
Abstract
β-Thalassemia (β-thal), a highly prevalent disease in tropical and subtropical regions of Southern China, is caused mainly by point mutations in the β-globin gene cluster. However, large deletions have also been found to contribute to some types of β-thal. We identified a novel 5 kb deletion in the β-globin cluster in a Chinese patient using multiplex ligation-dependent probe amplification (MLPA), and characterized it with single molecule real-time (SMRT) sequencing, gap-polymerase chain reaction (gap-PCR) and Sanger sequencing. The deletion was located between positions 5226189 and 5231091 on chromosome 11 (GRCh38), extending from 4 kb upstream of the 5' untranslated region (5'UTR) to the second intron of the β-globin gene. The patient with this deletion presented with microcytosis and hypochromic red cells, as well as relatively high Hb F and Hb A2 levels. Our research indicated that SMRT sequencing is a useful tool for accurate detection of large deletions. Our study broadens the spectrum of deletional β-thalassemias and provides a perspective for further study of the function of the β-globin cluster.
Introduction
β-Thalassemia (β-thal) mutations are carried by approximately 5.35% of the population of Southern China [Citation1]. This autosomal recessive inherited blood disease can lead to disability and death, and is caused mainly by point mutations on the β-globin genes. A total of 17 major point mutations, including codons 41/42 (HBB: c.126_129delCTTT), have been reported in Southern China. However, large deletions (longer than 1 kb) also contribute to some types of β-thal, including those in Southeast Asia [Citation2], China [Citation3], Yunnan [Citation4] and Taiwan [Citation5]. Carriers with any one of the above large deletions have elevated Hb F levels, with or without elevated Hb A2 levels [Citation5]. With the rapid development of the long read characterization method, single molecule real-time (SMRT) sequencing [Citation6], the precise breakpoints can now easily be distinguished. Herein, we identified a novel large deletion in the β-globin gene cluster in a Chinese patient and distinguished the breakpoints by using SMRT sequencing. The patient had a large deletion on the HBB gene, showed elevated Hb F and Hb A2 levels, and presented with microcytosis and hypochromic red cells.
Materials and methods
Patient
The proband was a 28-year-old man from Huizhou, Guangdong Province, People’s Republic of China (PRC). Genetic analysis detecting the common types of α- and β-thal mutations indicated that his α- and β-globin genes were normal. However, the hematological parameters detected in routine blood examination indicated hypochromic polyglobulia and microcytic red blood cells (RBCs) in his peripheral blood sample. In addition, hemoglobin (Hb) quantification indicated elevated Hb F and Hb A2 levels. Therefore, the patient came to our hospital for further study after written informed consent was obtained according to the Declaration of Helsinki.
Hematological analysis
A peripheral blood sample was collected, and hematological parameters were analyzed with a Sysmex XN5000 automated hematology analyzer (Sysmex Corporation, Kobe, Japan) that has high precision and reproducibility. Hemoglobin quantification was performed with an automated capillary electrophoresis (CE) system (CapillaryS2; Sebia, Lisses, France) that has high sensitivity.
Genetics detection
Common α-globin [–α3.7 (rightward), –α4.2 (leftward), – –SEA (Southeast Asian), Hb Constant Spring (Hb CS or HBA2: c.427T>C), Hb Quong Sze (Hb QS or HBA2: c.377T>C) and Hb Westmead or HBA2: c.369C>G], and β-globin [codons 41/42 (–TTCT) (HBB: c.126_129delCTTT), IVS-II-654 (C>T) (HBB: c.316–197C>T) −28 (A>G) (HBB: c.−78A>G), codons 71/72 (+A) (HBB: c.216_217insA), codon 17 (AAG>TAG) (HBB: c.52A>T), codon 26 (GAG>AAG) (Hb E or HBB: c.79G>A), codon 31 (–C) (HBB: c.94delC), codons 27/28 (+C) (HBB: c.84_85insC), IVS-I-1 (G>T) (HBB: c.92+1(G>T), codon 43 (GAG>TAG) (HBB: c.130G>T), −32 (C>A) (HBB: c.−82>A), −29 (A>G) (HBB: c.−79A>G), −30 (T>C) (HBB: c.−80T>C), codons 14/15 (+G) (HBB: c.45_46insG), Cap+40–43 (–AAACA) (HBB: c.−11_-8delAAACA), initiation codon (ATG>AGG) (HBB: c.2T>G) and IVS-I-5 (G>C) (HBB: c.92+5G>C)] [Citation7], mutations were detected with kits from Yaneng Biosciences, Shenzen, PRC. Multiple ligation-dependent probe amplification (MLPA) (SALSA MLPA KIT P102 D1 HBB; MRC Holland BV; Amsterdam, The Netherlands) [Citation8], was used to detect rearrangements of the β-globin gene cluster according to the manufacturer’s instructions. Deletions were determined with a ratio <0.7, whereas duplications were determined with a ratio>1.3.
Single molecule real-time sequencing
We used SMRT sequencing to detect the breakpoints of the deletion. According to the results of MLPA, we designed the following primers to amplify the target region, forward primer: 5′-CGT GAC TGA ATT TTA CCT TCA CAC CTA A-3′, reverse primer: 5′-AAG ACT AGG CAG TTA CAC TGG AGG-3′. Then the library was prepared by the addition of loop adapters to the DNA. Sequencing was performed on a PacBio Sequel System the from Berry Genomic Corporation (Beijing, PRC).
Breakpoint identification
We used gap-polymerase chain reaction (gap-PCR) to amplify the DNA encompassing the deletion breakpoints. The primers used were as follows, forward primer: 5′-CAC ACA GAC CAG CAC GTT G-3′, reverse primer: 5′-TGC CGA GCA CAC ACA ATT AC-3′. A unique 600 bp fragment was amplified in the proband and subsequently sequenced.
Results
The proband was a 28-year-old man with hypochromic polyglobulia and microcytic RBCs. Hematological analysis indicated a normal Hb level (13.8 g/dL), the mean corpuscular volume (MCV) and mean corpuscular Hb (MCH) values were 67.7 fL and 20.3 pg, respectively. Hemoglobin quantification indicated slightly elevated Hb F (4.3%) and Hb A2 (7.8%) (). However, genetic analysis detecting common β-thal mutations with PCR-reverse dot blot hybridization indicated a negative β-thal result. Therefore, MLPA was performed to detect the rearrangements of the β-globin gene cluster; the results revealed a long deletion similar to the Taiwanese deletion in β-thal (). To identify the breakpoint of the deletion, we performed SMRT sequencing, a long read, single molecule method. The SMRT analysis recapitulated the MLPA results and indicated an approximately 5 kb deletion extending from 4 kb upstream of the 5'UTR of the HBB gene to the second intron [. The proposed positions of the deletion were located between 5226189 and 5231091 on chromosome 11 (NC_000011.10: g.5226189_5231091del, GRCh38). To confirm the results of SMRT sequencing, we used gap-PCR to amplify the DNA encompassing the deletion and then performed Sanger sequencing [ and ]. The analysis of the breakpoints indicated that the 5′ breakpoint was within an AT-rich region that might play a role in recombination [Citation5] []. This novel deletion has been submitted to the dbVar database (http://www.ncbi.nlm.nih.gov/dbvar/), accession number nstd219.
Figure 1. Multiple ligation-dependent probe amplification analysis showed a heterozygous deletion in the β-globin cluster. Deletions were determined with a ratio <0.7. The deletion region extended from probe HBB up 337 bp to HBB EXON 2 196 bp.
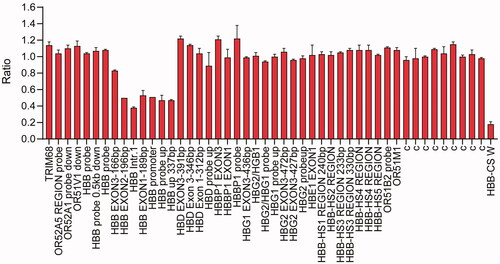
Figure 2. Molecular characterization of the 5 kb deletion. (A) Single molecule real-time sequencing analysis of the patient. The light yellow and blue regions indicate the two alleles of the β-globin gene cluster. The arrows indicate the region of the deletion. The blue dots indicate sequencing errors. The relative positions of the HBD, HBB and AC104389.7 genes on chromosome 16 are indicated by blue boxes. The vertical colored lines indicate nucleotides A (green), T (red), C (blue) and G (orange) discordant with alignment to the Hg38 reference sequence. (B) Gap-PCR analysis showing a unique 600 bp product in the proband. M: marker; N: normal control. (C) Sanger sequencing results of the deletion. The arrows indicate the breakpoints of the deletion.
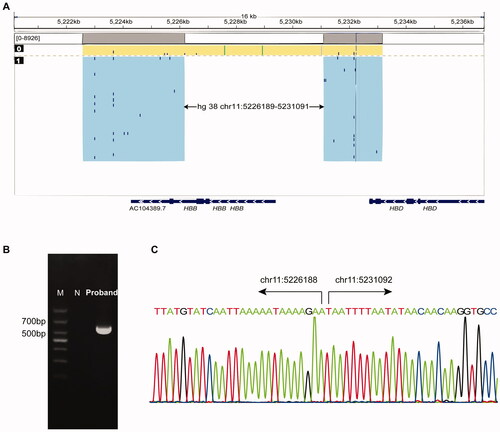
Table 1. The hematological data of the patient.
Discussion
Because of the technical limitations in previous studies, the precise breakpoints of many large deletions in the β-globin gene cluster have not been identified [Citation9]. Owing to the rapid development of next-generation sequencing and SMRT sequencing, the breakpoints of large deletions can now be precisely detected. In this study, we identified a novel 5 kb deletion covering the region upstream of the HBB promoter and the second intron by using MLPA and SMRT sequencing.
Many large deletions have been reported to be associated with elevated Hb F [Citation5,Citation10,Citation11]. According to the HbVar database and a literature search, large deletions in the β-globin gene cluster in Chinese patients are very rare, and only nine such types of deletions have been found [Citation3,Citation5,Citation11–17] (). Of these deletions, the Chinese Gγ(Aγδβ)0-thal and Southeast Asian-hereditary persistence of fetal Hb (SEA-HPFH), which also are associated with elevated Hb F, are the most common large deletions in the Chinese population. In addition, the Taiwanese deletion, a 1357 bp deletion extending from 548 bp upstream of the Cap site to +810 in the second intron on the β-globin gene, has been reported to be associated with relatively high Hb F levels. The underlying mechanism driving the increase in Hb F and Hb A2 in large deletional β-thal remains unclear. We propose that long distance regulation might be a mechanism potentially explaining the elevated expression of Hb F and Hb A2. Normally, the genes in the β-globin gene cluster, including HBB, HBG and HBD, competitively interact with the enhancer element in the 5'DNaseI hypersensitive sites (5'HS) region, which upregulates the expression of those genes. With HBB promoter deletion, the remaining HBG and HBD might have greater interaction with the enhancer element, thus increasing the expression of Hb F or Hb A2. This hypothesis appears to be supported by the Hb F and Hb A2 levels in the proband in our study, and two deletions (1605 and 1393 bp) [Citation5] reported in 1993 and 1988, respectively.
Figure 3. The known deletions found in the Chinese population. Large deletions are shown as horizontal red boxes. The red dotted lines indicate that the endpoint has not been determined.
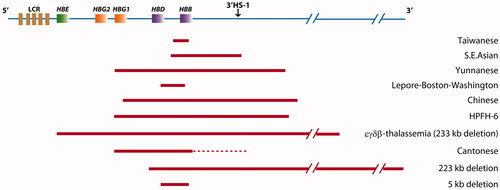
In this study, the proband presented with only hypochromic polyglobulia and microcytic RBCs. One limitation of this study is that we were unable to collect blood samples from the patient’s parents; therefore, understanding of the influence of this large deletion is incomplete. Furthermore, this is the first report of this large deletion; therefore, more cases might be required to confirm our findings. In conclusion, our study broadens the spectrum of deletional β-thalassemias and provides a perspective for further study of the function of the β-globin cluster.
Authors’ contributions
X-Q. Bao performed the experiments, analyzed the data and wrote the manuscript; J-C. Wang, D-Q. Qin, C-Z. Yao and J. Liang performed the experiments and analyzed the data. L. Du designed the study and revised the paper.
Acknowledgements
We thank the patient for his willingness to participate in this study.
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
Additional information
Funding
References
- Shang X, Peng Z, Ye Y, et al. Rapid targeted next-generation sequencing platform for molecular screening and clinical genotyping in subjects with hemoglobinopathies. EBioMedicine. 2017;23:150–159.
- Ko TM, Xu X. Molecular study and prenatal diagnosis of α- and β-thalassemias in Chinese. J Formos Med Assoc. 1998;97(1):5–15.
- Xu XM, Li ZQ, Liu ZY, et al. Molecular characterization and PCR detection of a deletional HPFH: application to rapid prenatal diagnosis for compound heterozygotes of this defect with beta-thalassemia in a Chinese family. Am J Hematol. 2000;65(3):183–188.
- Zhang XQ, Zhang JW. The 3' breakpoint of the yunnanese (Agammadeltabeta)0-thalassemia deletion lies in an L1 family sequence: implications for the mechanism of deletion and the reactivation of the Ggamma-globin gene. Hum Genet. 1998;103(1):90–95.
- Huang C-H, Chang Y-Y, Chen C-H, et al. Molecular characterization of a β-globin gene deletion of 1357 bp in a Taiwanese β-thalassemia carrier. Hemoglobin. 2008;32(5):498–504.
- Xu L, Mao A, Liu H, et al. Long-molecule sequencing: a new approach for identification of clinically significant DNA variants in α-thalassemia and β-thalassemia carriers. J Mol Diagn. 2020;22(8):1087–1095.
- Giardine B, Borg J, Viennas E, et al. Updates of the HbVar database of human hemoglobin variants and thalassemia mutations. Nucleic Acids Res. 2014;42(Database issue):D1063–D1069.
- Cui J, Azimi M, Baysdorfer C, et al. Application of multiplex ligation-dependent probe amplification to screen for β-globin cluster deletions: detection of two novel deletions in a multi ethnic population. Hemoglobin. 2013;37(3):241–256.
- Rooks H, Clark B, Best S, et al. A novel 506kb deletion causing εγδβ thalassemia. Blood Cells Mol Dis. 2012;49(3–4):121–127.
- Sankaran VG, Xu J, Byron R, et al. A functional element necessary for fetal hemoglobin silencing. N Engl J Med. 2011;365(9):807–814.
- Zhu F, Wei X, Cai D, et al. A novel 223 kb deletion in the beta-globin gene cluster was identified in a Chinese thalassemia major patient. Int J Lab Hematol. 2019;41(4):456–460.
- Hui ASY, Au PKC, Ting Y-H, et al. First report of a novel deletion due to εγδβ-thalassemia in a Chinese family. Hemoglobin. 2017;41(3):175–179.
- Lou J-W, Li Q, Wei X-F, et al. Identification of the linkage of a 1.357 KB β-globin gene deletion and Aγ-globin gene triplication in a Chinese family. Hemoglobin. 2010;34(4):343–353.
- Mager DL, Henthorn PS, Smithies O. A Chinese G gamma + (A gamma delta beta)zero thalassemia deletion: comparison to other deletions in the human beta-globin gene cluster and sequence analysis of the breakpoints. Nucleic Acids Res. 1985;13(18):6559–6575.
- Zhang JW, Song WF, Zhao YJ, et al. Molecular characterization of a novel form of (A gamma delta beta)zero thalassemia deletion in a Chinese family. Blood. 1993;81(6):1624–1629.
- Jiang F, Zuo L, Li D, et al. Molecular epidemiology and hematologic characterization of δβ-thalassemia and hereditary persistence of fetal hemoglobin in 125,661 families of greater Guangzhou area, the metropolis of southern China. BMC Med Genet. 2020;21(1):43.
- Huang G, Jiang W-L, Rong K-B, et al. Molecular characterization of a Chinese pedigree with β-thalassemia intermedia. Hemoglobin. 2010;34(2):179–183.