
Abstract
This year’s review on bioactivation and reactivity began as a part of the annual review on biotransformation and bioactivation led by Cyrus Khojasteh (see references). Increased contributions from experts in the field led to the development of a stand alone edition for the first time this year focused specifically on bioactivation and reactivity. Our objective for this review is to highlight and share articles which we deem influential and significant regarding the development of covalent inhibitors, mechanisms of reactive metabolite formation, enzyme inactivation, and drug safety. Based on the selected articles, we created two sections: (1) reactivity and enzyme inactivation, and (2) bioactivation mechanisms and safety (). Several biotransformation experts have contributed to this effort from academic and industry settings.
Table 1. Articles covered in this review.
Beyond bioactivation
Significant world events over the past year have continued to challenge the global community – from climate disasters and the ongoing COVID-19 pandemic to the suffering of marginalized communities and war. These events strengthen our resolve to be compassionate to others and to see each other as human. We thank all those who intentionally give back, provide opportunities for the next generation, and work to make a positive difference in the world around us. A special thank you to Cyrus Khojasteh and Namandje Bumpus for being excellent examples of giving back for the next generation of scientists.
We welcome your opinions, and we extend an invitation to anyone who would like to contribute to a future edition of this review.
Klarissa Jackson, on behalf of the authors.
Disclosure statement
Klarissa D. Jackson is co-investigator on a study funded by the Genentech Foundation. Jackson received a speaker honorarium from Genentech, Inc. to present research that is not related to this manuscript. No potential conflict of interest was reported by the author(s).
Source: Drug Metab Dispos. 2021;49:729–735
Synopsis
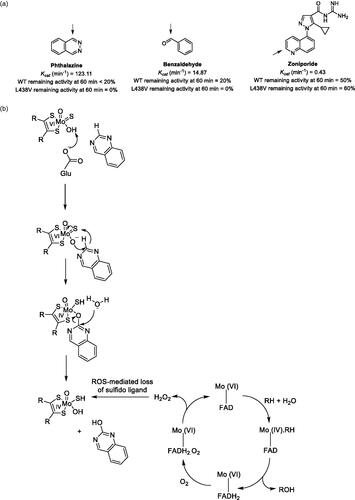
Aldehyde oxidase (AO) is a molybdo-flavoenzyme that catalyzes the transfer of oxygen to its substrates in a two-electron redox reaction. During substrate oxidation, the enzyme is reduced and then re-oxidized by electron transfer to molecular oxygen, producing reactive oxygen species (ROS) as a result. In this report, Garrido and Leimkuler evaluate the inactivation of human AO by hydrogen peroxide and superoxide as a consequence of AO catalytic activity (Garrido and Leimkühler Citation2021). Through studies comparing aerobic versus anaerobic incubation conditions and studies comparing the presence and absence of ROS scavengers, the authors concluded that ROS are responsible for enzyme inactivation over time. In addition, the rate of inactivation was found to be substrate dependent, suggesting that higher turnover substrates inactivate the enzyme more rapidly than slower turnover substrates (). In a comparison of the wild-type enzyme versus the L438V variant, hydrogen peroxide was reported to be predominantly responsible for inactivation of the wild-type enzyme, while both hydrogen peroxide and superoxide contributed to inactivation of the L438V variant. Finally, loss of the sulfido ligand from the molybdenum cofactor was determined to be the culprit in the ROS-mediated mechanism of inactivation, based upon experiments incubating recombinant human AO with substrate in the presence and absence of catalase, and then comparing the residual activity of chemically resulfurated enzyme versus a nonsulfurated control ().
Figure 1. (A) AO substrates demonstrating different degrees of inactivation of the wild-type and L438V enzymes, presumably due to different substrate turnover rates resulting in different rates of ROS production. Arrows indicate site of AO oxidation. (B) AO mechanism of substrate oxidation and catalytic cycle. The authors propose that ROS produced as a consequence of substrate oxidation inactivates the enzyme via desulfuration of the molybedenum cofactor.
Commentary
AO catalyzes the oxidation of a broad array of heteroaromatic rings, iminium ions and aldehydes. A number of drug candidates have been attrited in clinical trials due to unidentified or underpredicted AO metabolism, leading to poor exposure or accumulation of a toxic metabolite (Hutzler et al. Citation2013). Notably, the incidence of compounds exhibiting AO-mediated metabolism at the discovery stages has increased over the past several years with medicinal chemistry strategies to reduce P450 susceptibility (i.e. inclusion of heteroatoms in aromatic rings) and with the expansion of drug discovery programs targeting kinases, which typically require compounds containing aromatic heterocycles for target engagement. An increased awareness of chemical structures susceptible to AO metabolism, and a better understanding of species differences in AO expression and activity have aided in the prevention of drugs reaching clinical trials without prior knowledge of AO metabolism. Nevertheless, skepticism in the reliability of current approaches to predict human PK of AO substrates persists. The work reported here by Garrido and Leimkühler (Citation2021) represents another important contribution to the understanding of AO function and kinetics, and thus to the future development of more reliable approaches for predicting clinical AO metabolism.
Abbasi et al. first drew attention to the nonlinear time course exhibited by AO due to enzyme deactivation over time, highlighting a problem with the use of the substrate depletion (i.e. in vitro half-life) approach for estimating intrinsic clearance of AO substrates (Abbasi et al. Citation2019). Although the authors had different conclusions regarding the mechanism of enzyme inactivation (i.e. ROS-mediated inactivation), results obtained from these studies were consistent with those reported by Abassi et al. (Citation2019), in that the degree of inactivation was substrate dependent, with phthalazine (a ‘high turnover’ substrate) exhibiting greater inactivation than zoniporide (a ‘slow turnover’ substrate) over time. Consequently, these findings may indicate that enzyme inactivation is relatively inconsequential for very low turnover substrates. Notably, nearly all drugs that have failed out of clinical trials due to rapid AO-mediated metabolism were not recognized as AO substrates prior to human studies (Hutzler et al. Citation2013). Since the heightening in awareness of AO as a drug metabolizing enzyme, several drug candidates subject to AO metabolism have reached the market (e.g. idelalisib, lenvatinib, capmatinib) or been identified as AO substrates after reaching the market (e.g. lapatinib, imatinib), demonstrating that successful development of AO substrates into clinical drugs is achievable (Dick Citation2018). One notable characteristic is that each of these drugs, however, is not exclusively metabolized by AO, with P450 pathways also contributing (and in some cases predominating) the fractional metabolism.
In addition, the findings in this report may suggest that measures to reduce ROS production during AO assays could improve intrinsic clearance estimations. For example, studies comparing inactivation of the wild-type enzyme versus the L438V variant, which produces a greater proportion of superoxide relative to the wild-type enzyme, suggested that superoxide inactivates AO more rapidly than hydrogen peroxide. Accordingly, the authors noted that the incubation conditions (e.g. pH, temperature, and O2 concentration) can alter the ratio of superoxide/hydrogen peroxide production. Differences in incubation conditions were noted as a possible explanation for why Abassi et al. (Citation2019) did not observe a decrease in enzyme deactivation with ROS scavengers. Nevertheless, a discrepancy exists in the relationship between pO2 and superoxide production proposed by Garrido and Leimkühler (Citation2021) versus that documented by Lynch and Fridovich (Citation1979), warranting further investigation (i.e. whether high or low pO2 favors production of superoxide over hydrogen peroxide by molybedenum hydroxylases) (Lynch and Fridovich Citation1979). A possible factor in the study by Garrido and Leimkühler (Citation2021) was the use of a nearly 10-fold higher concentration of catalase and/or SOD versus Abassi et al. (Citation2019). Although ROS scavengers were ineffective at preventing enzyme inactivation for the high turnover substrate phthalazine, the scavengers reduced inactivation for the lower turnover substrates zoniporide and benzaldehyde under the conditions utilized in these studies (air-saturated EDTA-Tris buffer, pH 8.0 at 25 °C). However, these studies were conducted with purified recombinant human AO, and because catalase/SOD are already present in hepatocytes and cytosolic fractions, additional studies are needed to determine whether the addition of exogenous catalase/SOD would have any added effect on preventing AO deactivation in these systems. In addition, Abbasi et al. (Citation2019) unexpectedly observed a decline in activity in the presence of ROS scavengers for some substrates evaluated, but the authors did not speculate on an explanation for this observation. Further evaluation is needed to fully explain these discrepancies.
Taken together, the work reported by Abbasi et al. (Citation2019) along with the recent report by Garrido and Leimkühler (Citation2021) provide important contributions to aid the development of improved strategies for estimating intrinsic clearance of AO substrates. Furthermore, as a recent report suggested that extrahepatic AO is unlikely to explain the in vitro-in vivo disconnect in AO-mediated clearance (Kozminski et al. Citation2021), the notion that the observed discrepancy is related to incomplete modeling of the enzyme’s kinetics and/or suboptimal assay conditions may now be taking the lead as the most likely explanation, in addition to interindividual variability, as highlighted by differing inactivation rates between the wild-type and L438V enzymes.
References
- Abbasi A, Paragas EM, Joswig-Jones CA, Rodgers JT, Jones JP. 2019. Time course of aldehyde oxidase and why it is nonlinear. Drug Metab Dispos. 47(5):473–483.
- Dalvie D, Xiang C, Kang P, Zhou S. 2013. Interspecies variation in the metabolism of zoniporide by aldehyde oxidase. Xenobiotica. 43(5):399–408.
- Dick RA. 2018. Refinement of in vitro methods for identification of aldehyde oxidase substrates reveals metabolites of kinase inhibitors. Drug Metab Dispos. 46(6):846–859.
- Garrido C, Leimkühler S. 2021. The inactivation of human aldehyde oxidase 1 by hydrogen peroxide and superoxide. Drug Metab Dispos. 49(9):729–735.
- Hutzler JM, Obach RS, Dalvie D, Zientek MA. 2013. Strategies for a comprehensive understanding of metabolism by aldehyde oxidase. Expert Opin Drug Metab Toxicol. 9(2):153–168.
- Kozminski KD, Selimkhanov J, Heyward S, Zientek MA. 2021. Contribution of extrahepatic aldehyde oxidase activity to human clearance. Drug Metab Dispos. 49(9):743–749.
- Lynch RE, Fridovich I. 1979. Autoinactivation of xanthine oxidase: the role of superoxide radical and hydrogen peroxide. Biochim Biophys Acta. 571(2):195–200.
Source: Drug Metab Dispos. 2021;49(10):892–901
Synopsis
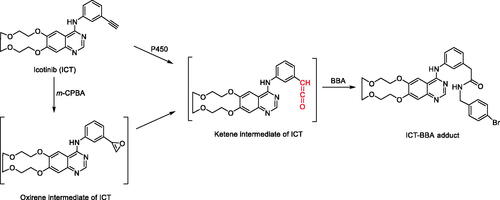
This manuscript describes efforts to elucidate the mechanism of time-, concentration-, and NADPH-dependent inhibition of CYP3A by icotinib (ICT) (Sun et al. Citation2021). The observed CYP3A inactivation was irreversible by dialysis in buffer and decreased when co-incubating with a CYP3A substrate, nifedipine, in human liver microsomes (HLM). Nucleophiles (glutathione and N-acetyl lysine) or reactive oxygen species scavengers (superoxide dismutase and catalase) did not provide protective effects on inactivation of CYP3A by ICT, indicating that the observed inactivation was not autoinactivation. Thus, the authors postulated that metabolically generated reactive intermediates inactivated CYP3A. Interestingly, a ketene intermediate was trapped by 4-bromobenzylamine (BBA) in HLM and human recombinant CYP3A incubations (). Ethyl and vinyl analogs of ICT did not inactivate CYP3A in HLM incubations, and as such, the authors concluded that the CYP3A inactivation of ICT was due to its acetylene moiety. Additionally, there was a significant loss of prosthetic heme content when ICT was incubated with human recombinant CYP3A4 without cytochrome b5. Overall, ICT is a mechanism-based inactivator of CYP3A4/5, and a metabolically generated ketene intermediate may be accountable for the observed CYP3A inactivation via destruction of the heme moiety.
Figure 2. Biomimetic chemical synthesis of ICT-BBA adduct and proposed bioactivation pathway for the formation of ICT-BBA adduct. BBA: 4-bromobenzylamine; m-CPBA: m-chloroperbenzoic acid.
Commentary
Icotinib (ICT), containing a phenyl terminal acetylene moiety, is an epidermal growth factor receptor tyrosine kinase inhibitor and was approved for the treatment of non-small cell lung cancer (NSCLC) in China. Patients treated with ICT are generally also concomitantly receiving other drugs due to associated disease conditions such as bone metastases or compromised immunity due to an organ transplant (Zhao et al. Citation2014; Wang et al. Citation2017). Therefore, it is important to understand the drug-drug interaction (DDI) potential of ICT as a victim or perpetrator, especially with respect to potential DDIs involving cytochrome P450 (CYPs) enzymes (Xu and Li Citation2019). The authors utilized various in vitro tools to determine the irreversible inhibition mechanism of ICT with CYP3A. It has been reported that oxidation of acetylenic groups can lead to inactivation of CYPs (Ortiz de Montellano Citation2019). From their mechanistic studies, the authors proposed that the acetylene moiety of ICT was metabolically activated, and the resulting ketene intermediate was responsible for heme destruction and the observed CYP3A enzyme inactivation.
When various concentrations of ICT were incubated with human liver microsomes (HLM) and NADPH, the CYP3A activity decreased in a time- and ICT concentration-dependent manner. Additionally, there was no time-dependent inhibition when ICT was incubated without NADPH in HLM. The authors postulated that reactive oxygen species (ROS) could be involved in the P450-catalyzed reaction, resulting in inactivation of host enzymes. Incubations with ROS scavengers, superoxide dismutase or catalase, did not decrease ICT-induced CYP3A inactivation, ruling out the possibility of autoinactivation. Nucleophilic agents, glutathione or Nacetyl lysine, did not demonstrate a protective effect on CYP3A inactivation by ICT, suggesting that reactive intermediates of ICT inactivated CYP3A before leaving the active site of the enzyme.
Based on the in vitro data, the mechanism of CYP3A inactivation was hypothesized to be via bioactivation of ICT generating a ketene intermediate (). When ICT was incubated with the trapping agent, 4-bromobenzylamine (BBA), and NADPH using HLM or human recombinant CYP3A, the ICT-BBA adduct was detected, confirming the hypothesis. The ICT-BBA adduct was also chemically synthesized by oxidation of ICT with m-chloroperbenzoic acid (m-CPBA) and then reacted with BBA (). Additionally, there was a decreased amount of prosthetic heme when ICT and human recombinant CYP3A4 were incubated with NADPH, indicating that heme was destroyed during CYP3A4 inactivation induced by ICT. In summary, a careful mechanistic investigation helps to elucidate the chemical moiety responsible for an enzyme inactivation. The knowledge gained from this study can help in designing new compounds and investigating other drugs with similar CYP inactivation.
References
- Ortiz de Montellano PR. 2019. Acetylenes: cytochrome P450 oxidation and mechanism-based enzyme inactivation. Drug Metab Rev. 51(2):162–177.
- Sun C, Zhao H, Li W, Jia Y, Yang Y, Peng Y, Zheng J. 2021. Icotinib induces mechanism-based inactivation of recombinant human CYP3A4/5 possibly via heme destruction by ketene intermediate. Drug Metab Dispos. 49(10):892–901.
- Wang W, Song Z, Zhang Y. 2017. Zoledronic acid as potential efficacy application combined with icotinib for non-small cell lung cancer with bone metastases. Transl Cancer Res. 6(1):129–135.
- Xu Z, Li J. 2019. Comparative review of drug-drug interactions with epidermal growth factor receptor tyrosine kinase inhibitors for the treatment of non-small-cell lung cancer. Onco Targets Ther. 12:5467–5484.
- Zhao Q, Wang Y, Tang Y, Peng L. 2014. Icotinib combined with rapamycin in a renal transplant recipient with epidermal growth factor receptor-mutated non-small cell lung cancer: a case report. Oncol Lett. 7(1):171–176.
Source: Clin Transl Sci. 2021;14(6):2420–2430
Synopsis
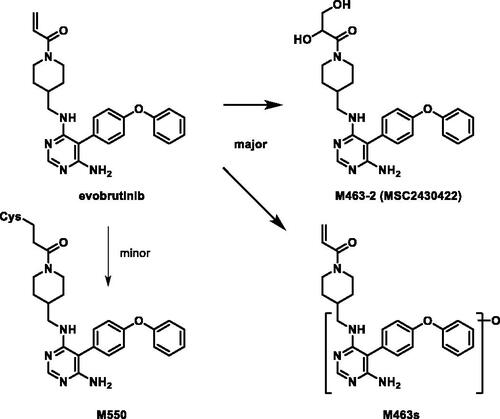
Evobrutinib is a piperidine acrylamide covalent inhibitor of Bruton’s tyrosine kinase (BTK) currently in clinical studies for the treatment of multiple sclerosis. Scheible et al. (Citation2021) reported a mass balance study of evobrutinib in six male healthy volunteers. From a single oral dose of 75 mg evobrutinib containing 100 µCi [14C]-evobrutinib in solution to healthy volunteers, evobrutinib showed rapid and close to complete absorption, extensive metabolism and relatively fast elimination. A median time to reach maximum concentration (Tmax) of 0.5 hr was observed and >85% of radioactivity were excreted in 72 hr. Evobrutinib and metabolite M463-2 represented 13 and 15% of total radioactivity (TRA) of plasma AUC0-24 with all other identified metabolites below 10% TRA. It is worth noting that a significant 41% TRA was non-extractable following multiple steps of ethanol and acetonitrile extraction, which indicated potential abundant covalent plasma protein adduct formation. This observation was reflected in the significant differences in terminal half-lives between evobrutinib (1.3 hr) and total radioactivity (∼100 hr). On the other hand, greater than 85% of radioactivity were excreted in the first 72 hr, indicating that the plasma protein adduct only accounted for a small portion of the total radioactivity dose. Radioactivity was mainly recovered in feces (71%) and lesser in urine (20%). Surprisingly, no unchanged drug was recovered in either urine or feces. Major metabolite M463-2 (6.2–9.3% dose in feces) was formed through oxidation to an epoxide intermediate on the acrylamide followed by hydrolysis (). Hydroxylation on the aromatic rings were the other major pathways with M445-1 and M445-3 (oxidation of parent) each representing ∼4–8% dose in feces and urine combined and M479-3 (oxidation of M463-2) representing ∼1–6% dose in feces. GSH and cysteine conjugation metabolism was a minor clearance pathway for evobrutinib.
Figure 3. Proposed metabolism of evobrutinib to its major metabolites in healthy volunteers.
Commentary
BTK has been a highly sought after target by research groups and pharmaceutical companies due to its critical role in B cell development (Singh et al. Citation2018). The first in class BTK covalent inhibitor ibrutinib was approved by the FDA in 2013 for B cell malignancies. Although targeting a different therapeutic area, evobrutinib is structurally very similar to ibrutinib; both possess a piperidine acrylamide warhead and a phenoxyphenyl group to interact with the Thr474 gatekeeper-mediated selectivity pocket (Caldwell et al. Citation2019). However, the subtle difference in 1,4 di-substitution of the piperidine in evobrutinib vs. 1,3 di-substitution in ibrutinib was hypothesized to significantly alter their selectivity toward a panel of kinases, especially EGFR. Interestingly, human ADME of evobrutinib also showed high similarity to that of ibrutinib (Scheers et al. Citation2015). Compound derived radioactivity was mainly excreted in feces (>70%) and both compounds were extensively metabolized with unchanged molecules barely detected in the excreta. The major metabolism reactions were oxidations including the dihydrodiol metabolite via an epoxide intermediate, which was the most abundant metabolite for evobrutinib, consistent with in vitro hepatocyte incubation findings (Li et al. Citation2019). Acrylamide, piperidine and aromatic ring oxidations inevitably complicated the interpretation of oxidative metabolites and the connections in the metabolic scheme. Based on the analyses, multiple metabolites with the same masses were captured including 9 mono-oxidation metabolites M445s, 12 di-oxidation metabolites M461s, and 16/10 tri-oxidation metabolites M479s/ M477s. GSH and cysteine conjugative metabolism was only a minor pathway for evobrutinib in vitro and in vivo, likely due to a slower reaction rate than P450 mediated oxidations. Lastly, amide hydrolysis and acrylamide reduction were also captured, but was not recapitulated from in vitro hepatocyte experiments, which indicated potential involvement of bacterial hydrolases and reductases (Wilson and Nicholson Citation2017).
References
- Caldwell RD, Qiu H, Askew BC, Bender AT, Brugger N, Camps M, Dhanabal M, Dutt V, Eichhorn T, Gardberg AS, et al. 2019. Discovery of evobrutinib: an oral, potent, and highly selective, covalent Bruton’s tyrosine kinase (BTK) inhibitor for the treatment of immunological diseases. J Med Chem. 62(17):7643–7655.
- Li Z, Zhang L, Yuan Y, Yang Z. 2019. Identification of metabolites of evobrutinib in rat and human hepatocytes by using ultra‐high performance liquid chromatography coupled with diode array detector and Q Exactive Orbitrap tandem mass spectrometry. Drug Test Anal. 11(1):129–139.
- Scheers E, Leclercq L, de Jong J, Bode N, Bockx M, Laenen A, Cuyckens F, Skee D, Murphy J, Sukbuntherng J, et al. 2015. Absorption, metabolism, and excretion of oral 14C radiolabeled ibrutinib: an open-label, phase I, single-dose study in healthy men. Drug Metab Dispos. 43(2):289–297.
- Scheible H, Dyroff M, Seithel-Keuth A, Harrison-Moench E, Mammasse N, Port A, Bachmann A, Dong J, van Lier JJ, Tracewell W, et al. 2021. Evobrutinib, a covalent Bruton’s tyrosine kinase inhibitor: mass balance, elimination route, and metabolism in healthy participants. Clin Transl Sci. 14(6):2420–2430.
- Singh SP, Dammeijer F, Hendriks RW. 2018. Role of Bruton’s tyrosine kinase in B cells and malignancies. Mol Cancer. 17(1):57.
- Wilson ID, Nicholson JK. 2017. Gut microbiome interactions with drug metabolism, efficacy, and toxicity. Transl Res. 179:204–222.
Source: Chem Res Toxicol. 2021;34(9):2135–2144
Synopsis
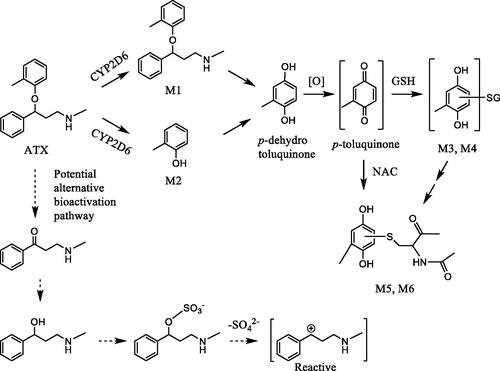
Atomoxetine (ATX) is a selective norepinephrine reuptake inhibitor, approved for treating attention deficit-hyperactivity disorder (ADHD). Several studies demonstrated that administration of ATX resulted in the idiosyncratic liver toxicity in humans (Lim et al. Citation2006; Erdogan et al. Citation2011). However, the underlying mechanism of ATX-induced liver toxicity remains unclear. You et al. (Citation2021) explored the potential mechanism of bioactivation of ATX (You et al. Citation2021). They hypothesized that ATX was metabolized to a p-toluquinone (methyl-p- benzoquinone), a reactive electrophile, which forms adducts with nucleophilic functional groups of biomolecules in the liver to elicit its toxicity. Incubation of ATX with rat liver microsomes (RLM) produced M1 and M2, 4-hydroxylated and O-dealkylated metabolites, respectively (). O-Dealkylation and oxidation of ATX generated p-hydrotoluquinone, which was further oxidized to p-toluquinone, a reactive metabolite. The presence of p-toluquinone was supported by GSH (M3 and M4) or NAC (M5, M6) conjugates of p-hydrotoluquinone using GSH or NAC trapping assay in RLM (). The in vivo relevance of these metabolites were confirmed by detection of GSH conjugates and NAC conjugates of p-hydrotoluquinone in rat bile and urine, respectively following an i.p. administration of ATX. Human CYP2D6 was found to play a dominant role in the formation of M5 and M6 using recombinant CYP and selective CYP inhibitors.
Figure 4. Proposed bioactivation pathway of atomoxetine.
Commentary
ATX exhibited idiosyncratic liver toxicity in humans with unclear underlying mechanism of toxicity (Erdogan et al. Citation2011). The current work provided the bioactivation pathway of ATX as a possible mechanism. The formation of p-toluquinone and its trapped reactive metabolites in this pathway were confirmed based on both in vitro and in vivo studies. This work was highlighted because the finding from the work may provide a basis to explain the idiosyncratic liver toxicity of ATX.
Quinone-type substructures are well known reactive electrophilic moieties which are believed to be involved in the liver toxicity of multiple drugs such acetaminophen and nevirapine (Chen et al. Citation1998; Sharma et al. Citation2012). The current study highlights bioactivation risks from the perspective of the parent drug, but unstudied ATX metabolites may play a direct or indirect role in that risk. An interesting future study would be to examine the hepatotoxicity of p-toluhydroquinone by administering p-toluhydroquinone in both in vitro (e.g. rat and human hepatocytes) and in vivo animal models. Additionally, if p-toluhydroquinone of ATX resulted in cytotoxicity in human hepatocytes, then the role of inhibition of CYP2D6 could be further examined to validate its role in bioactivation and cytotoxicity. The result from these studies would better support the proposed bioactivation pathway of ATX in humans. Another potential bioactivation pathway involves the product of ATX O-dealkylation to the benzylic ketone. This metabolite would likely be reduced to benzylic alcohol [3-(methylamino)-1-phenylpropan-1-ol] followed by sulfonation on the benzylic hydroxyl group (). Several studies reported that suflation on a benzylic hydroxyl group could lead to formation of an unstable reactive electrophilic species by loss of the suflate group that could react with biomolecules leading to carcinogenecity (Miller Citation1994; Watabe et al. Citation1994; Glatt Citation2000).
Lastly, the authors also determined the role of human CYP2D6 in the bioactivation of ATX and pointed out, the CYP2D6 activity exhibited large inter-individual variability due to genetic polymorphism (Zhou Citation2009). It was reported that CYP2D6 polymorphism was associated with gefitinib-induced liver injury (Takimoto et al. Citation2013). Therefore, it would be useful to determine if CYP2D6 polymorphism could be linked to ATX liver toxicity. Overall, You et al. (Citation2021) well described the potential mechanism of ATX bioactivation which may shed a light on understanding idiosyncratic liver toxicity of ATX.
References
- Chen W, Koenigs LL, Thompson SJ, Peter RM, Rettie AE, Trager WF, Nelson SD. 1998. Oxidation of acetaminophen to its toxic quinone imine and nontoxic catechol metabolites by baculovirus-expressed and purified human cytochromes P450 2E1 and 2A6. Chem Res Toxicol. 11(4):295–301.
- Erdogan A, Ozcay F, Piskin E, Karaman MG, Bilezikci B, Calik M, Tekin I, Haberal M. 2011. Idiosyncratic liver failure probably associated with atomoxetine: a case report. J Child Adolesc Psychopharmacol. 21(3):295–297.
- Glatt H. 2000. Sulfotransferases in the bioactivation of xenobiotics. Chem Biol Interact. 129(1–2):141–170.
- Lim JR, Faught PR, Chalasani NP, Molleston JP. 2006. Severe liver injury after initiating therapy with atomoxetine in two children. J Pediatr. 148(6):831–834.
- Miller JA. 1994. Sulfonation in chemical carcinogenesis–history and present status. Chem Biol Interact. 92(1–3):329–341.
- Sharma AM, Li Y, Novalen M, Hayes MA, Uetrecht J. 2012. Bioactivation of nevirapine to a reactive quinone methide: implications for liver injury. Chem Res Toxicol. 25(8):1708–1719.
- Takimoto T, Kijima T, Otani Y, Nonen S, Namba Y, Mori M, Yokota S, Minami S, Komuta K, Uchida J, et al. 2013. Polymorphisms of CYP2D6 gene and gefitinib-induced hepatotoxicity. Clin Lung Cancer. 14(5):502–507.
- Watabe T, Ogura K, Satsukawa M, Okuda H, Hiratsuka A. 1994. Molecular cloning and functions of rat liver hydroxysteroid sulfotransferases catalysing covalent binding of carcinogenic polycyclic arylmethanols to DNA. Chem Biol Interact. 92(1–3):87–105.
- You Y, Wang X, Ma K, Li J, Peng Y, Zheng J. 2021. Metabolic activation of atomoxetine mediated by cytochrome P450 2D6. Chem Res Toxicol. 34(9):2135–2144.
- Zhou SF. 2009. Polymorphism of human cytochrome P450 2D6 and its clinical significance: part I. Clin Pharmacokinet. 48(11):689–723.
Source: J. Med. Chem. 2021;64(12):8545−8563
Synopsis
Aromatic amines (Ar–NH2) are an important and attractive building block used in medicinal chemistry programs for their pharmacological and pharmacokinetic properties. Some secondary amines have the potential to undergo P450 catalyzed N-dealkylation reactions or amide hydrolysis to release the primary amine. Primary aromatic amines can cause mutagenicity and are proposed to undergo metabolism to hydroxylated amines as shown in which are known to be mutagenic (Inami Citation2009). The resulting aromatic hydroxylated amines are known to react with DNA bases which results in mutagenicity. There have been significant efforts to predict the mutagenic potential of aromatic amines, but it still remains a challenge. The authors (Shamovsky et al. Citation2021) propose that this issue reflects poor estimations of parameters such as electron affinity, resonance stabilization of the ArNH− intermediate, or fit into the active site of the metabolizing enzyme. The effort described here uses different approaches to investigate the energy profiles of pathways of hydroxylation for a diverse set of ArNH2 using DFT calculations. They also include an active site model of CYP1A2 with the goal of coming up with the best model which is consistent with known experimental results since many aromatic amines are bioactivated by CYP1A2 (Tureskey Citation2010). Their results suggest that the use of the active site of CYP1A2 in the model produces better results than the calculations which include the formation of the N-hydroxylations alone suggesting the active site of CYP1A2 plays a role. They demonstrate that by disrupting H-bonding, π−π, and van der Waals interactions or by adding disruptive interactions within the active site can distinguish between mutagenic compounds and nonmutagenic compounds. Examples of each of the disruption mechanisms can be found in along with many more examples found in the paper.
Figure 5. The proposed bioactivation pathway of aromatic amines (A) Structure activity relationship of mutagenic compounds versus using the removal of H-bond donors, introduction of steric repulsion groups, decreasing the stability of the ArNH- formation, introduction of electrostatic repulsion groups and disruption of π−π interaction with DNA bases (B).
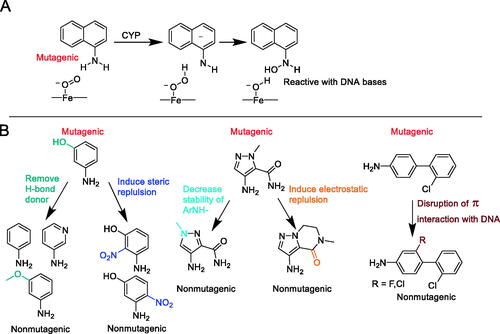
Commentary
The authors describe four methods to elucidate the mechanism of interaction of aromatic amines to form mutagenic intermediates. The use of DFT calculations with the inclusion of the active site of CYP1A2 has improved the predictability of their model instead of just using the reactivity of the aromatic amine because the bioactivation involves enzymes and not just the chemical reactivity of the ArNH2 (Ripa et al. Citation2014). The authors state that the most versatile way to reduce mutagenicity in the model is to disrupt the binding interactions in CYP1A2. Their work has helped define structure activity relationship tools to help remove potential mutagenicity which can be immensely helpful if one is on the medicinal chemistry design team which requires an aromatic amine as a required part of the pharmacophore. The structure activity relationship presented is also useful if a potentially mutagenic aromatic amine is produced due to an N-dealkylation reaction or amide hydrolysis in the course of normal metabolic pathways.
References
- Inami K, Okazawa M, Mochizuki M. 2009. Mutagenicity of aromatic amines and amides with chemical models for cytochrome P450 in Ames assay. Toxicol in Vitro. 23(6):986–991.
- Ripa L, Mee C, Sjö P, Shamovsky I. 2014. Theoretical studies of the mechanism of N-hydroxylation of primary aromatic amines by cytochrome P450 1A2: radicaloid or anionic? Chem Res Toxicol. 27(2):265–278.
- Shamovsky I, Ripa L, Narjes F, Bonn B, Schiesser S, Terstiege I, Tyrchan C. 2021. Mechanism-based insights into removing the mutagenicity of aromatic amines by small structural alterations. J Med Chem. 64(12):8545–8563.
- Turesky RJ. 2010. Aromatic amines and heterocyclic aromatic amines: from tobacco smoke to food mutagens. In: Geacintov NE and Broyde S, editors. The chemical biology of DNA damage. Weinheim, Germany: Wiley-VCH; p. 157–183.
Source: Toxicol Sci. 2021;180(1):17–25
Synopsis
Trimethoprim (TMP) is an orally administered antibiotic targeting formation of tetrahydrofolic acid. This drug is used as a single agent or co-administered with sulphamethoxazole (combination called co-trimoxazole). TMP is commonly prescribed and listed as one of the Essential Medicines by WHO Citation2019 (Lindberg et al. Citation2005). Yet it has significant side effects including skin reactions and in some rare cases, liver injuries (Lai et al. Citation1999). The general thought is that these injuries could be due to formation of reactive metabolites, and the investigators examined this concept with the focus on bioactivation of trimethoprim (Cao et al. Citation2021). One hypothesis was that a primary metabolite of TMP, mainly α-hydroxyTMP (TMP-OH; ), formed an electrophillic sulfate metabolite (TMP-S; ) and would then react with cellular protein to form adducts (called haptens). These haptens in turn could trigger immune responses that produce the observed immune-driven toxicity (hapten hypothesis; Uetrecht Citation2008). To test the hapten formation by TMP-S (sulfate metabolite), the investigators first needed to demonstrate that the primary metabolite TMP-OH is a substrate of human sulfotransferases (SULT) to form TMP-S. They examined 13 recombinant SULTs and to their surprise no TMP-S was detected. Synthetic TMP-S was made to examine if this conjugate was formed at all. The investigators were able to show that TMP-S was reactive and it did form covalent binding with liver and epidermal skin proteins. For this study, in addition to TMP-S, TMP-OH was examined. Unexpectedly, TMP-OH was shown to be electrophilic and it modified proteins but to a much lesser extent than TMP-S. As a control, TMP did not show covalent binding so the bridging hydroxyl in TMP-OH introduced a new reactivity center that based on the structure was not obvious. The covalent binding of TMP-OH was tested in the presence of various drug metabolizing enzyme inhibitors (CYP, UGT, SULT) to make sure that the covalent binding is not due to further metabolism that takes place during the incubation. The inhibitors did not change the extent of covalent binding, and therefore the reactivity of TMP-OH was called out as chemical reactivity and not enzymatic. Polyclonal antiserum was produced against TMP-modified BSA in rabbit. The modified BSA was synthesized by using the bridging position (alpha position) as the site of BSA attachment, so the two aromatic rings of TMP remain unmodified for antibody recognition. The antiserum was characterized using ELISA for its specificity and demonstrated it recognized TMP structural components. The antiserum was used to confirm the formation of adducted proteins from in vitro and in vivo samples studies using ELISA and immunoblot assay format. In vitro, it was shown that synthesized TMP-S covalently react to the liver S9 proteins and to a lesser extent to the epidermal skin proteins. However, it is not clear if this conjugate metabolite is actually formed since TMP-OH does not react to any of the SULT enzymes. As mentioned before, surprisingly TMP-OH was also shown to bind to the S9 fractions from liver and skin but to a much lesser extent than TMP-S. To examine the hypothesis of covalent binding of TMP result in both liver and skin toxicity, rats were dosed daily at 400 mg/kg for 3 days and up to 5 weeks. Covalent binding was detected but was limited to the liver, and not in the skin and several other organs examined. In addition, as assessed by ALT elevation no sign of hepatotoxicity was observed. Besides testing TMP, two other analogs were studied, TMP-OH and α-keto-TMP (TMP = O). Neither one of the two resulted in formation of covalent binding in any of the tissues examined including liver and skin. Further in vivo investigations were conducted using female PD-1-/- mice with co-treatment of anti-CTLA-4 with 2% TMP (wt/wt) in food. Similar to rats, no hepatotoxicity was observed using ALT marker and compared to the treated rat, significantly less hepatic covalent binding was detected. Finally, the investigators concludes that most likely TMP-OH could be causing the skin rash and not TMP-S.
Figure 6. (a) Overall reported trimethoprim (TMP) and its reactive metabolites. In vivo rat studies, TMP formed significant hepatic covalent binding but not with α-hydroxy-TMP (TMP-OH) and α-keto-TMP (TMP = O). In vitro TMP-OH and TMP-S formed covalent binding in S9 fractions with liver ≫ skin. Finally, no human SULT was found to convert TMP-OH to TMP-S. In the two lower boxes strucutres of TMP reactive metabolites and primary metabolites reported by Goldman et al. Citation2016 and Nolte et al. Citation2020. (b) (A) The reaction for the nevirapine (NVP) CYP-mediated metabolism to 12-hydroxy NVP (12-OH-NVP) and its subsequent metabolism by human sulfotransferases (SULT). (B) Minoxidil formation of pharmacologically active minoxidil sulfate.
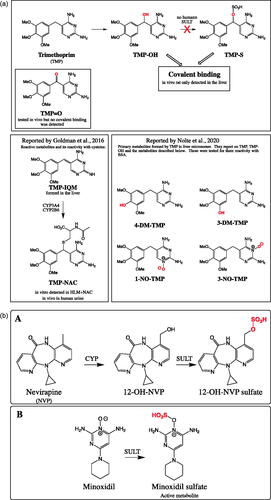
Commentary
TMP is an antibiotic drug approved over 60 years ago for a broad range of infections including bladder infection (Lindberg et al. Citation2005). Though broadly used, it has several side effects with one being skin rash, and its mechanism remains a mystery. Recently there are several publications focused on trimethoprim bioactivation and its role in toxicity. Goldman and colleagues reported on in vitro formation of an imine-quinone-methide reactive intermediate (TMP-IQM; ) that results in the formation of covalent binding through cysteine thiol (Goldman et al. Citation2016). Notle et al. also reported on reactivity of TMP and its primary metabolites with human serum albumin (Nolte et al. Citation2020). Here the investigators focused on the possible role of reactive metabolites that could result in skin toxicity. For this Cao et al. focus on TMP-OH and the possible formation of reactive metabolites in the skin.
One of the questions the investigators pursued was whether TMP-S could be formed in the skin. It is important to note that skin is one of the largest organs and it contains many drug metabolizing enzymes (Kazem et al. Citation2019). Many drugs are reported to be metabolized by the skin and this lends itself to possible species differences (Oesch et al. Citation2018). One particular group of drug metabolizing enzymes are SULTs, which are also present in the skin and sulfonate many types of drugs (Géniès et al. Citation2019). In the case of minoxidil, it locally converts to its sulfated form by SULT to exert its hair growth pharmacological activity (Buhl et al. Citation1990).
In the case of nevirapine, its skin toxicity has been shown to be through 12-hydroxynevirapine (12-OH-NVP; ; Chen et al. Citation2008). In the skin, this primary metabolite was conjugated by SULT to form a reactive metabolite 12-OH-NVP sulfate that covalently modified protein (Sharma et al. Citation2013; ). Here the investigators pursued a similar hypothesis with formation of reactive metabolite if TMP-S formed through TMP-OH. They reported that no TMP-S was formed in the incubation with SULT enzymes. This result puts in question if TMP-S is the culprit in the skin rash so this suggested a different mechanism than reported with NVP sulfate. On the other hand, is it possible that TMP-S was formed to a much lesser extent that was not really detectable but caused immune reaction?
Surprising TMP-OH was shown to form covalent binding to cellular proteins, which was also shown by Nolte et al. (Citation2020). What is not clear structurally, how the presence of the bridge α-hydroxyl activates TMP? One possible consideration is the loss of water molecule to form TMP-IQM intermediate () that results in the formation of an electrophilic bridging position, as described previously. The general reactivity of quinone-type intermediates was previously reported by Bolton and colleagues (Bolton et al. Citation1997), but this did not include TMP-OH-type molecules. It may be helpful to examine the chemical reaction of TMP-OH against various nucleophiles such as glutathione, lysine, etc.
The other structural information about TMP-OH is the fact that it has two enantiomers S and R. It is not clear if the enzymatic and chemical formation of TMP-OH is racemic or a pure enantiomer. Would each of these enantiomers exert a different property?
Finally, the investigators did a great job pursuing a few questions on TMP bioactivation role in skin rash. Though there are still unanswered questions, the TMP-OH seems to take a more prominent role in its possible direct covalent binding.
References
- Bolton JL, Turnipseed SB, Thompson JA. 1997. Influence of quinone methide reactivity on the alkylation of thiol and amino groups in proteins: studies utilizing amino acid and peptide models. Chem Biol Interact. 107(3):185–200.
- Buhl AE, Waldon DJ, Baker CA, Johnson GA. 1990. Minoxidil sulfate is the active metabolite that stimulates hair follicles. J Invest Dermatol. 95(5):553–557.
- Cao Y, Bairam A, Jee A, Liu M, Uetrecht J. 2021. Investigating the mechanism of trimethoprim-induced skin rash and liver injury. Toxicol Sci. 180(1):17–25.
- Chen J, Mannargudi BM, Ling Xu L, Uetrecht J. 2008. Demonstration of the metabolic pathway responsible for nevirapine-induced skin rash. Chem Res Toxicol. 21(9):1862–1870.
- Géniès C, Jamin EL, Debrauwer L, Zalko D, Person EN, Eilstein J, Grégoire S, Schepky A, Lange D, Ellison C, et al. 2019. Comparison of the metabolism of 10 chemicals in human and pig skin explants. J Appl Toxicol. 39(2):385–397.
- Goldman JL, Koen YM, Rogers SA, Li K, Leeder JS, Hanzlik RP. 2016. Bioactivation of trimethoprim to protein-reactive metabolites in human liver microsomes. Drug Metab Dispos. 44(10):1603–1607.
- Kazem S, Linssen EC, Gibbs S. 2019. Skin metabolism phase I and phase II enzymes in native and reconstructed human skin: a short review. Drug Discov Today. 24(9):1899–1910.
- Lai WG, Zahid N, Uetrecht JP. 1999. Metabolism of trimethoprim to a reactive iminoquinone methide by activated human neutrophils and hepatic microsomes. J Pharmacol Exp Ther. 291(1):292–299.
- Lindberg RH, Wennberg P, Johansson MI, Tysklind M, Andersson BA. 2005. Screening of human antibiotic substances and determination of weekly mass flows in five sewage treatment plants in Sweden. Environ Sci Technol. 39(10):3421–3429.
- Nolte WM, Tessman RT, Goldman JL. 2020. Screening trimethoprim primary metabolites for covalent binding to albumin. Med Chem Res. 29(7):1238–1246.
- Oesch F, Fabian E, Landsiedel R. 2018. Xenobiotica-metabolizing enzymes in the skin of rat, mouse, pig, guinea pig, man, and in human skin models. Arch Toxicol. 92(8):2411–2456.
- Sharma AM, Novalen M, Tanino T, Uetrecht JP. 2013. 12-OH-nevirapine sulfate, formed in the skin, is responsible for nevirapine-induced skin rash. Chem Res Toxicol. 26(5):817–827.
- Uetrecht J. 2008. Idiosyncratic drug reactions: past, present, and future. Chem Res Toxicol. 21(1):84–92.
- World Health Organization. 2019. World Health Organization model list of essential medicines: 21st list 2019. World Health Organization. https://apps.who.int/iris/handle/10665/325771.
Sources: Toxicology 2021;447:152628
Synopsis
Arzuk et al. (Citation2021) describe efforts to ascribe clozapine bioactivation in cardiac mitochondria as a potential molecular initiating event leading to reported cardiotoxicities in the clinic. After clozapine treatment, rats showed drug levels in heart tissues that were not as high as that in the liver but comparable to bone marrow, which is another site of clozapine metabolism. Based on reactions with microsomal and mitochondrial fractions, clozapine metabolism in the heart led to nonreactive norclozapine and clozapine N-oxide, and a reactive nitrenium ion, as reported for the liver. Although qualitative, these reactions implicated an overall lower metabolic capacity in the heart, but higher preference for clozapine bioactivation occurring in mitochondria that supports the proposed mechanism. Based on human cytochrome P450 (CYP) phenotyping inhibitors, rat orthologs of CYP1A2, 2D6 and 3A4 were main drivers in mitochondrial metabolism with CYP3A4 being the most important determinant of clozapine bioactivation. Follow up studies with intact rat mitochondria demonstrated that clozapine enters the organelle for metabolism to occur, although the transport mechanism was unclear. Upon entering mitochondria, clozapine seemed not to alter the mitochondrial permeability transition from in vitro experiments; however, at higher exposure levels, the drug suppressed oxygen consumption in CHO-K1 cells. In sum, these studies mark an important step forward in attempting to establish the plausibility of clozapine bioactivation in the heart as playing a role in observed cardiotoxicity for the drug.
Commentary
Clozapine is a highly effective second-generation antipsychotic used in the treatment of refractory schizophrenia (Remington et al. Citation2016). Nevertheless, its clinical use leads to adverse drug effects such as, agranulocytosis/neutropenia (Atkin et al. Citation1996) and cardiotoxicity (Kanniah and Kumar Citation2020). Knowledge of the corresponding toxicological mechanisms is then necessary for mitigating health risks (). As the most prominent adverse effect, agranulocytosis/neutropenia has undergone the most extensive mechanistic studies (review in Wiciński and Węclewicz Citation2018). The general paradigm involves myeloperoxidase bioactivation of clozapine into a nitrenium ion that forms protein adducts with neutrophils in bone marrow. The modified proteins compromise cellular functions leading to cell death and/or serve as haptens to elicit a toxic immune response. The insults, alone or in combination, then lead to the development of agranulocytosis/neutropenia. Such a mechanism cannot explain toxicities in other tissues due to limited expression of myeloperoxidase. Rather, cytochromes P450 (CYP) are responsible for clozapine metabolism into nonreactive metabolites and the reactive nitrenium ion based on human hepatic microsomal reactions (Pirmohamed et al. Citation1995; Dragovic et al. Citation2013). Specifically, CYP1A2 is primarily responsible for norclozapine and clozapine N-oxide, whereas CYP3A4 more effectively generates the nitrenium ion in the liver. The reactivity of the nitrenium ion leads to short half-life and rapid formation of covalent (non-reversible) adducts (Gardner et al. Citation2005), so that hepatically-generated reactive metabolites are not likely to migrate to the heart and play a role in cardiotoxicity. Rather, clozapine bioactivation more likely occurs in cardiac tissue to generate the reactive metabolite in situ and directly mediate damage. Given high ATP demands, cardiac mitochondria are possible sensitive targets for the damaging effects of reactive metabolites. The resulting mitochondrial dysfunction could then contribute to the etiology of cardiotoxicity. Despite precedence with other drugs, Arzuk et al. (Citation2021) were the first to explicitly investigate this toxicological mechanism for clozapine in cardiac tissue.
Figure 7. Proposed mechanism for clozapine bioactivation in cardiomyocytes leading to protein adduction and mitochondrial dysfunction as precursors to cell death or a toxic immune response resulting in cardiotoxicity.
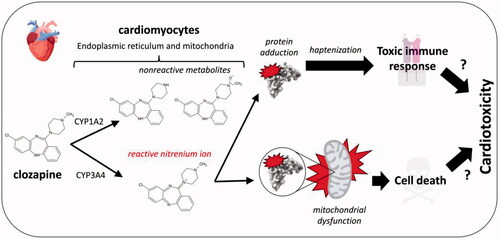
The authors showed that cardiac microsomal and mitochondrial fractions from mice were capable of metabolizing and bioactivating clozapine at a single drug concentration. As proposed for agranulocytosis/neutropenia, the nitrenium ion could react with proteins to form adducts that could lead to cell death and/or trigger a toxic immune response targeting the heart; however, further research is needed to demonstrate formation of clozapine-adducted cardiac proteins. The burden of those reactive metabolite adductions would depend on levels of Cyp3a11, the mouse ortholog of human CYP3A4 (Hrycay and Bandiera Citation2009). Much is known about factors impacting its activity, such as sex, concomitant medications, and genetic polymorphisms (Fujino et al. Citation2021), so that these findings provide a basis for predicting potential risk factors for the bioactivation and consequences for clozapine exposure. Another critical modulator of those outcomes is clozapine concentration that drives saturation of isozyme activity. Although not explored in the current studies, the experiments are feasible based on reported steady-state clozapine kinetics using rat liver microsomes (Dragovic et al. Citation2013). In that work, the specificity for clozapine bioactivation (Vmax/Km) was comparable to pathways leading to nonreactive metabolites demonstrating its potential significance in overall clearance. This impact could be even greater in cardiac mitochondria. A comparison of metabolism by mitochondrial and microsomal fractions requires careful preparation of subcellular fractions to avoid contamination and so the current study confirmed the purity of fractions based on the presence or absence of six protein markers selectively expressed in either organelle. The subsequent reactions by murine liver and cardiac subcellular fractions showed that the cardiac mitochondrial fractions had the highest bias toward clozapine bioactivation versus other elimination pathways (Arzuk et al. Citation2021). Preference for clozapine bioactivation and high reactivity of the nitrenium ion would increase the probability of mitochondrial damage in the heart.
Cardiac mitochondria are susceptible to damage from clozapine. Arzuk et al. (Citation2021) showed that the drug caused a suppression of rate of oxygen consumption in cell culture. In a later study, Hafez et al. (Hafez et al. Citation2021) reported a more thorough analysis of the impact of clozapine on cardiac mitochondrial function with rat cardiomyocytes. Drug treatment reduced cell viability and led to a shift from reduced to oxidized glutathione and formation of malondialdehyde implicating increased oxidative stress. Moreover, clozapine decreased succinate dehydrogenase activity for the organelle and led to swelling in response to mitochondrial damage. These outcomes are hallmarks of drug bioactivations that cause mitochondrial dysfunction implicating a possible role for the clozapine nitrenium ion. What current studies lack is the specific interrogation of clozapine metabolism and specifically, bioactivation, in contributing to observed compromised cell function. It is possible that the parent drug and/or nonreactive metabolites could play a role in the effects of clozapine treatment, so that further studies are necessary to resolve that ambiguity. Taken together, these findings describe important steps toward elucidating the mechanism(s) underlying clozapine-induced cardiotoxicity and as outlined here, point to future studies to ultimately realize that goal.
References
- Arzuk E, Karakuş F, Orhan H. 2021. Bioactivation of clozapine by mitochondria of the murine heart: possible cause of cardiotoxicity. Toxicology. 447:152628.
- Atkin K, Kendall F, Gould D, Freeman H, Liberman J, O’Sullivan D. 1996. Neutropenia and agranulocytosis in patients receiving clozapine in the UK and Ireland. Br J Psychiatry. 169(4):483–488.
- Dragovic S, Gunness P, Ingelman-Sundberg M, Vermeulen NPE, Commandeur JNM. 2013. Characterization of human cytochrome P450s involved in the bioactivation of clozapine. Drug Metab Dispos. 41(3):651–658.
- Fujino C, Sanoh S, Katsura T. 2021. Variation in expression of cytochrome P450 3A isoforms and toxicological effects: endo- and exogenous substances as regulatory factors and substrates. Biol Pharm Bull. 44(11):1617–1634.
- Gardner I, Popović M, Zahid N, Uetrecht JP. 2005. A comparison of the covalent binding of clozapine, procainamide, and vesnarinone to human neutrophils in vitro and rat tissues in vitro and in vivo. Chem Res Toxicol. 18(9):1384–1394.
- Hafez AA, Jamali Z, Khezri S, Salimi A. 2021. Thymoquinone reduces mitochondrial damage and death of cardiomyocytes induced by clozapine. Naunyn Schmiedebergs Arch Pharmacol. 394(8):1675–1684.
- Hrycay EG, Bandiera SM. 2009. Expression, function and regulation of mouse cytochrome P450 enzymes: comparison with human P450 enzymes. Curr Drug Metab. 10(10):1151–1183.
- Kanniah G, Kumar S. 2020. Clozapine associated cardiotoxicity: Issues, challenges and way forward. Asian J Psychiatr. 50(101950):101950.
- Pirmohamed M, Williams D, Madden S, Templeton E, Park B. 1995. Metabolism and bioactivation of clozapine by human liver in vitro. J Pharmacol Exp Ther. 272(3):984–990.
- Remington G, Lee J, Agid O, Takeuchi H, Foussias G, Hahn M, Fervaha G, Burton L, Powell V. 2016. Clozapine’s critical role in treatment resistant schizophrenia: ensuring both safety and use. Expert Opin Drug Saf. 15(9):1193–1203.
- Wiciński M, Węclewicz MM. 2018. Clozapine-induced agranulocytosis/granulocytopenia: mechanisms and monitoring. Curr Opin Hematol. 25(1):22–28.
Source: Biochemical Pharmacology 2021;194:114824
Synopsis
The work of Schleiff et al. (Citation2021) identified cytochromes P450 (CYPs) involved in bioactivation of seven diphenylamine nonsteroidal anti-inflammatory drugs (NSAIDs) into quinone-species metabolites and determined reaction specificities (Schleiff et al. Citation2021). Although diphenylamine NSAIDs are widely used for chronic pain condition treatments, their use may associate with hepatotoxicity risks. To date, the molecular mechanisms of diphenylamine NSAID-induced hepatotoxicity are unknown. In order to mitigate drug-induced hepatotoxicity risks and design drugs with favorable safety profiles, the knowledge of mechanisms underlying drug-induced hepatotoxicity is needed. In this work, the authors employed a novel quantitative kinetic approach and revealed opposing roles for CYP2C9 and CYP3A4 in diphenylamine NSAID bioactivation and detoxification, respectively. Importantly, the findings from this study have broader implications on bioactivation of other drug classes that contain diphenylamine scaffold.
Commentary
Diphenylamine non-steroidal anti-inflammatory drugs (NSAIDs) are used for chronic pain treatment (Bindu et al. Citation2020). However, it is known that diphenylamine NSAIDs can cause hepatotoxicity (Davis and Robson Citation2016; Bindu et al. Citation2020). Although the molecular mechanisms underlying the above drug-induced hepatotoxicity risks are not fully elucidated, bioactivation of drugs into reactive metabolites which compromise normal cellular functions can contribute to these adverse events. It has been reported that diphenyl NSAIDs are converted to reactive acyl glucuronides or quinone-species metabolites (Masubuchi et al. Citation1999; Regan et al. Citation2010). To date, the toxicological relevance of the above pathways is unclear largely due to the lack of quantitative data.
In this work, the investigators addressed the existing knowledge gap using a novel quantitative kinetics approach. They identified CYP isozymes responsible for diphenylamine NSAID bioactivation using chemical inhibitor phenotyping experiments. From these qualitative activity assessments, CYP2C8, 2C9, 2C19, and 3A4 enzymes were found to be involved in diphenylamine NSAID bioactivation. A metabolic pathway for the diphenyl NSAID diclofenac that undergoes bioactivation to form metabolites is shown in . Steady state kinetic analysis was employed to quantitatively measure rates of quinone-species metabolite formation. Additionally, the importance of halogenation and acidic group type on bioactivations were recapitulated. The authors measured overall kinetics for the elimination of drugs by the respective CYPs. With these, they were able to normalize bioactivation kinetics to overall drug metabolism and calculate its relative contribution. Further, they extrapolated the information on in vitro metabolism to in vivo clearance. It should be noted that this approach effectively identified the CYPs most likely to be clinically relevant in the bioactivation of diphenylamine NSAIDs.
Figure 8. Metabolic pathway for the diphenyl NSAID diclofenac that undergoes bioactivation to form quinone-species metabolites.
The contributions of CYP2C9 and CYP3A4 may drive the extent of diphenylamine NSAID bioactivation and detoxification, respectively. However, factors such as sex, age, genetic polymorphisms, and physiological conditions that drive either CYP2C9 bioactivations or CYP3A4 detoxifications might also play a role in clinical outcomes. Thus, future clinical studies are needed to validate the proposed roles of CYP2C9 and CYP3A4 in metabolic outcome for diphenyl NSAIDs. Interestingly, the diphenylamine moiety serves as a scaffold for other classes of drugs such as targeted cancer therapies. Therefore, the findings from this work have broader impacts on identifying and mitigating bioactivation risks associated with drug treatments.
References
- Bindu S, Mazumder S, Bandyopadhyay U. 2020. Non-steroidal anti-inflammatory drugs (NSAIDs) and organ damage: a current perspective. Biochem Pharmacol. 180:114147.
- Davis A, Robson J. 2016. The dangers of NSAIDs: look both ways. Br J Gen Pract. 66(645):172–173.
- Masubuchi Y, Yamada S, Horie T. 1999. Diphenylamine as an important structure of nonsteroidal anti-inflammatory drugs to uncouple mitochondrial oxidative phosphorylation. Biochem Pharmacol. 58(5):861–865.
- Regan SL, Maggs JL, Hammond TG, Lambert C, Williams DP, Park BK. 2010. Acyl glucuronides: the good, the bad and the ugly. Biopharm Drug Dispos. 31(7):367–395.
- Schleiff MA, Crosby S, Blue M, Schleiff BM, Boysen G, Miller GP. 2021. CYP2C9 and 3A4 play opposing roles in bioactivation and detoxification of diphenylamine NSAIDs. Biochem Pharmacol. 194:114824.
Source: Chem Res Toxicol. 2021;34(6):1612–1618
Synopsis
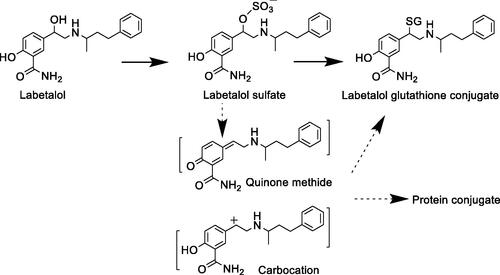
Labetalol hydrochloride (LHCl) is an α- and β-adrenoreceptor blocker widely used for the treatment of hypertension and angina pectoris. Yang et al. reported metabolic bioactivation through sulfonation of the benzylic alcohol group in LHCl, which provides a mechanism to possibly explain its liver toxicity (Yang et al. Citation2021). The results presented in the article are consistent with sulfonation of the benzylic hydroxyl group followed by replacement with glutathione (GSH). A conjugate with GSH replacing the benzylic hydroxyl group was identified in incubation of LHCl in rat and human hepatocytes. The GSH conjugate was subsequently confirmed with a synthetic standard. This GSH conjugate was also identified as a major metabolite in bile of rats following oral administration of LHCl. More importantly, formation of this GSH conjugate was inhibited in hepatocyte incubations and in rats by 2,6-dichloro-4-nitrophenol (DCNP), a sulfotransferases inhibitor. Conjugation by a SULT enzyme made the benzylic hydroxyl group a good leaving group leading to formation of a carbocation or quinone methide intermediate (). The reactivity of these sulfonate or quinone methide intermediates toward large molecules like proteins may elicit toxicity in the liver or other organs where SULTs are expressed. Cytotoxicity in hepatocytes suggested the toxicity potential of bioactivated LHCl, which was attenuated by co-incubation with the DCNP. The result further supported sulfonation of the benzylic hydroxyl group as a potential contributor to hepatotoxicity risk. It is interesting to point out that the GSH conjugate was also detected in incubation of LHCl and GSH in a buffer in the absence of enzymes. However, the level of GSH conjugate was much lower than that found in hepatocyte incubations. The results support the major role of sulfotransferase catalysis in the bioactivation.
Figure 9. Bioactivation pathways of labetalol.
Commentary
Adverse effects of labetalol include liver toxicity and acute renal failure. Because of its severe liver injury, including hepatitis, fulminant hepatic failure, and even hepatic necrosis, LHCl has been withdrawn from some European countries. This work provided bioactivation data to develop a possible mechanism underlying liver toxicity. Sulfonation of the benzylic alcohol activated the carbon oxygen bond to lead to formation of a carbocation or a quinone-methide intermediate (), which can form conjugates with a thiol in GSH or a protein. The authors performed an elegant experiment showing that DCNP inhibited GSH adduct formation in vitro in hepatocytes and in vivo in rats. In addition reduced drug toxicity was observed in the hepatocyte incubations.
Similarly, sulfonation of benzyl hydroxyl group in aloe-emodin, 1-hydroxymethylpyrene, 1-hydroxysafrole, hydroxyl methyleugenol, liguzinediol, and nevirapine could lead to formation of reactive intermediates that may contribute to their toxicities (Popovic et al. Citation2010; Yang et al. Citation2021). The resulting sulfate is a good leaving group to generate an electrophilic species (Glatt Citation2000). Therefore, benzyl alcohol and benzyl carbon upon hydroxylation could present a bioactivation liability in drug designs.
This is another example of drug bioactivation through a phase II conjugation reaction. Other examples of conjugation leading to bioactivation include the carboxylic glucuronidation of GDC-0810 activating α,β-double bond for nucleophilic conjugate addition (Mulder et al. Citation2020), and the carboxylic glucuronidation of gemfibrozil and clopidogrel leading to bioactivation via CYP2C8 (Ma et al. Citation2017). Gan et al. summarized the bioactivation mechanisms that were catalyzed by non-P450 enzymes (Popovic et al. Citation2010; Gan et al. Citation2016).
References
- Gan J, Ma S, Zhang D. 2016. Non-cytochrome P450-mediated bioactivation and its toxicological relevance. Drug Metab Rev. 48(4):473–501.
- Glatt H. 2000. Sulfotransferases in the bioactivation of xenobiotics. Chem Biol Interact. 129(1–2):141–170.
- Ma Y, Fu Y, Khojasteh SC, Dalvie D, Zhang D. 2017. Glucuronides as potential anionic substrates of human cytochrome P450 2C8 (CYP2C8). J Med Chem. 60(21):8691–8705.
- Mulder T, Bobba S, Johnson K, Grandner JM, Wang W, Zhang C, Cai J, Choo EF, Khojasteh SC, Zhang D. 2020. Bioactivation of α,β-unsaturated carboxylic acids through acyl glucuronidation. Drug Metab Dispos. 48(9):819–829.
- Popovic M, Shenton JM, Chen J, Baban A, Tharmanathan T, Mannargudi B, Abdulla D, Uetrecht JP. 2010. Nevirapine hypersensitivity. Handb Exp Pharmacol. (196):437–451.
- Yang L, Xin L, Shi J, Li W, Tian M, Hu Z, Peng Y, Zheng J. 2021. Metabolic activation and cytotoxicity of labetalol hydrochloride mediated by sulfotransferases. Chem Res Toxicol. 34(6):1612–1618.
Source: Drug Metab Dispos. 2022:50(1):49–57; doi: 10.1124/dmd.121.000642. Epub 2021 Oct 4.
Synopsis
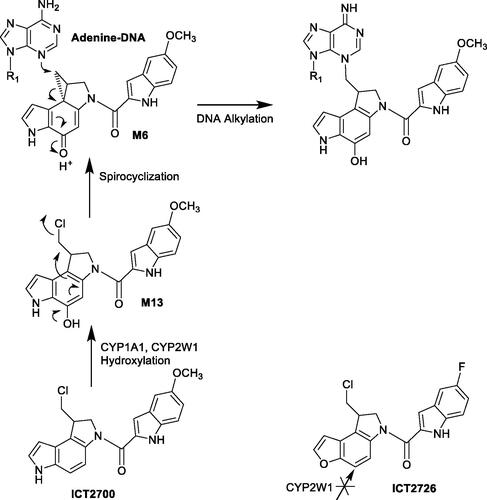
Duocarmycins are natural products that have anticancer activities. The authors (Bart et al. Citation2022) investigated the molecular interactions of duocarmycin analogs with P450 CYP1A1 and CYP2W1 enzymes to support applying tissue-specific P450-mediated bioactivation to activate the prodrugs. CYP1A1 binds and metabolizes enantiomers of both compounds ICT2700 and ICT2726; CYP2W1 selectively binds the S enantiomer of ICT2726. In contrast, CYP2W1 differentially binds the R isomer of the indole based ICT2700 over the S stereoisomer. Thus, the ICT2700 R configuration warrants further investigation as a scaffold to favor CYP2W1-selective bioactivation. Less selectivity of CYP1A1 bioactivation can be de-emphasized in duocarmycin prodrug design. The metabolite profile of ICT2726 by CYP2W1 showed selective chemical modifications and could potentially be used as a biomarker to identify CYP2W1 metabolic activity. This work defines the binding and metabolism by both P450 enzymes CYP1A1 and CYP2W1 to support the design of duocarmycins selectively activated by only one human P450 enzyme CYP2W1. This provides an excellent example of applying selective catalytic activity of P450 enzymes in activating prodrugs.
Commentary
The authors used several experimental approaches including ligand binding, metabolite profiling, and kinetic analysis to demonstrate applications of selective P450 biactivation in prodrug designs. Binding of ligands with purified and expressed CYP1A1 and 2W1 enzymes was monitored by shifts in the absorbance maximum of the heme Soret peaks. ‘Type I’ binding associated with decreases in absorbance at ∼430 nm and increases at ∼393 nm were observed for CYP1A1 and CYP2W1 when titrated with ICT2700 or ICT2726 isomers. Metabolites of ICT2700 or ICT2726 isomers catalyzed by CYP1A1 and CYP2W1 were profiled and characterized by LC/MS. CYP1A1 is an extrahepatic enzyme expressed in tumors, lung, placenta, skin, intestine, and bladder with large variation between tissues and individuals (Lang et al. Citation2019; Sutherland et al. Citation2013). In contrast, CYP2W1 is a poorly understood P450 that is expressed in colon cancer tissues (Stenstedt et al. Citation2014). CYP1A1 hydroxylates duocarmycin ICT2700 at the position on the chloromethyl indoline leading to formation of hydroxyl seco-duocarmycin (M13) that undergoes spontaneous spirocyclization, releasing the chlorine and generating a highly constrained cyclopropane (M6) () (Sheldrake et al. Citation2013; Wang et al. Citation2019). The indole at the opposite end of duocarmycins facilitates sequence-selective DNA minor groove binding, the constrained cyclopropane reacts with N3 of adenine leading to cytotoxicity. Although high yield of metabolites of ICT2726 were formed by both CYP1A1 and 2W1, no cytotoxic metabolites such as M13- or M6-equivalent metabolites were identified, consistent with observations that ICT2726 was cytotoxic due to lack of bioactivation (Travica et al. Citation2013).
Figure 10. Bioactivation pathways of duocarmycin ICT2700.
References
- Bart AG, Morais G, Vangala VR, Loadman PM, Pors K, Scott EE. 2022. Cytochrome P450 binding and bioactivation of tumor-targeted duocarmycin agents. Drug Metab Dispos. 50(1):49–57.
- Lang D, Radtke M, Bairlein M. 2019. Highly variable expression of CYP1A1 in human liver and impact on pharmacokinetics of Riociguat and Granisetron in humans. Chem Res Toxicol. 32(6):1115–1122.
- Sheldrake HM, Travica S, Johansson I, Loadman PM, Sutherland M, Elsalem L, Illingworth N, Cresswell AJ, Reuillon T, Shnyder SD, et al. 2013. Re-engineering of the duocarmycin structural architecture enables bioprecursor development targeting CYP1A1 and CYP2W1 for biological activity. J Med Chem. 56(15):6273–6277.
- Stenstedt K, Hallstrom M, Lédel F, Ragnhammar P, Ingelman-Sundberg M, Johansson I, Edler D. 2014. The expression of CYP2W1 in colorectal primary tumors, corresponding lymph node metastases and liver metastases. Acta Oncol. 53(7):885–891.
- Sutherland M, Gill JH, Loadman PM, Laye JP, Sheldrake HM, Illingworth NA, Alandas MN, Cooper PA, Searcey M, Pors K, et al. 2013. Antitumor activity of a duocarmycin analogue rationalized to be metabolically activated by cytochrome P450 1A1 in human transitional cell carcinoma of the bladder. Mol Cancer Ther. 12(1):27–37.
- Travica S, Pors K, Loadman PM, Shnyder SD, Johansson I, Alandas MN, Sheldrake HM, Mkrtchian S, Patterson LH, Ingelman-Sundberg M. 2013. Colon cancer-specific cytochrome P450 2W1 converts duocarmycin analogues into potent tumor cytotoxins. Clin Cancer Res. 19(11):2952–2961.
- Wang S, Chen B, Dragovich P, Pillow T, Staben L, Guo J, Su D, Zhang C, Bobba S, Ma Y, et al. 2019. A novel depurination methodology to assess DNA alkylation of chloro-bis-seco-cyclopropylbenzoindoles allowed for comparison of minor-groove reactivity. Drug Metab Dispos. 47(5):547–555.
Source: Xenobiotica. 2021;51(8):933–948
Synopsis
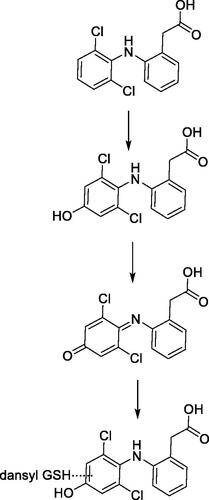
Chacko et al. (Chacko et al. Citation2021) describe the in vitro and in vivo biotransformation pathways of a novel macrocyclic, reversible FXIa inhibitor. FXIa-6f, Methyl N-[(10R,14S)-14-[4-(3-Chloro-2,6-difluorophenyl)-6-oxo1,2,3,6-tetrahydropyridin-1-yl]-10-methyl-9-oxo-8,16-diazatricyclo[13.3.1.02,7]nonadeca-1(19), 2,4,6,15,17-hexaen-5-yl]-carbamate, is in development as an antithrombotic agent. In vivo, FXIa-6f showed high oral bioavailability of rat and monkey. In the present study 3H-FXIa-6f was dosed in bile-duct cannulated rats and intact rats for biotransformation studies, whereas unlabeled metabolite identification was carried out in toxicokinetic samples. In vitro metabolic species differences were similar to those observed in circulating metabolites. A descarbamoylated aniline metabolite M6 which predominant in rat plasma and a desfluoro ipso-substituted glutathione conjugate M2 identified via structural elucidation, are of interest to this commentary because they resulted from bioactivation of FXIa-6f.
Commentary
M2 was identified as an oxidative ipso-substituted glutathione conjugate of FXIa-6f at 6.9 and 34.7% UV abundance in rat and dog liver microsomal incubations, respectively.
The mechanism for formation of M2 is thought to proceed via arene oxide formation shown in . Once the arene oxide is formed, the mechanism can proceed via either (a) or (b) pathways upon nucleophilic attack by glutathione. The result is the opening of the arene oxide to an unstable intermediate which could spontaneously lose HF to restore aromaticity in the ring. It is possible that M2 is one of two positional isomers as shown in , or even a mixture of both, i.e. two separate metabolites which could co-elute under the current chromatographic conditions. Oxidative ipso-substitution of F atom is a relatively uncommon biotransformation reaction, although some precedence exists. The reaction under discussion for aryl fluorides proceed via arene oxide formation, a slightly different mechanism than those documented for alkyl flourides which could involve hydrogen atom abstraction of the ipso hydrogen atom or even oxidation at a remote site (Zhang et al. Citation2005), leading to subsequent losses of HF. Loss of fluorine atom via oxidative ipso-substitution has been previously shown at a difluorobenzylic group for an inhibitor of hedgehog pathway (Gunduz et al. Citation2012) and at a fluoropyrimidyl group for a BACE inhibitor (Mandal et al. Citation2018), and is sometimes referred to as oxidative defluorination of aryl fluorides. Interestingly, in the in vitro experiments carried out to determine covalent binding to microsomal protein across species, the binding without NADPH was similar but varied with species. Rat and dog liver microsomal binding were approximately 20-fold higher in the presence of NADPH as compared to 2- and 4-fold higher for monkey and human liver microsomes, respectively. Addition of 5 mM glutathione alleviated the covalent binding in rat and dog liver microsomes.
Figure 11. Structure of FXla-6f along with putative bioactivation pathways of oxidative ipso-substituted metabolite M2 and aniline metabolite M6.
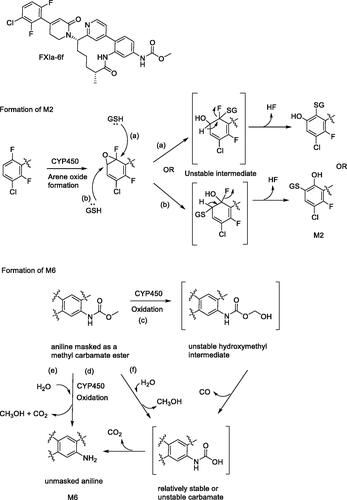
Another interesting bioactivation pathway of FXIa-6f involved formation of the descarbamoylated aniline metabolite, M6. In rat, M6 accounted for ∼5% of the total plasma radioactivity AUC(0-48h). M6 levels in circulation quantitatively increased after repeat dosing, but the metabolite to parent ratio remained unchanged. M6 was a predominant metabolite (12% of all UV related peaks) in rat liver microsomal incubations and was also identified in rat hepatocytes. However, it was not detected in hepatocyte incubations from other species, indicating inter-species differences in metabolism of FXIa-6f. M6 was also formed in trace amounts rather than abundantly present in incubations with Aroclor-induced rat liver S9, a kinetic setting where AhR induced CYPs and other AhR-inducible enzymes are higher. CYP phenotyping data is not available yet. M6 is an aniline metabolite formed via oxidation of FXIa-6f followed by loss of methyl carbamoyl moiety, in the presence of NADPH. The concentration dependency and kinetics of the metabolite formation with respect to varying concentrations of FXIa-6f were not reported and a stable carbamate metabolite of FXIa-6f post cleavage of the methyl ester was not identified either. Collectively, without direct evidence via mechanistic investigations with either stable labeled water or oxygen to understand the source of oxygen atom or pH dependent hydrolytic stability of FXIa-6f, the role of hydrolysis, albeit minor, cannot be completely ruled out. The potential mechanisms for formation of M6 are shown in . Pathways (c) and (d) involve CYP450 mediated oxidation of the methyl group, potentially resulting in an unstable hydroxymethyl intermediate that would undergo an instantaneous loss of CO and CO2 directly or via an unstable carbamate (in this specific case) to result in the aniline metabolite. Note that while some amines are stable as their carbamic acid derivatives or metabolites (Hayakawa et al. Citation2003; Shaffer et al. Citation2005), others are not and were identified only as their carbamoyl glucuronides (Tremaine et al. Citation1989; Gunduz et al. Citation2010). Pathways (e) and (f), if present as minor pathways, putatively proceed via hydrolysis and are shown herein in the spirit of the lacking direct evidence to support chemical or enzymatic hydrolysis, as discussed earlier.
References
- Chacko SA, Yang W, Wang Y, Tian Y, Hong Y, Wallace M, Wang B, Ewing WR, Luettgen JM, Shu Y-Z, et al. 2021. Preclinical metabolism and disposition of an orally bioavailable macrocyclic FXIa inhibitor. Xenobiotica. 51(8):933–948.
- Gunduz M, Argikar UA, Baeschlin D, Ferreira S, Hosagrahara V, Harriman S. 2010. Identification of a novel N-carbamoyl glucuronide: in vitro, in vivo, and mechanistic studies. Drug Metab Dispos. 38(3):361–367.
- Gunduz M, Argikar UA, Kamel A, Colizza K, Bushee JL, Cirello A, Lombardo F, Harriman S. 2012. Oxidative ipso substitution of 2,4-difluoro-benzylphthalazines: identification of a rare stable quinone methide and subsequent GSH conjugate. Drug Metab Dispos. 40(11):2074–2080.
- Hayakawa H, Fukushima Y, Kato H, Fukumoto H, Kadota T, Yamamoto H, Kuroiwa H, Nishigaki J, Tsuji A. 2003. Metabolism and disposition of novel des-fluoro quinolone garenoxacin in experimental animals and an interspecies scaling of pharmacokinetic parameters. Drug Metab Dispos. 31(11):1409–1418.
- Mandal M, Mitra K, Grotz D, Lin X, Palamanda J, Kumari P, Buevich A, Caldwell JP, Chen X, Cox K, et al. 2018. Overcoming time-dependent inhibition (TDI) of cytochrome P450 3A4 (CYP3A4) resulting from bioactivation of a fluoropyrimidine moiety. J Med Chem. 61(23):10700–10708.
- Shaffer CL, Gunduz M, O’Connell TN, Obach RS, Yee S. 2005. Biotransformation of a GABAA receptor partial agonist in Sprague-Dawley rats and cynomolgus monkeys: identification of two unique N-carbamoyl metabolites. Drug Metab Dispos. 33(11):1688–1699.
- Tremaine LM, Stroh JG, Ronfeld RA. 1989. Characterization of a carbamic acid ester glucuronide of the secondary amine sertraline. Drug Metab Dispos. 17(1):58–63.
- Zhang D, Krishna R, Wang L, Zeng J, Mitroka J, Dai R, Narasimhan N, Reeves RA, Srinivas NR, Klunk LJ. 2005. Metabolism, pharmacokinetics, and protein covalent binding of radiolabeled MaxiPost (BMS-204352) in humans. Drug Metab Dispos. 33(1):83–93.
Source: Biochem Pharmacol. 2021;183:114303. doi: 10.1124/dmd.121.000563. doi: 10.1016/j.bcp.2020.114303. Epub 2020 Oct 27. PMID: 33121928
Synopsis
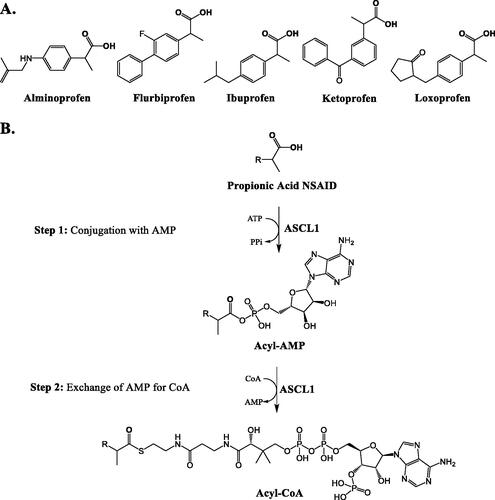
Hashizume et al. (Citation2021) investigated the acyl-CoA synthetase (ACS) enzyme activity toward non-steroidal anti-inflammatory drugs (NSAIDs) in the human liver. The authors surveyed the formation of acyl-CoA conjugates in human hepatocytes from propionic acid (5 compounds, ), anthranilic acid (1 compound), phenyl acetic acid (1 compound), and indole acetic acid (2 compounds) classes of NSAIDs and only detected acyl-CoA conjugate formation from the propionic acid NSAIDs. In an effort to determine the relevant ACS isoforms in the human liver, the authors measured the mRNA expression of the 26 human ACS enzymes in human liver tissue, for which two enzymes, ACSL1 (ACS long-chain 1) and ACSM2B (ACS medium-chain 2B), accounted for ∼60% of the mRNA transcript copies. These enzymes were recombinantly expressed and their activity toward propionic acid NSAIDs was measured; only ASCL1 demonstrated acyl-CoA conjugation of these drugs (). Further, the expression of ASCL1 in human liver tissue from 28 donors was significantly correlated with the rate of propionic acid conjugate formation in tissue homogenates. Finally, through incubations of pure standards of acyl-CoA and -glucuronide metabolites of ibuprofen with human liver homogenate, the authors demonstrated that the CoA conjugate was less stable in the prescence of homogenate than the glucuronide, and the authors also repeated these results using crude metabolite isolates of the other four propionic acid NSAIDs (Hashizume et al. Citation2021).
Figure 12. The five propionic acid NSAIDs investigated in the work by Hashizum et al. (A) and the steps of propionic acid NSAID acyl-CoA conjugation by ASCL1 (B).
Commentary
NSAIDs, which often contain carboxylic acids, make up 5–10% of the total medications prescribed each year (Wongrakpanich et al. Citation2018). The acyl-glucuronide metabolites of carboxylic acid-containing drugs are proposed indicators of bioactivation potential, as these conjugates have been demonstrated to undergo acyl-migration and are less stable than simple O– and N– linked glucuronides (Bailey and Dickinson Citation2003). The conjugation of carboxylic acid drugs with coenzyme A (CoA) is a growing area of interest, with work to suggest that the formation of CoA conjugates may also indicate bioactivation potential, perhaps even more so than acyl-glucuronide formation (Grillo et al. Citation2012). This two-step reaction is mediated by acyl-CoA synthetase enzymes (ACS), which act endogenously in lipid biosynthesis. The first step consists of acid conjugation with AMP, followed by the substitution of AMP with CoA () (Darnell and Weidolf Citation2013).
Through this investigation, it was demonstrated that propionic acid NSAIDs undergo CoA conjugation in human hepatocytes, while the other NSAID acid classes did not. These results can inform future carboxylic acid drug design, especially if CoA conjugation is established to be an indicator of xenobiotic bioreactivity. The authors determined ACSL1 and ACSM2B to be the most abundantly transcribed ASCs in human hepatocytes, work that should be further bolstered through subsequent analysis of ACS protein expression. Of the two enzymes, ACSL1 was more abundant and was able to catalyze NSAID CoA conjugation, while ASCMB2 did not. This work gives other researchers a starting point for enzyme phenotyping of CoA drug conjugations and supports the need for an in-depth analysis of natural variations in activity and expression of ACSL1, as has been done for other drug metabolizing enzymes. Variants of ACSL1 in humans have been identified and correlated to metabolic disease markers (Manichaikul et al. Citation2016), and ASCL1 has been shown to be induced by rosuvastatin treatment through sterol regulatory element binding protein 2 activation in mice and HepG2 cells (Singh et al. Citation2016). The presence of ACSL1 enzyme variants and induction potential may impact NSAID CoA conjugation and therefore bioactivation risk. Finally, it was demonstrated that acyl-CoA conjugates were less stable in the presence of human liver homogenate than acyl-glucuronide metabolites, giving further evidence to support a role for CoA conjugates in carboxylic acid drug bioreactivity. Work is still needed to isolate and characterize drug-protein conjugates formed from CoA metabolites, probe the differences in bioreactivity between CoA and glucuronide conjugates, and ultimately directly correlate these findings with drug toxicity.
References
- Bailey MJ, Dickinson RG. 2003. Acyl glucuronide reactivity in perspective: biological consequences. Chem Biol Interact. 145(2):117–137.
- Darnell M, Weidolf L. 2013. Metabolism of xenobiotic carboxylic acids: focus on coenzyme A conjugation, reactivity, and interference with lipid metabolism. Chem Res Toxicol. 26(8):1139–1155.
- Grillo MP, Tadano Lohr M, Wait JC. 2012. Metabolic activation of mefenamic acid leading to mefenamyl-S-acyl-glutathione adduct formation in vitro and in vivo in rat. Drug Metab Dispos. 40(8):1515–1526.
- Hashizume H, Fukami T, Mishima K, Arakawa H, Mishiro K, Zhang Y, Nakano M, Nakajima M. 2021. Identification of an isoform catalyzing the CoA conjugation of nonsteroidal anti-inflammatory drugs and the evaluation of the expression levels of acyl-CoA synthetases in the human liver. Biochem Pharmacol. 183:114303.
- Manichaikul A, Wang XQ, Zhao W, Wojczynski MK, Siebenthall K, Stamatoyannopoulos JA, Saleheen D, Borecki IB, Reilly MP, Rich SS, et al. 2016. Genetic association of long-chain acyl-CoA synthetase 1 variants with fasting glucose, diabetes, and subclinical atherosclerosis. J Lipid Res. 57(3):433–442.
- Singh AB, Kan CF, Dong B, Liu J. 2016. SREBP2 activation induces hepatic long-chain acyl-CoA synthetase 1 (ACSL1) expression in vivo and in vitro through a sterol regulatory element (SRE) Motif of the ACSL1 C-promoter. J Biol Chem. 291(10):5373–5384.
- Wongrakpanich S, Wongrakpanich A, Melhado K, Rangaswami JA. 2018. Comprehensive review of non-steroidal anti-inflammatory drug use in the elderly. Aging Dis. 9(1):143–150.
Source: Anal. Methods. 2021;13:399–410
Synopsis
Tandutinib (TND) is a small molecule inhibitor of multiple receptor tyrosine kinases, including FMS-like tyrosine kinase-3 (FLT3), platelet-derived growth factor (PDGFR), and c-KIT (Pandey et al. Citation2002). This drug was under clinical investigation for treatment of acute myelogenous leukemia (DeAngelo et al. Citation2006); however, it has been associated with dose-limiting toxicities (muscle weakness/neuromuscular toxicity). The objective of the study by Al-Shakliah et al. (Citation2021) was to determine the biotransformation and bioactivation pathways of tandutinib using in silico, in vitro, and in vivo methods. Al-Shakliah et al. (Citation2021) used in silico approaches to predict the sites of tandutinib biotransformation and identify the structural features potentially associated with drug toxicity. The WhichP450 module of StarDrop software was used to propose the sites of oxidative metabolism by cytochrome P450 enzymes, and XenoSite reactivity module software was used to identify the potential sites of bioactivation. The piperazine and piperidine rings of tandutinib were predicted to undergo bioactivation to iminium ions that can react with cyanide to form cyano adducts. To confirm the in silico predictions, cyanide trapping studies were conducted in vitro with tandutinib incubated with rat liver microsomes in the presence of potassium cyanide (KCN). Five cyano adducts were identified by LC-MS/MS analysis: two cyano adducts at the piperidine ring and three cyano adducts at the piperazine ring (). Metabolic incubations were also conducted with rat liver microsomes and hepatocytes to characterize tandutinib oxidative and conjugative metabolites. The in vitro metabolites identified were predicted to be products of O-deisopropylation, O-demethylation, hydroxylation of the piperidine and piperazine rings, reduction of the aminocarbonyl, α-carbonyl formation, and N-glucuronidation. The metabolites identified in vivo in rats were similar to those found in vitro. The O-desisopropyl metabolite was further metabolized to a sulfate conjugate.
Figure 13. Proposed bioactivation pathway of tandutinib. Figure adapted from Al-Shakliah et al. (Citation2021). KCN: potassium cyanide. Predicted structures of four cyano adducts (TNDCN588, TNDCN546, TNDCN574, and TNDCN602) are shown.
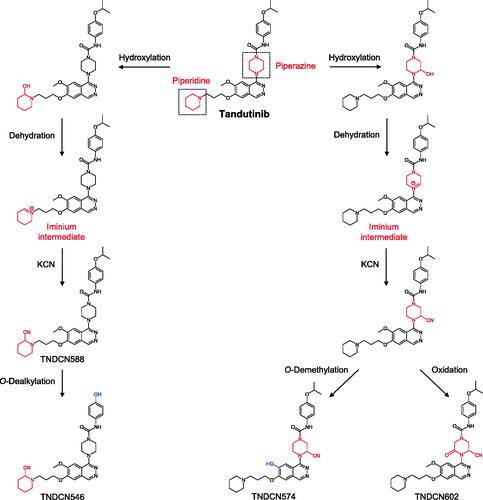
Commentary
Al-Shakliah et al. (Citation2021) proposed that tandutinib metabolic activation may be involved in the toxicities associated with tandutinib. This hypothesis was supported by the in silico predictions of drug toxicity; however, the actual mechanisms are unknown. Tandutinib is a 4-piperazinylquinazoline kinase inhibitor. Cyclic tertiary amines, such as piperidine and piperazine groups, have been shown to undergo metabolic activation to iminium ion intermediates via α-carbon oxidation (Bolleddula et al. Citation2014). Cyanide trapping experiments are conducted to provide evidence for iminium ion intermediates trapped as cyano adducts (Argoti et al. Citation2005). In the present study, the in silico predictions for tandutinib bioactivation at the piperidine and piperazine rings were consistient with the in vitro findings, as demonstrated by the detection of multiple cyano adducts (). Nonetheless, the link between tandutinib bioactivation and drug toxicity remains unclear. Kalgutkar and Driscoll (Citation2020) recently discussed whether cycloalkyl amines are true structural alerts associated with drug toxicity. As noted, it is important to consider the detoxication pathways of iminium ion intermediates, such as aldehyde oxidase-mediated metabolism (Kalgutkar and Driscoll Citation2020).
Other kinase inhibitors containing cyclic tertiary amines (e.g. piperazine and morpholine groups), such as imatinib (Li et al. Citation2014) and pemigatinib (Tang et al. Citation2022), respectively, have been shown to undergo bioactivation to form iminium ion intermediates trapped as cyano adducts. For example, reactive iminium ions derived from pemigatinib were predicted to react with lysine residues of P450 3 A enzymes leading to mechanism-based inactivation of the enzymes (Tang et al. Citation2022). Imatinib is also associated with mechanism-based inactivation of P450 3A4 (Filppula et al. Citation2012), which may involve reactive metabolite formation (Kenny et al. Citation2012). Although P450 3A4 was predicted to play a role in tantunib metabolism (Al-Shakliah et al. Citation2021), the actual P450 enzyme(s) involved in tandutinib bioactivation have not been reported. This knowledge gap requires further investigation.
References
- Al-Shakliah NS, Attwa MW, AlRabiah H, Kadi AA. 2021. Identification and characterization of in vitro, in vivo, and reactive metabolites for tandutinib using liquid chromatography ion trap mass spectrometry. Anal Methods. 13(3):399–410.
- Argoti D, Liang L, Conteh A, Chen L, Bershas D, Yu CP, Vouros P, Yang E. 2005. Cyanide trapping of iminium ion reactive intermediates followed by detection and structure identification using liquid chromatography-tandem mass spectrometry (LC-MS/MS). Chem Res Toxicol. 18(10):1537–1544.
- Bolleddula J, DeMent K, Driscoll JP, Worboys P, Brassil PJ, Bourdet DL. 2014. Biotransformation and bioactivation reactions of alicyclic amines in drug molecules. Drug Metab Rev. 46(3):379–419.
- DeAngelo DJ, Stone RM, Heaney ML, Nimer SD, Paquette RL, Klisovic RB, Caligiuri MA, Cooper MR, Lecerf J, Karol MD, et al. 2006. Phase 1 clinical results with tandutinib (MLN518), a novel FLT3 antagonist, in patients with acute myelogenous leukemia or high-risk myelodysplastic syndrome: safety, pharmaokinetics, and pharmacodynamics. Blood. 108(12):3674–3681.
- Filppula AM, Laitila J, Neuvonen PJ, Backman JT. 2012. Potent mechanism-based inhibition of CYP3A4 by imatinib explains its liability to interact with CYP3A4 substrates. Br J Pharmacol. 165(8):2787–2798.
- Kalgutkar AS, Driscoll JP. 2020. Is there enough evidence to classify cycloalkyl amine substituents as structural alerts? Biochem Pharmacol. 174:113796.
- Kenny JR, Mukadam S, Zhang C, Tay S, Collins C, Galetin A, Khojasteh SC. 2012. Drug-drug interaction potential of marketed oncology drugs: in vitro assessment of time-dependent cytochrome P450 inhibition, reactive metabolite formation and drug-drug interaction prediction. Pharm Res. 29(7):1960–1976.
- Li AC, Yu E, Ring SC, Chovan JP. 2014. Structural identification of imatinib cyanide adducts by mass spectrometry and elucidation of bioactivation pathway. Rapid Commun Mass Spectrom. 28(1):123–134.
- Pandey A, Volkots DL, Seroogy JM, Rose JW, Yu JC, Lambing JL, Hutchaleelaha A, Hollenbach SJ, Abe K, Giese NA, et al. 2002. Identification of orally active, potent, and selective 4-piperazinylquinazolines as antagonists of the platelet-derived growth factor receptor tyrosine kinase family. J Med Chem. 45(17):3772–3793.
- Tang LWT, Wei W, Verma RK, Koh SK, Zhou L, Fan H, Chan ECY. 2022. Direct and sequential bioactivation of pemigatinib to reactive iminium ion intermediates culminate in mechanism-based inactivation of cytochrome P450 3A. Drug Metab Dispos. 50(5):529–540.
Source: Int J Mol Sci. 2021;22:2954. doi:10.3390/ijms22062954
Synopsis
In this paper, the authors’ goal is to highlight the current evidence for the mechanistic hypotheses and the role of the immune system behind idiosyncratic drug-induced liver injury (IDILI) using a number of examples from the clinic. Based on the highlighted evidence, the authors propose that the examples could provide opportunities in the area of diagnosis, prevention or treatment of IDILI. The authors list numerous examples which they feel support the strong connection of immune response with IDILI. Further supporting the immunology role, the genetic association with IDILI and other IDRs is discussed throughout the review, in particular focusing on human leucocyte antigen (HLA) haplotypes. The circumstantial evidence that reactive metabolites (RM) play a significant role in the mechanisms of IDILI, and that understanding these RM associated mechanisms could lead to the development of assays for their detection, prevention or treatment are of particular interest for this biotransformation review manuscript. As with most aspects of drug development, attempts have been made to develop in vitro assays in an attempt to predict the risk presented by a molecule, but it is said that none have been reliable, and are unlikely to be improved upon. This is likely to be due to our incomplete understanding of the mechanisms involved in IDRs. Use of animal models have been attempted, but by its very idiosyncratic nature, this makes it very difficult, coupled with the likelihood that toxicity in a preclinical model is caused by high doses of the drug rather than a mechanism of IDILI. In addition to this, the authors’ assertion is that hypotheses of IDILI will need to be consistent with the clinical characteristics, and without this, they are unlikely to be true. The trigger for the immune response mostly appears to be either drug related (flucloxacillin) or due to reactive metabolites (Halothane and many more) covalently binding to proteins forming neoantigens, which then results in a cascade of immune related responses and ultimately onset of IDILI or immune tolerance (). Plenty of evidence of RM association is provided to support a strong connection but the authors rightly point out that association does not prove causation, and there are examples where this does not appear to be the case. It is also proposed that the other mechanisms postulated as being responsible for IDILI are complementary to an immune mechanism rather than excluding it. Clinical challenges of assessing the impact of IDILI are discussed and are used to highlight mechanisms and possible avenues to explore the ability to monitor for IDILI. The authors conclude that evidence continues to grow to support the assertion that IDILI is mediated by the adaptive immune system, especially CD8+ T cells. Due to similarities between humans and animals in terms of metabolism and immune response, certain factors could be carefully studied in preclinical species and extended to humans, to confirm or otherwise that the mechanisms are similar. The authors acknowledge the complexity of the whole system but reflect that the current superficial understanding of early-stage immune responses to drugs could significantly advance our understanding of the mechanisms of IDILI. This in turn could help improve drug development and safety.
Figure 14. Proposed mechanism of idiosyncratic drug-induced liver injury (IDILI).
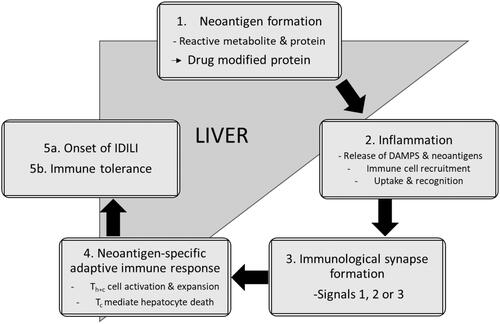
Commentary
This review article collates accumulated evidence describing the link between drugs or reactive metabolites (RM) and an adaptive immune mediated idiosyncratic reaction. The review constructively builds on the known complex picture of the mechanisms that lead to IDILI and highlights how these understandings need to be developed further to allow better predictions from preclinical assays, better patient profiling, or improved diagnosis and treatment.
Throughout the review, and from the many other papers in this area such as the review by Sernoskie et al. (Citation2021), there are often useful examples to support a mechanistic theory, but examples are also referenced where this is not always so clear, although in these cases the authors do propose how the mechanistic theory may still hold true. This highlights how incomplete or possibly incorrect our understanding of this complex area is.
The majority of routine assays available for preclinical evaluation of the propensity of drugs and metabolites to cause idiosyncratic liver injury are based on the proposed mechanism of RM formation and subsequent haptenization or neoantigenesis of proteins. This in turn may result in adaptive immune responses. Nevertheless, as the cited review states, association does not prove causation, even through that is the best the industry has to work with at present. These assays are ideally human based, although preclinical models are also often used relying on IVIV translatable metabolic and mechanistic pathways. New assays being developed utilize a range of these systems from single cell systems, immortalized cell-lines, co-cultures, and the relatively new area of 3D models incorporating microfluidics (Segovia-Zafra et al. Citation2021).
Not only do the assays need to generate RMs in a humanized system, but they also need to induce a detectable relevant cell-stress event. These could be events such as neoantigen formation via covalent binding, or cell markers such as GSH and bile acid levels, ROS or mitochondrial disfunction. These often single point biomarkers are important, but the development of in vitro models with a relevant immunological system, representing the complexity of in vivo systems and their genetic and immunological components could be as, if not more important.
Recent work in the area of bioactivation, subsequent neoantigen formation triggering an adaptive immune response is exemplified in publications from Prof. Jack Uetrecht’s (Sernoskie et al. Citation2021) or Prof. Dean Naisbitt’s (Naisbitt et al. Citation2020) research groups. Based on the understanding generated in this area, a number of recent papers have been published describing development of preclinical (Segovia-Zafra et al. Citation2021) and clinical models (Hammond et al. Citation2021) to aid prediction and improve our understanding of the mechanisms involved.
If preclinical and clinically based in vitro assays can be developed to provide an improved correlation with IDILI in human, then a significant step will have been made in making safer drugs or improved treatment when they do occur. However, the idiosyncratic element will continue to pose considerable challenges and therefore is still an area where the development of translatable and predictive pre-clinical and clinical assays to improve our understanding of IDILI in order to reduce drug attrition and improve patient safety due to IDILI, remains an important worthy goal.
References
- Hammond S, Thomson P, Meng X, Naisbitt D. 2021. In-vitro approaches to predict and study T-cell mediated hypersensitivity to drugs front. Front Immunol. 12:630530.
- Naisbitt D, Olsson-Brown A, Gibson A, Meng X, Ogese M, Tailor A, Thomson P. 2020. Immune dysregulation increases the incidence of delayed-type drug hypersensitivity reactions. Allergy. 75(4):781–797.
- Segovia-Zafra A, Zeo-Sánchez D, Lopez-Gomez C, Perez-Valdes Z, Garcıa-Fuentes E, Andrade R, Lucena M, Villanueva-Paz M. 2021. Preclinical models of idiosyncratic drug-induced liver injury (iDILI): moving towards prediction. Acta Pharm Sin B. 11(12):3685–3726.
- Sernoskie S, Jee A, Uetrecht J. 2021. The emerging role of the innate immune response in idiosyncratic drug reactions. Pharmacol Rev. 73(3):861–896.