 10 or 30 mg/kg bw AAIa. The level of other AAI metabolites including aristolochic acid Ia (AA1a) and aristolactam I (ALI) were quantified as well. The metabolite patterns obtained revealed that AA1a and AL1 accounted for about 89.3 and 7.4%, respectively, of the metabolites detected in serum. The thiol metabolites accounted for 3.3% for AAI-Cys and 0.03% for AAI-NAC, while AAI-SG was detected only in trace amounts in urine together with trace amounts of AAI-NAC. The authors relate this low level of urinary excretion to potential biliary excretion of the adducts, which remains open for future research. Nevertheless, low levels may still reflect an important detoxification pathway because bioactivation pathways may already raise a concern even when they represent only a small fraction of the administered dose.
Figure 14. Metabolic pathways for AAI, including the newly identified detoxification of the aristolactam nitrenium ion intermediate by thiols.
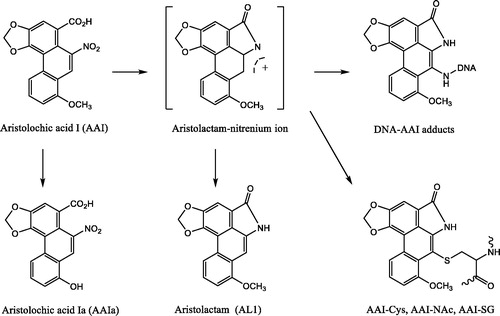
References
- Arlt VM, Stiborova M, Schmeiser HH. 2002. Aristolochic acid as a probable human cancer hazard in herbal remedies: a review. Mutagenesis. 17(4):265–277.
- Arlt VM, Stiborová M, vom Brocke J, Simões ML, Lord GM, Nortier JL, Hollstein M, Phillips DH, Schmeiser HH. 2007. Aristolochic acid mutagenesis: molecular clues to the aetiology of Balkan endemic nephropathy-associated urothelial cancer. Carcinogenesis. 28(11):2253–2261.
- Mengs U, Lang W, Poch JA. 1982. The carcinogenic action of aristolochic acid in rats. Arch Toxicol. 51(2):107–119.
- Nortier JL, Vanherweghem JL. 2002. Renal interstitial fibrosis and urothelial carcinoma associated with the use of a Chinese herb (Aristolochia fangchi). Toxicology. 181–182:577–580.
- Schmeiser HH, Bieler CA, Wiessler M, deStrihou CV, Cosyns JP. 1996. Detection of DNA adducts formed by aristolochic acid in renal tissue from patients with Chinese herbs nephropathy. Cancer Res. 56(9):2025–2028.
- Vanhaelen M, Vanhaelenfastre R, But P, Vanherweghem JL. 1994. Identification of aristolochic acid in Chinese herbs. Lancet. 343(8890):174–174.
- Vanherweghem J-L, Tielemans C, Abramowicz D, Depierreux M, Vanhaelen-Fastre R, Vanhaelen M, Dratwa M, Richard C, Vandervelde D, Verbeelen D, et al. 1993. Rapidly progressive interstitial renal fibrosis in young-women – association with slimming regimen including Chinese herbs. Lancet. 341(8842):387–391.
- Zhang JY, Chan CK, Ham YH, Chan W. 2020. Identifying cysteine, N-acetylcysteine, and glutathione conjugates as novel metabolites of aristolochic acid I: emergence of a new detoxification pathway. Chem Res Toxicol. 33(6):1374–1381.
Significance of multiple bioactivation pathways for meclofenamate as revealed through modeling and reaction kinetics
Mary Alexandra SchleiffNoah R. FlynnSasin PayakachatBenjamin Mark SchleiffAnna O. PinsonDennis W. ProvinceS. Joshua SwamidassGunnar BoysenGrover P. MillerSources: Drug Metab Dispos. 2021;49:133–141
SYNOPSIS
Meclofenamate (MCF; trade name Meclomen) is a nonsteroidal anti-inflammatory (NSAID) used for treatment of pain (Wolfe et al. Citation1999). MCF is a member of a class of fenamates targeting COX that were discovered to alleviate NSAID gastrointestinal adverse events. Nevertheless, the compound MCF carried the risk of hepatotoxicity that limited its clinical usage (Aronson Citation2016). It is thought that this toxicity is initiated as a result of cytochrome P450 bioactivation that forms covalent adducts to hepatic proteins and glutathione (GSH) (Galati et al. Citation2002). Here the investigators (Schleiff et al. Citation2021) used computational and experimental approaches to analyze possible key reactive metabolite formations.
Through computational methods, 19 possible reactive metabolites highlighted possible reactions including metabolites resulting from monooxidation, dioxidation and/or oxidation followed by the resulting dechlorination. These metabolites led to the formation of quinone-species reactive metabolites. The position of these oxidation sites could be on either of the two aromatic rings, and based on computational calculations, it favoured the acidic aromatic ring. These reactive metabolites were assessed using computational models for their propensity for reacting with GSH.
Schleiff et al. followed these analyses with metabolite identification and kinetic incubations with liver microsomes. They used dansyl GSH for trapping reactive metabolites by employing the fluorescent characteristics of the dansyl moiety, which allows the analysis to be quantitative, unlike mass spectrometric based GSH conjugates measurements that are highly selective but far from quantitative. They reported four major GSH conjugates with three being fully characterized. Based on kinetic studies, the two major conjugates were formed from mono-oxidation (1 & 2) and oxidative dechlorination (3) processes (). They also concluded that the total bioactivation pathways account for about 13% of the total oxidative metabolism.
Commentary
Drug induced liver injury (DILI) remains as one of the major causes of hepatotoxicity and contributes significantly to drug attrition. DILI is a multifactorial condition that involves mainly direct cytotoxicity and/or immune-mediate toxicity (Li et al. Citation2009). In this article, the investigators used computational and experimental methods to quantitate potential liability of MCF that contributes to initiation of DILI. In fact, this method may be useful during drug discovery stages to generate hypotheses and mitigate bioactivation mechanisms. With dansyl GSH instead of GSH, the investigators were able to quantitatively measure GSH conjugate formation. This strategy is a more direct measurement using dansyl fluorescence properties that is not affected by conjugation, whereas quantification using mass spectrometric responses is not quantitatively reliable. Though the presence of dansyl should not change the reactivity of the free thiol to the reactive centers, in this case quinone type intermediates, the dansyl interferes with glutathione S-transferase (GST) mediated conjugation (Gan et al. Citation2005). This outcome may change the total kinetics of the reaction. Another open question is whether the major reactive metabolite pathway causes the toxicity. Taking together both computation and experimental methods allows for further assessment of bioactivation that augment the current drug discovery paradigm.
Figure 15. Predicted sites of oxidations of meclofenamate to form reactive metabolites.
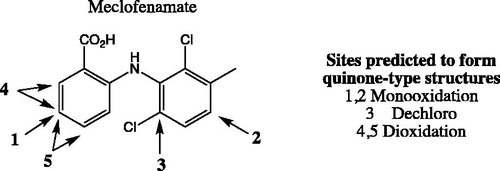
References
- Aronson JK, editor 2016. Flufenamic acid and meclofenamic acid. Meyler's side effects of drugs. 16th ed. Oxford: Elsevier; p. 361.
- Galati G, Tafazoli S, Sabzevari O, Chan TS, O’Brien PJ. 2002. Idiosyncratic NSAID drug induced oxidative stress. Chem Biol Interact. 142:25–41.
- Gan J, Harper TW, Hsueh M-M, Qu Q, Humphreys WG. 2005. Dansyl glutathione as a trapping agent for the quantitative estimation and identification of reactive metabolites. Chem. Res. Toxicol. 18(5):896–903.
- Li Y, Qi XM, Xue X, Wu XF, Wu YF, Chen M, Xing GZ, Luan Y, Ren J. 2009. The relationship between diphenylamine structure and NSAIDs-induced hepatocytes injury. Toxicol Lett. 186:111–114.
- Schleiff MA, Flynn NR, Payakachat S, Schleiff BM, Pinson AO, Province DW, Swamidass SJ, Boysen G, Miller GP. 2021. Significance of multiple bioactivation pathways for meclofenamate as revealed through modeling and reaction kinetics. Drug Metab Dispos. 49:133–141.
- Wolfe MM, Lichtenstein DR, Singh G. 1999. Gastrointestinal toxicity of nonsteroidal anti-inflammatory drugs. N Engl J Med. 340:1888–1899.
Detoxication versus bioactivation pathways of lapatinib in vitro: UGT1A1 catalyzes the hepatic glucuronidation of debenzylated lapatinib
Dasean T. Nardone-WhiteJennifer E. BissadaArsany A. AboudaKlarissa D. JacksonSource: Drug Metab Dispos. 2021;49(3):233–244
SYNOPSIS
This paper by Nardone-White et al. (Citation2021) describes the biotransformation pathways of lapatinib’s major metabolite, debenzylated laptinib (M1), and the observed interspecies differences in the in vitro metabolism of M1. Non-P450 biotransformation pathways such as glucuronidation, sulfonation, and aldehyde oxidase mediated oxidation were identified as disposition pathways for M1. It is noteworthy that these pathways are competitive with respect to bioactivation of M1 via quinone-imine formation. For glucuronidation, reaction phenotyping studies with Uridine diphosphoglucuronosyltransferases (UGTs) revealed key contributions from UGT1A1 and UGT1A8. UGT1A10, followed by UGT1A3 and UGT1A9 to the conversion of M1 to M1-glucuronide, suggesting glucuronidation of M1 in liver, intestines, and possibly kidney. Species differences were also observed in the metabolism of M1.
Commentary
CYP3A4 metabolizes the tyrosine kinase inhibitor lapatinib (Castellino et al. Citation2012) including oxidative O-debenzylation, which has been implicated in lapatinib-induced hepatotoxicity. The authors of this present article further deconvoluted competing biotransformation pathways of the M1 metabolite as shown in . Metabolic reactions included a quinone-imine intermediate that was trapped to form an M1 GSH conjugate. M1 was also found to be metabolized to a glucuronide conjugate, a sulfate conjugate, and an aldehyde oxidase mediated metabolite on the substituted aminoquinazoline ring (M3). Although M3 has been identified as a direct metabolite of M1, evidence is lacking for an alternative competing putative metabolic pathway arising from lapatinibto form M3. The authors showed that the uridine diphosphoglucuronosyltransferase isoforms (UGTs) 1A1, 1A3, 1A8, 1A9, and 1A10 play a role in glucuronidation of M1 to form M1-glucuronide. Interestingly, the same isoforms are also involved in the metabolism of lapatinib (US Food and Drug Administration). Furthermore, as discussed earlier in this compiled review, kinase inhibitors without terminal electronegative groups show a general tendency toward inhibiting UGT1A1, UGT1A8, UGT1A9, and UGT1A10 (Miners et al. Citation2017; Korprasertthaworn et al. Citation2019). Therefore, in vitro studies with UGTs investigating interactions between lapatinib and M1, as well as UGT inhibition by M1 (which contains an unmasked electronegative phenolic group), will be of interest. Species differences were also observed in the metabolism of M1 in in vitro experiments with various sub-cellular fractions. While M1 glucuronidation was higher in dog than rat and human, M1 sulfonation was higher in human compared with rat and dog. M3 formation was higher in human than rat and was not observed at all in dog, a species which lacks functional aldehyde oxidase. Interspecies differences in the metabolism of M1 may impact the extent to which hepatotoxicity is/was observed during preclinical development and may even influence the ability to accurately estimate the risk of hepatotoxicity in human.
Figure 16. Metabolic pathways of debenzylated metabolite of lapatinib, M1. Metabolic pathways of lapatinib are not shown.
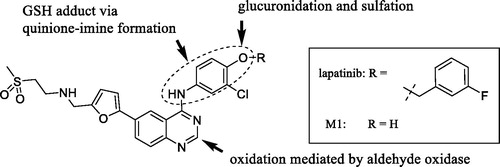
References
- Castellino S, O’Mara M, Koch K, Borts DJ, Bowers GD, MacLauchlin C. 2012. Human metabolism of lapatinib, a dual kinase inhibitor: implications for hepatotoxicity. Drug Metab Dispos. 40:139–150.
- Korprasertthaworn P, Chau N, Nair PC, Rowland A, Miners JO. 2019. Inhibition of human UDP-glucuronosyltransferase (UGT) enzymes by kinase inhibitors: effects of dabrafenib, ibrutinib, nintedanib, trametinib and BIBF 1202. Biochem Pharmacol. 169:113616.
- Miners JO, Chau N, Rowland A, Burns K, McKinnon RA, Mackenzie PI, Tucker GT, Knights KM, Kichenadasse G. 2017. Inhibition of human UDP-glucuronosyltransferase enzymes by lapatinib, pazopanib, regorafenib and sorafenib: implications for hyperbilirubinemia. Biochem Pharmacol. 129:85–95.
- Nardone-White DT, Bissada JE, Abouda AA, Jackson KD. 2021. Detoxication versus bioactivation pathways of lapatinib in vitro: UGT1A1 catalyzes the hepatic glucuronidation of debenzylated lapatinib. Drug Metab Dispos. 49(3):233–244.
- US Food and Drug Administration. 2021. TykerbTM Clinical Pharmacology and Biopharmaceutics NDA Review. [accessed 2021 Feb 22]. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2007/022059s000_ClinPharmR.pdf.
Discovery of JNJ-63576253: a clinical stage androgen receptor antagonist for F877L mutant and wild-type castration-resistant prostate cancer (mCRPC)
Zhuming ZhangPeter J. ConnollyHeng Keang LimVineet PandeLieven MeerpoelChristopher TelehaJonathan R. BranchJanine OndrusIan HicksonTammy BushLeopoldo LuistroKathryn PackmanJames R. BischoffSalam IbrahimChristopher ParrettYolanda ChongMarco M. GottardisGilles BignanSource: J Med Chem. 2021;4(2):909–924
SYNOPSIS
Oncogenic activation of the androgen receptor (AR) is a vital process in the progression of prostate cancer. Marketed antiandrogens such as apalutamide and enzalutamide (), which have demonstrated improved metastasis-free survival for both castration-sensitive and -resistant prostate cancer, elicit their pharmacologic action by blocking the binding of androgens to the AR. Although apalutamide and enzalutamide are the standard-of-care for advanced prostate cancer treatment, acquired resistance almost invariably emerges, resulting in disease progression to metastatic castration-resistant prostate cancer. Recent studies have shown that mutation of phenylalanine to leucine at position 877 (F877L) in the AR confers resistance to both drugs, leading to antagonist-to-agonist switch in response to preclinical treatment in vivo. In an attempt to identify AR antagonists with activity against wild-type as well as F877L and other clinically relevant mutant forms of the AR, Zhang et al. (Citation2021) recently reported on compound 1 () as a favorable lead with attractive disposition and pharmacological properties. However, repeat (14-day) oral dose safety studies with 1 in dogs led to severe liver necrosis, bile degeneration and considerable elevations in liver enzymes. Examination of circulating metabolites in dog plasma led to the detection of a phenolic metabolite 2, which raised the possibility that 2 could undergo bioactivation to a reactive ortho-quinone intermediate. Incubations of an authentic standard of 2 in NADPH- and glutathione (GSH)-fortified dog liver microsomes led to the detection of the corresponding GSH conjugate 5, which can be formed during the bioactivation of 4 via the catechol (3) → ortho-quinone (4) reaction sequence (). Because of the low metabolic turnover of parent compound 1, no GSH conjugates were detected in corresponding NADPH- and GSH-supplemented dog liver microsomal incubations with 1. The hypothesis that the hepatotoxicity of 1 in dogs originated from its bioactivation to a protein-reactive ortho-quinone led to medicinal chemistry efforts towards abrogation of the bioactivation pathway. The basic piperidine nitrogen was needed for full F877L AR antagonism, so that emphasis was placed on modifications at the phenyl ring to which the piperidine group was tethered. Out of these efforts emerged the corresponding 2-pyridylphenol 6 (JNJ-63576253, ), which was not bioactivated (absence of GSH adduct formation) in dog liver microsomes. Resistance towards bioactivation was likely due to the diminished propensity of 6 towards the two-electron oxidation process to the ortho-quinone species as a result of the resonance stabilization between the phenolic OH group and the pyridyl nitrogen. Compound 6 demonstrated no hepatotoxicity in single and/or repeat dosing 14-day dog safety studies, which reaffirmed the relevance of the bioactivation pathway as a causative factor in the hepatotoxicity with 1. From a structure–metabolism relationship perspective, the phenyl ring variant of 6 (i.e. compound 7) was bioactivated to an ortho-quinone in dog liver microsomes as evident from the formation of the corresponding GSH conjugate, and moreover, 7 was hepatotoxic in chronic dog safety studies in a manner similar to 1, further strengthening the link between bioactivation and the observed hepatotoxicity.
Commentary
The formation of reactive metabolites during the metabolism of innocuous new chemical entities is considered to be an undesired outcome in drug discovery, given the association between reactive metabolites and adverse drug reactions (particularly of the idiosyncratic kind). Case studies demonstrating successful resolution of reactive metabolite liabilities are abundant in the medicinal chemistry literature and often involve the following strategies: (a) removal of the functional group susceptible to bioactivation, (b) modulation of electronic character (e.g. reduction in electron-richness of aromatic/heteroaromatic rings), (c) strategic placement of functional groups to block bioactivation and (d) introduction of alternate metabolic soft spots (Kalgutkar Citation2020). The lack of bioactivation of 2-pyridylphenol 6 relative to phenol 7 is analogous to the observations with the hepatotoxic antidepressant nefazodone and its non-hepatotoxic counterpart buspirone () (Kalgutkar et al. Citation2005). para-Hydroxynefazodone is a principal circulating metabolite of nefazodone in humans and is bioactivated by human CYP3A4 to electrophilic quinone-imine and 1,4-benzoquinone intermediates in human liver microsomes, a process that also causes significant protein covalent binding in human hepatic tissue (liver microsomes, S-9 and hepatocytes) in an NADPH-dependent fashion. The metabolism of buspirone is similar to nefazodone in that para-hydroxybuspirone (a para-hydroxypyrimidine derivative) represents a major circulating metabolite in human plasma. In contrast to nefazodone however, absence of GSH adducts in human liver microsomes of buspirone implies that quinone-imine species are not formed during oxidation by CYP3A4. Consistent with the experimental observations, quantum mechanical calculations suggest that the greater acidity and poor resonance stabilization of the oxidation products of para-hydroxybuspirone disfavor the oxidation of para-hydroxybuspirone to the quinone-imine species.
In the work of Zhang et al. a simple bioisosteric C/N exchange from phenyl to pyridyl was able to abolish the bioactivation to protein-reactive ortho-quinone intermediates, thus yielding a clinical stage AR antagonist with improved safety margins and a balanced pharmacodynamic and pharmacokinetic profile in preclinical species. However, a few questions remain unanswered. For example, was the bioactivation of 2 in dog liver microsomes also observed in corresponding incubations in human liver microsomes or an alternate non-rodent toxicology species (e.g. non-human primates)? If 2 was resistant to CYP mediated bioactivation in human and non-human primate liver microsomes, then the discovery team would have had the choice of replacing the dog with the non-human primate as the second toxicology species. This point is important for consideration, because the parent lead compound 1 appeared to be devoid of a hepatotoxic response in repeat dose toxicology studies in rats. It is reasonable to use metabolism-driven arguments (similarities and/or differences in metabolic (including bioactivation) profile) to justify selection of an appropriate animal species to assess toxicology.
Figure 17. (a) Structure–metabolism relationships to eliminate the bioactivation liability associated with the phenol metabolite 2 of AR antagonist 1. (b) Structures of hepatotoxin nefazodone and non-hepatotoxin buspirone and their primary para-hydroxylated metabolites in human.
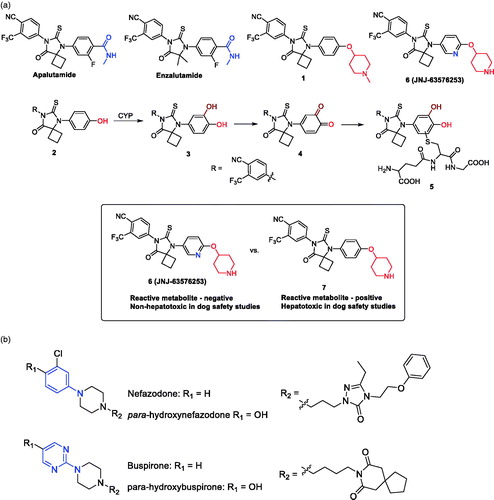
References
- Kalgutkar AS, Vaz AD, Lame ME, Henne KR, Soglia J, Zhao SX, Abramov YA, Lombardo F, Collin C, Hendsch ZS, et al. 2005. Bioactivation of the nontricyclic antidepressant nefazodone to a reactive quinone-imine species in human liver microsomes and recombinant cytochrome P450 3A4. Drug Metab Dispos. 33(3):243–253.
- Kalgutkar AS. 2020. Designing around structural alerts in drug discovery. J Med Chem. 63(12):6276–6302.
- Zhang Z, Connolly PJ, Lim HK, Pande V, Meerpoel L, Teleha C, Branch JR, Ondrus J, Hickson I, Bush T, et al. 2021. Discovery of JNJ-63576253: a clinical stage androgen receptor antagonist for F877L mutant and wild-type castration-resistant prostate cancer (mCRPC). J Med Chem. 64(2):909–924.
Bioactivation of α,β-unsaturated carboxylic acids through acyl-glucuronidation
Teresa MulderSudder BobbaKevin M. JohnsonJessica M. GrandnerWei WangChenghong ZhangJingwei CaiEdna F. ChooS. Cyrus KhojastehDonglu ZhangSources: Drug Metab Dispos. 2020;48:819–829
SYNOPSIS
Mulder et al. (Citation2020) published a new bioactivation mechanism through Phase II conjugation of α,β-unsaturated carboxylic acids. They observed low plasma extraction radioactivity recovery following oral administration of [14C]GDC-0810 to monkeys; the parent and its acyl-glucuronide metabolite, M6, were the major circulating drug-related materials. In the same study, cysteine conjugate of M6 (M11) was a minor metabolite detected in plasma and excreta (monkey urine and bile). The observations were consistent with protein covalent binding of radioactivity. The authors proposed a unique bioactivation mechanism for this compound through acyl glucuronidation (). Several in vitro experiments were designed to investigate this new mechanism. The synthetic acyl glucuronide metabolite M6, but not the parent, efficiently reacted with cysteine and glutathione in buffer to form a cysteine and glutathione adduct. Incubations of GDC-0810 or M6 with monkey or human liver microsomes in the presence of NADPH and GSH did not produce any oxidative GSH adducts and the respective substrates were qualitatively recovered. The later experiment ruled out the potential of oxidation mediated bioactivation of parent and M6. In silico analysis also supported the change of β-carbon electron density of α,β-unsaturated carboxylic acids that allows a nucleophilic addition following acyl glucuronidation. In silico analysis indicated the inherent reactivity differences between the glucuronide and its acid precursor.
Commentary
Bioactivation is an important factor that affects drug safety, and therefore, the success of drug development. Specifically, bioactivation produces reactive metabolites that could lead to conjugation with large molecules causing drug induced liver injuries (DILI) through direct toxicities and/or immune-mediate toxicity (Li et al. Citation2009). Most bioactivation reactions are mediated by oxidative enzymes such as P450, while bioactivation through non-P450 pathways has been summarized by Gan et al. (Citation2016). High doses of carboxylic acid-containing nonsteroidal anti-inflammatory (NSAIDs) are often associated with toxicities that are attributed to instability and reactivity of their acyl glucuronides or formation of reactive thioesters (Galati et al Citation2002; Grillo et al Citation2003). In this article, the authors reported a new bioactivation mechanism through acyl glucuronidation of α,β-unsaturated carboxylic acids that generates a reactive site at the β-carbon instead of at the carboxylic carbon. Their results did not support the typical bioactivation through thioester formation. The α,β-unsaturated carboxylic acid functionality is present in only a limited number of drugs, but it is included in some critical ones such as monomethyl fumarate, which is in the front line for treatment of multiple sclerosis.
Figure 18. Bioactivation pathway of GDC-0810.
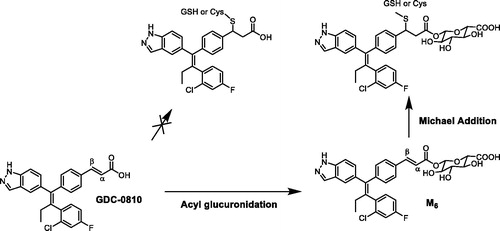
References
- Galati G, Tafazoli S, Sabzevari O, Chan TS, O’Brien PJ. 2002. Idiosyncratic NSAID drug induced oxidative stress. Chem Biol Interact. 142:25–41.
- Gan J, Ma S, Zhang D. 2016. Non-P450 mediated drug bioactivation pathways and its toxicological relevance. Drug Metab. 48:473–501.
- Grillo MP, Hua F, Knutson CG, Ware JA, Li C. 2003. Mechanistic studies on the bioactivation of diclofenac: Identification of diclofenac-S-acyl-glutathione in vitro in incubations with rat and human hepatocytes. Chem Res Toxicol. 16:1410–1417.
- Li Y, Qi XM, Xue X, Wu XF, Wu YF, Chen M, Xing GZ, Luan Y, Ren J. 2009. The relationship between diphenylamine structure and NSAIDs-induced hepatocytes injury. Toxicol Lett. 186:111–114.
- Mulder T, Bobba S, Johnson KM, Grandner JM, Wang W, Zhang C, Cai J, Choo EF, Khojasteh SC, Zhang D. 2020. Bioactivation of α,β-Unsaturated carboxylic acids through acyl-glucuronidation. Drug Metab Dispos. 48:819–829.
Strategies to mitigate the bioactivation of aryl amines
Chenghong ZhangJames J. CrawfordMatthew L. LandryHuifen ChenJane R. KennyS. Cyrus KhojastehWendy LeeShuguang MaWendy B. YoungSource: Chem Res Tox. 2020;33:1950 − 1959
SYNOPSIS
The aniline moiety is considered a toxicophore and can undergo bioactivation to reactive intermediates. The resulting electrophilic reactive intermediates are susceptible towards conjugation with nucleophiles including glutathione (GSH) or cellular proteins (). Depletion of intracellular GSH or addition to cellular protein can lead to toxicity. In the work presented here, Zhang et al. performed experiments detailing the effect of adding nitrogen to the aromatic ring of aniline and incubating the fragment-based compounds with microsomal proteins in the presence of NADPH and GSH (Zhang et al. Citation2020). GSH adducts were identified using a mixture of stable labelled GSH and naturally occurring GSH. A semiquantitative method was used to give a measure of the amount of GSH adduct formed using the peak height. Seeking to mimic the electron-withdrawing effects of the nitrogen, they also describe the effects of electron-withdrawing groups on simple fragment molecules. Their results show that in general adding nitrogen to aromatic rings mitigated the bioactivation when compared to the aniline structure with the trend being consistent as shown in with the bioactivation of pyrazine being similar to that of aniline followed by pyrimidine and pyridine. The pyridazine ring system was devoid of detectable bioactivation in all the molecules tested. With the exception of a fluorine substituent, the electron-withdrawing groups had little to no effect on the bioactivation potential of the substituted aniline.
Commentary
Addition of nitrogens to aniline rings in this report reduces the formation of reactive intermediates measured by trapping with GSH. The structure–activity relationship they found is very useful in a medicinal chemistry setting. Carbon-containing aromatic rings can often be replaced with a ring containing a nitrogen, which can sometimes improve potency (Pennington and Moustakas Citation2017). Interestingly, in a previous report, Kalgutkar et al. compared the bioactivation of nefazodone and buspirone. The aniline-containing nefazodone was shown to form a quinone-imine and was trapped with GSH. By contrast, buspirone contains a pyrimidine and did not form GSH adducts through a quinone-imine (Kalgutkar et al. Citation2005). Ab initio calculations by Kalgutkar et al. the authors showed that the nitrogen-containing ring system required more energy to form the destabilizing quinone-imine intermediate. In this report, they found that calculating the energy for the hydrogen atom abstraction step in the formation of the reactive quinoid intermediate did a reasonable job of predicting and binning the compounds.
Figure 19. (a) The bioactivation pathway for aniline-containing compounds. (b) Ranking of aromatic rings in terms of their bioactivation potential.
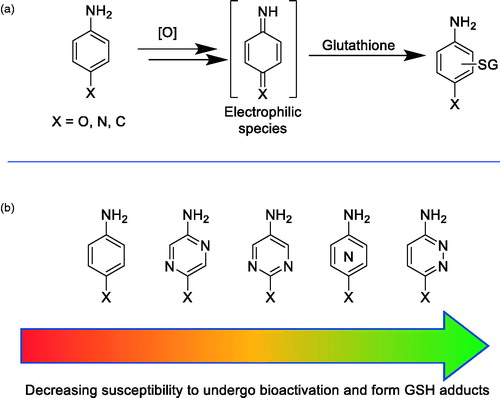
References
- Kalgutkar AS, Vaz ADN, Lame ME, Henne KR, Soglia J, Zhao SX, Abramov YA, Lombardo F, Collin C, Hendsch ZS, et al. 2005. Bioactivation of the nontricyclic antidepressant nefazodone to a reactive quinone-imine species in human liver microsomes and recombinant cytochrome P450 3A4. Drug Metabol Dispos. 33:243–253.
- Pennington LD, Moustakas DT. 2017. The necessary nitrogen atom: a versatile high-impact design element for multiparameter optimization. J Med Chem. 60:3552–3579.
- Zhang C, Crawford JJ, Landry ML, Chen H, Kenny JR, Khojasteh SC, Lee W, Ma S, Young WB. 2020. Strategies to mitigate the bioactivation of aryl amines. Chem Res Toxicol. 33(7):1950–1959.
Investigation of clozapine and olanzapine reactive metabolite formation and protein binding by liquid chromatography-tandem mass spectrometry
Timon GeibMadhuranayaki ThulasingamJesper Z. HaeggströmLekha SlenoSource: Chem Res Toxicol. 2020;33:2420–2431
SYNOPSIS
Clozapine (CLZ) and olanzapine (OLZ) are antipsychotic drugs that are used in the treatment of schizophrenia and paranoia (). While CLZ is a more effective treatment, it is not the favored treatment due to its association with a number of toxic outcomes including idiosyncratic agranulocytosis, which occurs in 0.8% of the patient population. Both CLZ and OLZ undergo metabolism to structurally similar nitrenium ions via oxidative metabolism in the liver. These studies investigated oxidative metabolism of CLZ and OLZ as well as the two major CLZ metabolites, desmethyl-CLZ (DCLZ) and CLZ-N-oxide (CLZ-NO) in vitro in rat liver microsomes. The results identified multiple CLZ reactive metabolite-related GSH adducts that result from either a nitrenium intermediate or a possible arene oxide intermediate (). Similar adducts were formed in incubations with OLZ, DCLZ and CLZ-NO. Metabolite binding to selected peptides and recombinant purified human proteins was also evaluated. Modification sites were characterized on human glutathione (hGST) S-transferase alpha 1 (OLZ at Cys112), hGST mu 2 (OLZ at Cys115), and hGST pi (CLZ, DCLZ, CLZ-NO and OLZ at Cys170), human microsomal GST 1 (hMGST1, CLZ, and OLZ at Cys50), and human serum albumin (hSA, CLZ at Cys34). Furthermore, two modified rat proteins, microsomal GST 1 (CLZ and OLZ at Cys50), and one CYP (OLZ-modified, multiple possible isoforms), were also characterized from rat liver microsomes.
Commentary
The findings from this work are of interest from a bioactivation perspective. Covalent binding of reactive metabolites has been associated with CLZ toxicity in many previous in vitro and in vivo studies, and much work has been done to understand the mechanism of this toxicity; however, the mechanism of clozapine-induced agranulocytosis remains elusive. Two possible mechanisms include the nitrenium ion binding to essential cellular proteins resulting in disruption of neutrophil function, or it may act as a hapten and initiate an immune reaction resulting in immune-mediated destruction of the neutrophil. The ultimate aim of research into reactive metabolites generated via CLZ metabolism would be to prospectively predict which individuals are going to develop agranulocytosis or to understand why other analogues, like OLZ, may offer a safer alternative. CLZ and OLZ, though structurally similar, exhibit differences in toxicity at equimolar doses in rat (Ng et al. Citation2014). In the study described above, both drugs formed multiple GSH and protein adducts in vitro under oxidative conditions. Thus, the authors speculate that it is unlikely that a difference in the metabolites formed is the source of the difference in toxicity. Instead, the lower turnover in vitro and higher hydrophilicity of OLZ may contribute to the safer drug profile of OLZ. Elucidating the source of toxicity from clinical data becomes more complex as the therapeutic ranges of plasma concentrations of CLZ and OLZ in humans are 50–500 ng/ml and 20–40 ng/ml, respectively (Urban et al. Citation2017). Disparity in daily dose to meet these plasma concentrations may result in vastly different exposure, or body burden, to reactive species that are formed (Dalvie et al. Citation2015). Regardless, the evidence provided in this publication can be useful to support further in vitro and in vivo binding studies to elucidate the mechanism of CLZ toxicity.
Figure 20. (a) Structures of clozapine and olazapine. (b) The bioactivation pathway of clozapine.
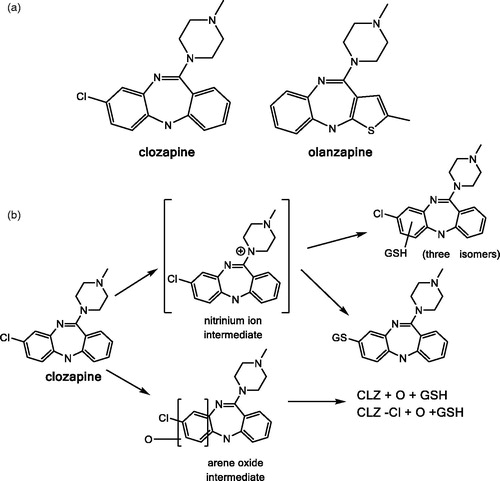
References
- Dalvie D, Kalgutkar AS, Chen W. 2015. Practical approaches to resolving reactive metabolite liabilities in early discovery. Drug Metab Rev. 47(1):56–70.
- Ng W, Kennar R, Uetrecht J. 2014. Effect of clozapine and olanzapine on neutrophil kinetics: implications for drug-induced agranulocytosis. Chem Res Toxicol. 27(7):1104–1108.
- Urban AE, Cubała WJ. 2017. Therapeutic drug monitoring of atypical antipsychotics. Psychiatr Pol. 51(6):1059–1077.
Application of a rat liver drug bioactivation transcriptional response assay early in drug development that informs chemically reactive metabolite formation and potential for drug-induced liver injury
James J. MonroeKeith Q. TanisAlexei A. PodtelezhnikovTruyen NguyenSam V. Machotka Donna LynchRaymond EversJairam PalamandaRandy R. MillerTodd PippertTamara D. CabaluTimothy E. JohnsonAmy G. AslamkhanWen KangAlex M. TamburinoKaushik Mitra Nancy G. B. AgrawalFrank D. SistareSource: Toxicol Sci. 2020;177(1):281–299
SYNOPSIS
The objective of this study was to identify a transcriptomic gene signature that is predictive of chemically reactive metabolite (CRM) mediated drug-induced liver injury (DILI) for toxicological risk assessment in early drug development. Monroe et al. (Citation2020) describe the development and implementation of the bioactivation liver response assay (BA-LRA) using an in vivo rat model to differentiate compounds associated with clinical DILI (DILI-positive) from compounds considered liver safe (DILI-negative). The BA-LRA gene panel consisted of 46 genes regulated by the NRF2/Keap1 electrophilic stress response pathway and the NRF1 proteosomal endoplasmic reticulum stress pathway. These genes were upregulated in response to reactive metabolites. A total of 116 DILI-positive and DILI-negative compounds were tested to develop and validate the in vivo BA-LRA approach. The study design used 4-day repeat oral dosing in rats, and BA-LRA gene expression changes were compared to vehicle control. The major finding of this study was that covalent protein binding by chemically reactive metabolites alone did not predict clinical DILI. Consideration of clinical dose, hepatic available dose, hepatic exposure to metabolites, and cellular response to reactive metabolites (quantified by the BA-LRA score) improved DILI risk prediction. The authors present case examples of drug candidates for which use of the BA-LRA score helped inform structure–activity relationships (SAR) to design drug candidates with less bioactivation potential. The advantages and limitations of using the BA-LRA approach for DILI risk assessment are discussed.
Commentary
Four nonexclusive mechanisms of drug-induced liver injury (DILI) have been identified: chemically reactive metabolite (CRM) formation, disruption of bile acid homeostasis, mitochondrial toxicity, and immune-mediated injury (Mosedale and Watkins Citation2017). Formation of chemically reactive metabolites is hypothesized to be a key initiating event and common underlying mechanism for many cases of clinical DILI. While nucleophilic trapping studies and covalent protein binding assays adequately assess the bioactivation potential of a drug (Evans et al. Citation2004; Obach et al. Citation2008; Bauman et al. Citation2009; Usui et al. Citation2009), these approaches do not define the link between reactive metabolite formation and downstream cellular responses that lead to clinical DILI. Thus, more comprehensive in vitro and in vivo models are needed to predict idiosyncratic DILI in drug development to reduce late-stage clinical failures and mitigate adverse drug reactions in patients. The bioactivation liver response assay (BA-LRA) described by Monroe et al. (Citation2020) focuses on CRM-mediated DILI. This approach quantitively assesses the extent to which reactive metabolites activate cell stress pathways. The BA-LRA genes were used as transcriptional biomarkers of bioactivation. A threshold BA-LRA score of 0.22 was set to predict clinical DILI. The term ‘BA-LRA burden’ was used to account for the combined influence of BA-LRA score and maximum daily dose on DILI risk. The sensitivity and specificity of the BA-LRA burden to detect true DILI-positive and DILI-negative compounds were determined as 32 and 92%, respectively, for the set of 116 total compounds tested. In addition, studies were conducted to distinguish compounds that directly activate NRF2 (bardoxolone) versus compounds that activate NRF2 through chemically reactive metabolite formation. A limitation of the study was the false negative finding for acetaminophen liver injury, which showed a low BA-LRA score in rats after 4-day repeat dosing, compared to a higher BA-LRA score for acetaminophen after 24-hour dosing. Another limitation observed was that BA-LRA scores were biased toward P450-mediated bioactivation compared to drugs that form reactive CoA-thioesters or unstable acyl-glucuronides. Species differences in drug metabolism and transcriptional response to electrophilic stress are also important to consider when using the in vivo rat BA-LRA model for predicting human DILI risk.
Case example: The authors evaluated the ability of compounds designed as reversible covalent inhibitors of Bruton’s tyrosine kinase, the pharmacological target, to cause a BA-LRA response. The BA-LRA score was used to inform structure–activity relationships (SAR) and to assess the bioactivation potential and DILI risk for these compounds. Semicarbazide adducts were detected from NADPH-supplemented human and rat liver microsomal incubations with MRK-A (1) and MRK-B (2) (). The BA-LRA response was positive for compounds 1 and 2. The predicted bioactivation pathway was formation of an aldehyde from the piperidine moiety (). Based on this analysis, MRK-C (3) was designed to reduce the bioactivation potential. The result was that compound 3 was negative for BA-LRA response, and no semicarbazide adducts were detected in human and rat liver microsomes. A remaining question is, would drugs designed as irreversible covalent modifiers of Bruton’s tyrosine kinase, such as ibrutinib and acalabrutinib, elicit a high BA-LRA score based on their chemical reactivity?
In a related article, Kang et al. (Citation2020) established an in vitro BA-LRA system using rat and human HepatoPac, a hepatocyte micropatterned co-culture model. A total of 93 DILI-positive and DILI-negative compounds were tested in this in vitro model, and hepatocytes were cultured over a 9-day period. A subset of BA-LRA genes, which were associated with the NRF2/Keap1 and NRF1/proteasome pathways, were selected for the rat and human HepatoPac assays. DILI-positive drugs showed dose-dependent increases in BA-LRA scores. Similar to the findings by Monroe et al. (Citation2020), this study demonstrated that clinical dose and hepatic drug exposure (liver exposure index) are key factors that help predict cellular response to reactive metabolites and DILI risk. Given the advantages of in vitro transcriptional biomarkers to detect CRM-mediated DILI compared to traditional cytotoxicity assays, future studies applying the in vitro BA-LRA approach with other organotypic hepatocyte culture systems, such as 3D spheroid hepatocyte cultures (Proctor et al. Citation2017; Bell et al. Citation2018), could be promising. In summary, the studies by Monroe et al. (Citation2020) and Kang et al. (Citation2020) present novel in vivo and in vitro tools to assess CRM-mediated DILI. These approaches have the potential to improve DILI risk assessment in early drug development and reduce late-stage drug candidate attrition.
Figure 21. SAR analysis of Bruton’s tyrosine kinase reversible covalent inhibitors. (a) Chemical structures and bioactivation assessment of MRK-A (1), MRK-B (2), and MRK-C (3). (b) Proposed bioactivation pathway of 1 and 2; semicarbazide-adduct formation. (Figures re-drawn and adapted from Monroe et al. Citation2020).
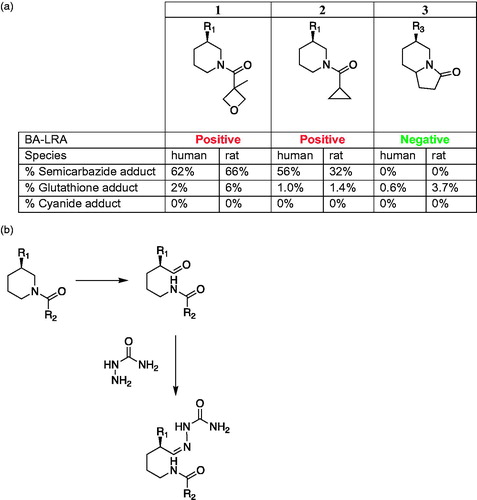
References
- Bauman JN, Kelly JM, Tripathy S, Zhao SX, Lam WW, Kalgutkar AS, Obach RS. 2009. Can in vitro metabolism-dependent covalent binding data distinguish hepatotoxic from nonhepatotoxic drugs? An analysis using human hepatocytes and S-9 fraction. Chem Res Toxicol. 22(2):332–340.
- Bell CC, Dankers ACA, Lauschke VM, Sison-Young R, Jenkins R, Rowe C, Goldring CE, Park K, Regan SL, Walker T, et al. 2018. Comparison of hepatic 2D sandwich cultures and 3D spheroids for long-term toxicity applications: a multicenter study. Toxicol Sci. 162(2):655–666.
- Evans DC, Watt AP, Nicoll-Griffith DA, Baillie TA. 2004. Drug-protein adducts: an industry perspective on minimizing the potential for drug bioactivation in drug discovery and development. Chem Res Toxicol. 17(1):3–16.
- Kang W, Podtelezhnikov AA, Tanis KQ, Pacchione S, et al. 2020. Development and application of a transcriptomic signature of bioactivation in an advanced in vitro liver model to reduce drug-induced liver injury risk early in the pharmaceutical pipeline. Toxicol Sci. 177(1):121–139.
- Monroe JJ, Tanis KQ, Podtelezhnikov AA, Nguyen T, et al. 2020. Application of a rat liver drug bioactivation transcriptional response assay early in drug development that informs chemically reactive metabolite formation and potential for drug-induced liver injury. Toxicol Sci. 177(1):281–299.
- Mosedale M, Watkins PB. 2017. Drug-induced liver injury: advances in mechanistic understanding that will inform risk management. Clin Pharmacol Ther. 101(4):469–480.
- Obach RS, Kalgutkar AS, Solgia JR, Zhao SX. 2008. Can in vitro metabolism-dependent covalent binding data in liver microsomes distinguish hepatotoxic from nonhepatotoxic drugs? An analysis of 18 drugs with consideration of intrinsic clearance and daily dose. Chem Res Toxicol. 21(9):1814–1822.
- Proctor WR, Foster AJ, Vogt J, Summer C, et al. 2017. Utility of spherical human liver microtissues for prediction of clinical drug-induced liver injury. Arch Toxicol. 91(8):2849–2863.
- Usui T, Mise M, Hashizume T, Yabuki M, Komuro S. 2009. Evaluation of the potential for drug-induced liver injury based on in vitro covalent binding to human liver proteins. Drug Metab Dispos. 37(12):2383–2392.