Abstract
Soybean mosaic virus (SMV; Potyvirus, Potyviridae) is a plant pathogenic virus that infects many commercially important plants worldwide, including soybean, and impairs their production and quality. SMV can be classified into strains based on differences in virulence. Many of these strains have been sequenced; however, only a few complete genomic sequences of virus strains and isolates from soybean grown in China have been reported. In this study, the complete genomic sequences of three SMV isolates of different virulence from plants in China were determined and compared with the sequences of 54 other SMV strains and isolates. Genomic nucleotide diversity analysis and the Neutrality Test showed that the sequence of the 5′ untranslated regions (5′UTR) of the virus genome-linked protein (VPg) and first protein (P1) genes had been altered by negative selection pressure. Phylogenetic analyses indicated that sequence variation in the cytoplasmic inclusion (CI) cistron of the virus was correlated closely with the level of virulence and with the source of the virus isolate. The CI gene was found to be crucial for classification and characterization of the SMV strains, and may be a determining factor in the level of SMV virulence.
Résumé
Le virus de la mosaïque du soja (SMV : potyvirus, famille des potyviridés) est un virus pathogène qui infecte plusieurs cultures commerciales importantes partout dans le monde, y compris le soja, et qui en réduit la production et la qualité. Le SMV peut être classé en différentes souches, selon sa virulence. Plusieurs de ces souches ont été séquencées, toutefois, on ne connaît que quelques séquences génomiques complètes des souches du virus et des isolats qui s'attaquent aux variétés de soja cultivées en Chine. Dans cette étude, les séquences génomiques complètes de 3 souches de SMV de virulence différente et de leurs isolats, provenant de plants cultivés en Chine, ont été déterminées et comparées aux séquences de 54 autres souches et isolats de SMV. L'analyse de la diversité des nucléotides génomiques et le test de neutralité ont montré que la séquence des régions 5′ non traduites (5′ UTR) de la protéine du virus lié au génome (VPg) et des gènes de la première protéine (P1) a été altérée par pression de sélection négative. Les analyses phylogénétiques ont indiqué que la variation de la séquence du cistron de l'inclusion cytoplasmique (CI) du virus était étroitement corrélée avec le degré de virulence et la source de l'isolat du virus. Le gène du CI s'est avéré essentiel pour la classification et la caractérisation des souches de SMV et peut être un facteur déterminant du degré de virulence de ce virus.
Introduction
Soybean mosaic virus (SMV) belongs to the genus Potyvirus and the family Potyviridae (King et al., Citation2011) and is one of the most prevalent virus diseases of soybean; it infects crops globally and severely impairs their production yield and quality. SMV is transmitted by aphids or infected seed. After infection by SMV, the soybean plants exhibit the characteristics of a dwarf plant, with narrow leaves and, in severe conditions, necrosis and tip death occurs. Consequently, yield is decreased dramatically. These symptoms mainly result from the interaction between SMV strains and the cultivars (Eggenberger et al., Citation2008; Seo et al., 2009a); however, environmental conditions also have a certain influence on symptoms.
Like other potyviruses, SMV is a single-stranded positive-sense RNA virus of approximately 10 000 nucleotides (nt) in length, and with a poly(A) tail at the 3′-terminus. The genome is translated from a single open reading frame (ORF) into a polyprotein, which is cleaved further and processed to form 11 maturation proteins (i.e. P1, HC-Pro, P3, P3-PiPo, 6K1, CI, 6K2, NIa-Pro, NIa-VPg, NIb and CP) that have different functions (Adams et al., Citation2005; Chung et al., Citation2008).
The virulence of SMV has diverged during the virus's long-term co-evolution with its host to produce groups of viruses that show different levels of virulence, each group being considered as a different strain. In the USA, SMV has been classified into seven strains (G1–G7), based on the symptoms found following inoculation of differential types of soybean cultivars (Cho & Goodman, Citation1979). Scientists in the Republic of Korea and in the USA follow the same SMV strain differential cultivar system. In China, the National Center for Soybean Improvement (NCSI) uses its own differential cultivar system, which classifies SMV found in China into 21 strains (Li et al., 2010).
Currently, scientists in the USA have completed the sequencing of SMV G1−G7 and some other strains and isolates, and Korean researchers also have reported the complete sequence of certain strains and of 24 isolates of SMV (Seo et al., 2009b). In contrast, in China, only a limited number of complete genomic sequences, namely from isolates HZ1 (GenBank no. AJ628750) (Shi et al., 2005), HZ (GenBank no. AJ312439), HH5 (GenBank no. AJ310200) (Chen et al., 2004), SC6 (GenBank no. HM590054), 4469-4 (GenBank no. HM590055) and 4547 (GenBank no. HQ396725) (Yang et al., 2011) have been deposited in the database.
Determination and analysis of the complete genomic sequence of SMV strains will provide insight into variation in its structure and genes and help to identify mechanisms involved in infection and virulence. In addition, analysis of the complete sequence of the SMV population will provide useful information on gene stability and evolution (Desbiez & Lecoq Citation2008; Ogawa et al., Citation2008; Seo et al., 2009b). Such analysis has become possible as more data accumulate in the gene databases. In this study, we characterized the disease reactions of three isolates of different virulence (strains) from 10 differential resistant soybean cultivars and sequenced the three genomes. These sequences were compared with the complete genome sequences of all SMVs in the National Center for Biotechnology Information database (NCBI) to identify molecular differences among strains and to explore the association between virus genome sequence and its virulence and geographical distribution.
Materials and methods
Virus isolates and origin
Three representative isolates (SC3, 6202-2 and 6067-1) that showed typical symptoms were selected for this study. All three isolates were provided by the National Center for Soybean Improvement, Nanjing Agricultural University. Isolates 6202-2 and 6067-1 were collected from Guangdong Province and Guangxi Zhuang Autonomous Region in China, respectively. The isolates were part of 201 found by Li et al. (Citation2010) and isolates 6202-2 and 6067-1 were classified as SC21 and SC15 strain groups, respectively (Li et al., 2010). Prior to sequence determination, we performed local-lesion isolation and purification of the three isolates both using ‘Topcrop’ and DAS-ELISA serum identification to ensure each culture contained a pure SMV. The virus isolates were propagated on the soybean cultivar ‘Nannong1138-2’; strain differentiation was based the protocol established by Zhan et al. (Citation2006). Inoculation and strain identification were carried out inside an insect-proof greenhouse.
Determination of complete genomic sequences
We performed multiple-sequence alignment of the full SMV sequences of strains and isolates deposited in the NCBI database using ClustalX 1.83 software. Conserved domains and regions suitable for primer design were identified. Primers were synthesized by Invitrogen (Shanghai) Co., Ltd; primer sequences and predicted amplified product size are listed in .
Table 1. Primers used in this study
SMV-infected leaves were harvested. Total RNA was extracted using the RNeasy Plant Mini Kit (Qiagen) and first strand cDNA was synthesized using M-MLV reverse transcriptase (TaKaRa) in accordance with manufacturer's instructions. The intermediate genomic sequences were amplified with five primer pairs (named Pr-1 to Pr-5) using Ex Taq enzyme (TaKaRa). Primers were designed such that adjacent sequences had at least a 150-bp overlap to ensure that samples originated from the same genome. Polymerase chain reaction (PCR) was performed for 35 thermocycles with the following steps: denaturation at 94ºC for 30 s, annealing at 55ºC for 30 s and polymerization at 72ºC for 60 s, and with the final extension step at 72ºC for 5 min.
Rap5′-RACE (rapid amplification of cDNA ends) was performed using a RACE kit (TaKaRa), and 3′-RACE was completed using the RNA PCR kit (AMV) (TaKaRa). All use of primers () and the procedures were carried out in accordance with the kit manual.
All amplification products were detected by 1% agarose gel electrophoresis; fragments were purified with the Qiagen's Qiaquick kit and then cloned into a pMD-19 vector. Sequence determination was completed by BGI-Shenzhen (China). Sequencing was performed at least twice for each fragment to ensure accuracy; more than two clones were sequenced completely for each fragment.
Complete sequence analysis
Sequences were assembled using BioXM software (version 2.6). Searches of the complete sequences were carried out using the NCBI database and the BLAST program to identify homologous sequences. Multiple-sequence alignments of nucleotide and amino acids were completed using ClustalX software (version 1.83) and BioEdit (version 7.0.5). Genomic nucleotide diversity analysis and the Neutrality Test were performed using DNASP 5.0. Phylogenetic analysis was based on the neighbour-joining (NJ) method, using MEGA software (version 4.0) and PAUP; the assembled phylogenetic tree was viewed using TREEVIEW software (version 3.2).
Results
Biological features of SMV isolates
The disease reaction of three isolates (SC3, 6202-2 and 6067-1) were verified on 10 differential soybean cultivars (), and findings compared with those of Li et al. (Citation2010). Isolate 6067-1 had the highest virulence and could infect all 10 cultivars. Furthermore, the symptoms shown by ‘Nannong 1138-2’ plants following strain 6067-1 virus infection were systemic necrosis and, on occasion, even tip death. Isolate 6202-2 exhibited moderate virulence and infected five differential cultivars (‘Nannong 1138-2’, ‘Youbian 30’, 8101, ‘Davis’ and ‘Buffalo’). Isolate SC3 had the lowest virulence and infected only three cultivars (‘Nannong 1138-2’, ‘Youbian 30’ and 8101). These three strains were representative of a range of virulence and therefore were chosen for use in this study.
Table 2. Responses of 10 differential soybean cultivars to the SC3, 6067-1(SC15) and 6202-2(SC21) Soybean mosaic virus (SMV) strains in China
Molecular biological features of SMV strains
The genomes of strains SC3, 6202-2 and 6067-1 were 9589, 9589 and 9588 nt in length, respectively. Each genome contained a single ORF, which started at nt 132, 133 or 133; untranslated regions (UTRs) were present at both the 5′- and 3′-termini. In addition, each genome had a poly(A) tail sequence at the 3′-terminus. All three strains encoded a polyprotein of 3067 amino acids (aa) in length. These sequences were submitted to the NCBI database, and accession numbers are listed in .
Table 3. SMV strains and isolates analyzed in this study
Genomic nucleotide diversity analysis and Neutrality Test
DNA polymorphism provides richer information on the nature of genetic evolution compared with that from amino acid polymorphisms; therefore, the analysis of DNA polymorphisms is the preferred option for evolutionary geneticists. In this study, we carried out nucleotide diversity analysis and Neutrality Tests to investigate SMV genome evolution and genetic variation under natural selection pressure.
We assessed the nucleotide variability of the SMV population by comparison of the complete genomic sequences of 57 SMV strains and isolates. The Pi value was calculated using DNASP 5.0 software and nucleotide diversity was estimated. We used the sliding windows approach to graph the Pi values of the 57 SMV strains and isolates () in order to understand clearly the genome polymorphism of various regions. The results showed that the 5′UTR, the P1 N-terminus 300-bp segment and the third protein (P3) C-terminus 200-bp segment exhibited the highest variability in the whole genome, while the nuclear inclusion ‘a’ protein (NIa-Pro), nuclear inclusion ‘b’ protein (NIb), 6K2 and coat protein (CP) genes had lower polymorphism. We estimated the SMV genetic variability for each gene region, based on the Tajima Neutrality Test (). The purpose of this test was to identify if the target DNA sequence followed the neutral evolution model (Tajima, Citation1989). Tajima's D value was used to estimate the deviation from neutral model expectation. We found that the D values of all genes were negative, which suggested that each gene was under negative selection pressure to varying degrees. The 5′UTR and P1 and helper component-proteinase (HC-Pro) genes may be under greater selection pressure, as they had the lowest D values.
Fig. 1. Pi values of the 57 Soybean mosaic virus (SMV) strains and isolates.
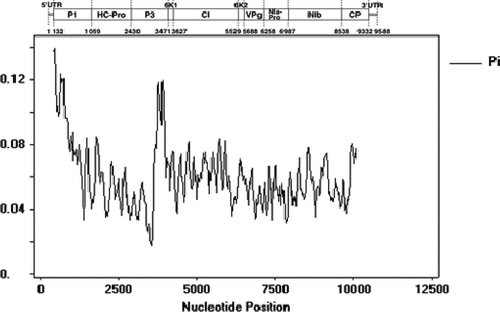
Table 4. Tajima's Neutrality test and Fu and Li's test for SMV gene region of 57 strains and isolates
The Fu and Li test method combines coalescent theory and can be used to track mutations in chronological order (Fu & Li, Citation1993); therefore, we also used this test to determine whether the SMV population met the neutral evolution model. We calculated Tajima's D values and Fu and Li's D* and F* values for different genes in the SMV, and test results are summarized in . Results showed that all the Fu and Li's D* values for the sequences were negative, which was in line with the Tajima's D values. Furthermore, Fu and Li's D* values for the 5′UTR and the VPg gene were negative significantly and P1 gene value was negative at a highly significant level. Therefore, these data indicated that these three genes had been influenced by negative selection pressure. Other genes were also shown to be affected by negative selection and to conform to the neutral evolution model. We found that Fu and Li's D* values were all lower than Tajima's D values, which indicated that the rate of mutation in SMV may have increased more recently.
Phylogenetic analyses
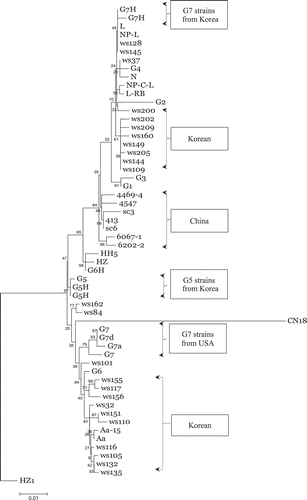
Phylogenetic analysis was performed based on the complete genomic sequences of the three SMV strains used in the present study and on 54 SMV sequences reported previously (). A phylogenetic tree was constructed based on the NJ method. The Pinellia HZ1 virus sequence (GenBank no. AJ628750) was used as a control outgroup (Shi et al., 2005), as it exhibited high variability compared with the SMV population and had high homology with Watermelon mosaic virus based BLAST analysis. The phylogenetic tree showed that the complete SMV genome nucleotide or polyprotein amino acid sequence alignments had no significant geographical association. In addition, SMV virulence did not correlate significantly with the complete genomic sequences. Phylogenetic analysis was also performed on the nucleotide and predicted amino acid sequences of the SMV genes. This analysis also showed no significant association with SMV strain, virulence or sample source. However, phylogenetic analysis of the CI gene alone showed that its nucleotide sequence correlated significantly with SMV virulence and sample source (). A NJ phylogenetic tree that was constructed based on the CI nucleotide sequence, six SMV strains and isolates from China, except for the SMV isolate HZ1, could be classified into one subgroup. Similarly, four isolates from Canada could be classified as one subgroup. Korean strains and isolates were classified into two subgroups; however, strains from the USA did not cluster as one group, due to their high sequence variation. The phylogenetic tree showed that SMV strains and isolates with the same virulence were grouped into the same subgroup and that SMV G2, G4, G5, G6, G7 and G7H could be classified into different subgroups. These results suggested that the CI gene is related closely to virulence and to the SMV sample source, and therefore this gene could serve as an aid to the taxonomic classification of SMV strains. The findings of NJ phylogenetic tree of the CI amino acid sequence were consistent with that of the nucleotide phylogenetic tree ().
Fig. 2. Neighbour-joining tree of the aligned cytoplasmic inclusion (CI) cistron nucleotides sequences of 57 Soybean mosaic virus (SMV) strains and isolates.
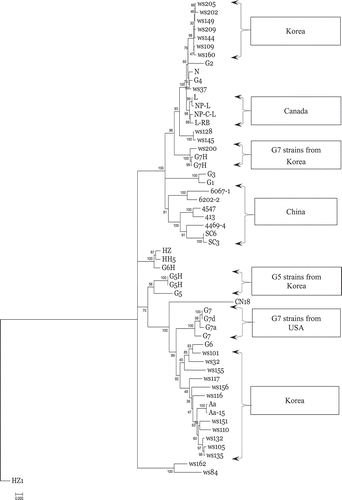
Fig. 3. Neighbour-joining tree of the aligned cytoplasmic inclusion (CI) cistron predicted amino acid sequences of 57 SMV strains and isolates.
Discussion
At the present time, the classification of SMV strains is based mainly on symptoms induced by isolates on the differential cultivars. The choice of cultivars used by different researchers varies both in China and worldwide; therefore, no uniform SMV strain classification system is available currently. Such diverse methods of classification severely limit international exchange of information, and pose problems for the breeding of disease-resistant soybean crops. Gene/allele identification has been reported for those cultivars used in the US-Korea system (Zheng et al., Citation2005). In this study, we determined the complete sequence of three SMV strains from China and found them to be homologous to those from other parts of the world. However, different strains had differences at both the nucleotide and amino acid levels. These three strains exhibited remarkably different disease reactions in differential cultivars.
The 5′UTR and the P1 gene had the least conserved SMV sequences, which may be related to their functions. Previous reports (Suehiro et al., Citation2004; Rajamkia et al., Citation2005) have demonstrated that the P1 and P3 proteins have functions that are related to disease dissemination and resistance, and sequences had been relatively loosely constrained during evolution (Seo et al., 2009b). The NIa-Pro, NIb, 6K2 and CP gene sequences were conserved, and had the lowest variability at the nucleotide level. Satendra et al. (Citation2008) found that for Papaya ringspot virus (PRSV), a significant proportion of recombination hotspots were located in the P1 gene, followed by P3 (Satendra et al., 2008). Furthermore, Yang et al. (Citation2011) found that the SMV 4469-4 strain, which is approximately 400 nt longer than strain Sc6 and other SMV strains, may result from recombination of SMV and Bean common mosaic virus (BCMV) or a BCMV-like virus at the N-terminus of the genome (Yang et al., 2011). Tajima test analysis showed that SMV genes were under negative selection pressure to a varying degree, and that the 5′UTR, and P1 and HC-Pro genes may be under greater selection pressure. Fu and Li's test results indicated that the 5′UTR and P1 and VPg genes were influenced by negative selection. Other genes were also affected by negative selection, but they conformed to the neutral evolution model. Fu and Li's D* values were all lower than the Tajima D values, which indicated that SMV may have a rate of mutation that has increased more recently. The cultivation of disease-resistant soybeans may speed up variation in SMV strains, especially for the 5′UTR and P1 and VPg genes.
Soybean is the natural host of SMV. This crop originated in China and was introduced to Europe and America after the year 1700 (Hymowitz & Harlan, Citation1983), and then later to Africa. The origin of SMV is believed to be consistent with that of soybean. In this study, we explored the relationship between the SMV complete genomic sequence and the classification of strains, sample sources and virus virulence. Phylogenetic analysis indicated that the SMV genome sequence had no significant correlation with geographical distribution at either the amino acid or nucleotide levels. However, a phylogenetic tree based on the nucleotide homology of the SMV CI gene, showed that its nucleotide sequence correlated significantly with the source of the SMV sample. These results showed that the Korean strains and isolates could be classified into two subgroups and may originate from different ancestors. Seo et al. (Citation2009b) reported previously that a NJ tree based on the CI sequence separated all 44 SMV strains into two major subgroups, probably based on response to cultivars that carried the Rsv3 gene. Our results support this view (Seo et al., 2009b) as our NJ tree did not separate the strains from China. This finding suggested the cultivars used to classify the viruses by virulence may not carry this gene. The United States SMV strains could be divided into several subgroups and their nucleotide homology was low. Soybean was not introduced to the USA from other countries until the late 1700s, which unavoidably facilitated the spread of SMV. This situation has resulted in a diverse genetic background of the SMV genome in the USA and may be the reason why some strains or isolates are classified into subgroups from other countries. Jain et al. (Citation1992) listed the countries of origin of the soybean germplasm and of any seed-borne virus from which many of the United States SMV strains reported by Cho & Goodman (Citation1979) were isolated. Thus, these strains may not be useful in determining geographic origin but should be valuable for detecting diversity.
The phylogenetic tree described in this study and based on the nucleotide homology of the SMV CI gene allowed the classification of strains G5, G6 and G7 into different subgroups, and led to the classification of two strains with low virulence (G1 and G3) into the same subgroup. In China, SC3 and SC6 (two strains with low virulence) belong to the same subgroup, while 6067-1 and 6202-2 (two strains with moderate virulence) are also classified into the same subgroup. We found that there were only four differences in the amino acid sequence of the CI protein between the two isolates. This situation may be the reason why the two isolates were classified into the same subgroup despite the fact that virulence differed. The G7 strain from Korea and the G7 strain from the USA were classified into different groups. We speculate therefore that the two G7 strains have undergone different evolutionary paths in these two countries. Different ecological factors may cause differences in host resistance and virus virulence in different geographical regions. The long-term co-evolution between SMV and its host has resulted in differences in SMV infectivity in different regions of the world. SMV strains from different geographical regions have different molecular and biological features and even the same strain from different regions can have different levels of virulence. The rapid variation in the virus sequence and the effect of the ecological environment have resulted in complex changes in the SMV genome during long-term co-evolution; therefore, it is difficult to classify strains simply based on the complete genome. Interestingly, this study has shown that the CI gene is a good aid to the classification of SMV strains, due to its high sequence conservation and its relationship with virulence. Interestingly, Hunst & Tolin (Citation1983) found that the accumulation of the CI protein in pinwheel inclusion bodies is a unique feature of SMV infection.
The functional products of the CI gene exhibit NTPase and RNA helicase activities and can facilitate intercellular transport and genome replication (Carrington et al., Citation1998; Gómez de Cedrón et al., 2006). The C-terminus of the CI is involved in the long-distance transport of the virus (Spetz & Valkonen, Citation2004). SMV strain Ws200 (GenBank no.:FJ548849) is regarded as an Rsv-3-mediated resistance-breaking strain, and its resistance results from a mutation in CI (Seo et al., 2009a). There is also speculation that CI may be an Rsv-3-mediated resistance-breaking determinant (Seo et al., 2009a). Zhang et al. (Citation2009) constructed chimeras by exchanging fragments between avirulent SMV-G7 and the virulent SMV-N to identify virulence determinant(s) of SMV on an Rsv3-genotype soybean. Analysis of the chimeras showed that both the N- and C-terminus regions of the CI cistron are required for Rsv3-mediated resistance (Zhang et al., 2009). In addition, Jenner et al. (Citation2000, Citation2002) demonstrated that mutations in the CI gene could overcome Turnip mosaic virus (TuMV)'s resistance mediated by TuRB01. CI genes from other members of the Potyvirus, including Plum pox virus (PPV) and Tobacco etch virus (TEV), have been reported to be related to host determinants (Bilgin et al., Citation2003; Jiménez et al., Citation2006). These findings by other groups provided support for the results of our current study.
Acknowledgements
The study was supported by grants from the National Natural Science Foundation of China (Nos 30971815, 31171574), the MOE 111 Project (No. B08025), the National Soybean Industrial Technology System of China (No. CARS-004), and the Fund for Transgenic Breeding of Soybean Resistant to Soybean Mosaic Virus (No. 2008ZX08004-004).
Notes
†Both authors contributed equally to this work.
References
- Adams , M.J. , Antoniw , J.F. and Beaudoino , F. 2005 . Overview and analysis of the polyprotein cleavage sites in the family Potyviridae . Mol. Plant Pathol , 6 : 471 – 487 .
- Bilgin , D.D. , Liu , Y. , Schiff , M. and Dinesh-Kumar , S.P. 2003 . P58IPK, a plant ortholog of double-stranded RNA-dependent protein kinase PKR inhibitor, functions in viral pathogenesis . Dev. Cell , 4 : 651 – 661 .
- Carrington , J.C. , Jensen , P.E. and Schaad , M.C. 1998 . Genetic evidence for an essential role for Potyvirus CI protein in cell-to-cell movement . Plant J. , 14 : 393 – 400 .
- Chen , J. , Zheng , H.Y. , Lin , L. , Adams , M.J. , Antoniw , J.F. Zhao , M.F. 2004 . A virus related to Soybean mosaic virus from Pinellia ternata in China and its comparison with local soybean SMV isolates . Arch. Virol , 149 : 349 – 363 .
- Cho , E.K. and Goodman , R.M. 1979 . Strains of Soybean mosaic virus: classification based on virulence in resistant soybean cultivars . Phytopathology , 69 : 467 – 470 .
- Choi , B.K. , Koo , J.M. , Ahn , H.J. , Yum , H.J. , Choi , C.W. Ryu . 2005 . Emergence of Rsv-resistance breaking Soybean mosaic virus isolates from Korean soybean cultivars . Virus Res. , 112 : 42 – 51 .
- Chowda-Reddy , R.V. , Sun , H. , Chen , H. , Poysa , V. , Ling , H. , Gijzen , M. and Wang , A. 2011 . Mutations in the P3 protein of Soybean mosaic virus G2 isolates determine virulence on Rsv4-genotype soybean . Mol. Plant-Microb. Interact. , 24 : 37 – 43 .
- Chung , B.Y.W. , Miller , W.A. , Atkins , J.F. and Firth , A.E. 2008 . An overlapping essential gene in the . Potyviridae. Proc. Nat. Acad. Sci. U.S.A , 105 : 5897 – 5902 .
- Desbiez , C. and Lecoq , H. 2008 . Evidence for multiple intraspecific recombinants in natural populations of Watermelon mosaic virus (WMV, Potyvirus) . Arch. Virol. , 153 : 1749 – 1754 .
- Eggenberger , A.L. , Hajimorad , M.R. and Hill , J.H. 2008 . Gain of virulence on Rsv1-genotype soybean by an avirulent Soybean mosaic virus requires concurrent mutations in both P3 and HC-Pro . Mol. Plant-Microb. Interact , 21 : 931 – 936 .
- Eggenberger , A.L. , Stark , D.M. and Beachy , R.N. 1989 . The nucleotide sequence of a Soybean mosaic virus coat protein-coding region and its expression in Escherichia coli, Agrobacterium tumefaciens and tobacco callus . J. Gen. Virol. , 70 : 1853 – 1860 .
- Fu , Y.X. and Li , W.H. 1993 . Statistical tests of neutrality of mutations . Genetics , 133 : 693 – 709 .
- Gagarinova , A.G. , Babu , M. , Poysa , V. , Hill , J.H. and Wang , A. 2008 . Identification and molecular characterization of two naturally occurring Soybean mosaic virus isolates that are closely related but differ in their ability to overcome Rsv4 resistance . Virus Res. , 138 : 50 – 56 .
- Gómez De Cedrón , M. , Osaba , L. , López , L. and García , J.A. 2006 . Genetic analysis of the function of the Plum pox virus CI RNA helicase in virus movement . Virus Res. , 116 : 136 – 145 .
- Hajimorad , M.R. , Eggenberger , A.L. and Hill , J.H. 2003 . Evolution of Soybean mosaic virus-G7 molecularly cloned genome in Rsv1-genotype soybean results in emergence of a mutant capable of evading Rsv1-mediated recognition . Virology , 314 : 497 – 509 .
- Hunst , P.L. and Tolin , S.A. 1983 . Ultrastructural cytology of soybean infected with mild and severe strains of . Soybean mosaic virus. Phytopathology , 73 : 615 – 619 .
- Hymowitz , T. and Harlan , J.R. 1983 . Introduction of soybean to North America by Samuel Bowen in 1765 . Econ. Bot. , 37 : 371 – 379 .
- Jain , R.K. , Mckern , N.M. , Tolin , S.A. , Hill , J.H. , Barnett , O.W. Tosic , M. 1992 . Confirmation that fourteen Potyvirus isolates from soybean are strains of one virus by comparing coat protein peptide profiles . Phytopathology , 82 : 294 – 299 .
- Jayaram , C. , Hill , J.H. and Miller , W.A. 1992 . Complete nucleotide sequences of two Soybean mosaic virus strains differentiated by response of soybean containing the Rsv resistance gene . J. Gen. Virol. , 73 : 2067 – 2077 .
- Jenner , C.E. , Sánchez , F. , Nettleship , S.B. , Foster , G.D. , Ponz , F. and Walsh , J.A. 2000 . The cylindrical inclusion gene of Turnip mosaic virus encodes a pathogenic determinant to the Brassica resistance gene TuRB01 . Mol. Plant–Microbe Interact , 13 : 1102 – 1108 .
- Jenner , C.E. , Tomimura , K. , Ohshima , K. , Hughes , S.L. and Walsh , J.A. 2002 . Mutations in Turnip mosaic virus P3 and cylindrical inclusion proteins are separately required to overcome two Brassica napus resistance genes . Virology , 300 : 50 – 59 .
- Jiménez , I. , López , L. , Alamillo , J.M. , Valli , A. and García , J.A. 2006 . Identification of a Plum pox virus CI-interacting protein from chloroplast that has a negative effect in virus infection . Mol. Plant–Microbe Interact. , 19 : 350 – 358 .
- King , A.M.Q. , Adams , M.J. , Carstens , E.B. and Lefkowitz , E.J. 2011 . Virus Taxonomy. 9th report of the International Committee on Taxonomy of Viruses , San Diego : Elsevier Academic Press .
- Li , K. , Yang , Q.H. , Zhi , H.J. and Gai , J.Y. 2010 . Identification and distribution of Soybean mosaic virus strains in Southern China . Plant Dis. , 94 : 351 – 357 .
- Lim , W.S. , Kim , Y.H. and Kim , K.H. 2003 . Complete genome sequences of the genomic RNA of Soybean mosaic virus strains G7H and G5 . Plant Pathol. J. , 19 : 171 – 176 .
- Ogawa , T. , Tomitaka , Y. , Nakagawa , A. and Ohshima , K. 2008 . Genetic structure of a population of Potato virus Y inducing potato tuber necrotic ring spot disease in Japan; comparison with North American and European populations . Virus Res. , 131 : 199 – 212 .
- Rajamkia , M.L. , Kelloniemia , J. , Alminaitea , A. , Kekarainen , T. , Rabenstein , F. and Valkonen , J.P.T. 2005 . A novel insertion site inside the Potyvirus P1 cistron allows expression of heterologous proteins and suggests some P1 functions . Virology , 342 : 88 – 101 .
- Satendra , K. , Mangrauthia , B. , Parameswari , R. , Jain , K. and Praveen , S. 2008 . Role of genetic recombination in the molecular architecture of . Papaya ringspot virus. Biochem Genet. , 46 : 835 – 846 .
- Seo , J.-K. , Lee , S.-H. and Kim , K.-H. 2009a . Strain-specific cylindrical inclusion protein of Soybean mosaic virus elicits extreme resistance and a lethal systemic hypersensitive response in two resistant soybean cultivars . Mol. Plant–Microbe Interact. , 22 : 1151 – 1159 .
- Seo , J-K. , Ohshima , K. , Lee , H-G. , Son , M. , Choi , H-S. Lee , S-HE. 2009b . Molecular variability and genetic structure of the population of Soybean mosaic virus based on the analysis of complete genome sequences . Virology , 393 : 91 – 103 .
- Shi , Y.H. , Hong , X.Y. , Chen , J. , Adams , M.J. , Zheng , H.Y. Lin , L. 2005 . Further molecular characterisation of potyviruses infecting aroid plants for medicinal use in China . Arch. Virol. , 150 : 125 – 135 .
- Spetz , C. and Valkonen , J.P. 2004 . Potyviral 6K2 protein long-distance movement and symptom-induction functions are independent and host-specific . Mol. Plant–Microbe Interact. , 17 : 502 – 510 .
- Suehiro , N. , Natsuaki , T. , Watanabe , T. and Okuda , S. 2004 . An important determinant of the ability of Turnip mosaic virus to infect Brassica spp. and/or Raphanus sativus is in its P3 protein . J. Gen. Virol. , 85 : 2087 – 2098 .
- Tajima , F. 1989 . Statistical methods to test for nucleotide mutation hypothesis by DNA polymorphism . Genetics , 123 : 585 – 595 .
- Yang , Y.Q. , Gong , J.W. , Li , H.W. and Zhi , H.J. 2011 . Identification of a novel Soybean mosaic virus isolate in China that contains a unique 5′ terminus sharing high sequence homology with Bean common mosaic virus. Virus Res . 157 : 13 – 18 .
- Zhan , Y. , Zhi , H.J , Yu , D.Y. and Gai , J.Y. 2006 . Identification and distribution of SMV strains in Huang-Huai Valleys . Sci. Agric. Sin , 39 : 2009 – 2015 .
- Zhang , C.Q. , Hajimorad , M.R. , Eggenberger , A.L. , Tsang , S. , Whitham , S.A. and Hill , J.H. 2009 . Cytoplasmic inclusion cistron of Soybean mosaic virus serves as a virulence determinant on Rsv3-genotype soybean and a symptom determinant . Virology , 391 : 240 – 248 .
- Zheng , C. , Chen , P. and Gergerich , R. 2005 . Characterization of resistance to Soybean mosaic virus in diverse soybean germplasm . Crop Sci. , 45 : 2503 – 2509 .