
Abstract
MicroRNAs (miRNAs) regulate gene expression at the post-transcriptional level and play a critical role in many important biological processes of plants. Wheat stripe rust is one of the most destructive fungal diseases of wheat worldwide, yet the roles of wheat miRNAs in response to Puccinia striiformis f. sp. tritici (Pst) are largely unknown. Here, we report a simple array platform that could detect 188 plant miRNAs in 95 miRNAs families from eight plant species. We identified two new members of conserved miRNAs families and five known miRNAs using the platform and RNA gel blot analysis. The transcript accumulation of seven miRNAs was detected in wheat leaves ‘Suwon 11’ inoculated with Pst using stem-loop real-time quantitative PCR (RT-qPCR). By analysing their predicted target genes, we discuss and propose the basal roles for miRNAs in the interaction between wheat and Pst, with most of the target genes being stress-related.
Résumé
Les microARN (miARN) régulent l’expression génique après la transcription et jouent un rôle primordial dans de nombreux processus biologiques chez les plantes. La rouille jaune est une des maladies fongiques les plus destructrices du blé, et ce, à l’échelle de la planète, mais, malgré cela, les rôles joués par les miARN chez cette céréale quant à leur réaction à Puccinia striiformis f. sp. tritici (Pst) sont très peu connus. Dans cet article, on fait état d’une biopuce qui pourrait détecter 188 miARN de plantes appartenant à 95 familles de miARN provenant de 8 espèces de plantes. À l’aide de la biopuce et par transfert de type Northern, nous avons identifié deux nouveaux membres de familles de miARN conservées et cinq miARN connus. L’accumulation des transcrits de sept miARN a été détectée par PCR quantitative en temps réel (RT-qPCR), avec amorces tige-boucle, dans des feuilles de blé ‘Suwon 11’ inoculées avec Pst. En analysant leurs gènes cibles prévus, nous examinons et clarifions les rôles fondamentaux des miARN dans les interactions blé-Pst relativement à la plupart des gènes cibles liés au stress.
Introduction
Wheat (Triticum aestivum L.) is a widely grown crop plant whose yield and grain quality are greatly impacted by various biotic and abiotic stresses (Xin et al. Citation2010). Wheat stripe rust, caused by Puccinia striiformis Westend. f. sp. tritici Eriks. (Pst), is one of the most destructive diseases of wheat, which significantly affects yield. Understanding the interactions between wheat and Pst is important for devising strategies to improve disease resistance. To reduce the damage caused by various types of stresses, plants have evolved many adaptive response mechanisms to improve their tolerance and resistance capacity at the transcriptional, post-transcriptional and post-translational levels (Shukla et al. Citation2008). For example, a number of wheat genes were significantly induced or repressed in response to Pst (Ma et al. Citation2009; Wang et al. Citation2009). Although microRNAs (miRNAs) have recently emerged as important post-transcriptional regulators in plant stress responses (Xin et al. Citation2010), the roles of miRNAs in the wheat response to Pst are largely unknown.
miRNAs are a class of endogenous, small, non-coding and fundamental regulatory RNAs that act at the post-transcriptional level in plants and animals (Bartel Citation2004). Since 2002, many miRNAs have been identified in various plants, such as Arabidopsis thaliana (Liu et al. Citation2008), strawberry (Li et al. Citation2009), rice (Sunkar et al. Citation2005), soybean (Zeng et al. Citation2010), maize (Mica et al. Citation2006) and wheat (Yao et al. Citation2007). Many plant miRNAs are highly conserved between the monocotyledonous and dicotyledonous species (Axtell & Bartel Citation2005; Zhang et al. Citation2006) and also share tissue-specific, developmental stage-specific or disease-specific patterns (Yin et al. Citation2008). These miRNAs have been shown to play necessary roles in plants by negatively regulating target genes during various biological processes, such as organ development, phase change, signal transduction and in response to a variety of biotic and abiotic stresses (Jin Citation2008; Ma et al. Citation2009; Padmanabhan et al. Citation2009; Lv et al. Citation2010).
At present, RNA sequencing is the state-of-the art method to detect novel miRNAs, but the large amount of data requires a lot of time to analyse. The miRNAs can be detected using microarrays and RNA gel blots if the sequences of the homologous species are known, since miRNAs share high homology among species. In recent studies, these technologies have been used for detecting miRNAs successfully. Undoubtedly, microarray has made important contributions to both basic and applied research (Ekins & Chu Citation1999; Katagiri & Glazebrook Citation2009), and several studies have demonstrated that miRNA microarray can be successfully applied to assess the expression of hundreds of miRNAs on a global scale in a single assay. miRNA arrays are now being developed to explore the biogenesis of miRNAs, tissue distribution and differential miRNA expression between normal and abnormal states (Yin & Zhao Citation2007; Xu et al. Citation2012). The recent development of stem-loop primers for RT-qPCR provides another tool for detecting and characterizing mature miRNAs (Chen et al. Citation2005). Due to the convenience and sensitivity of this method, it has often been used in studies involving the expression of small RNAs (Peltier & Latham Citation2008; Galiveti et al. Citation2010).
Since 2006, several studies have shown that small RNAs play a critical role in bacterial disease resistance responses in Arabidopsis (Navarro et al. Citation2006; Jin Citation2008; Padmanabhan et al. Citation2009). However, research on the plant miRNAs involved in fungal disease resistance, especially for wheat-Pst interactions, has been scarce. The purpose of this study was to identify the conserved miRNAs involved in the wheat-Pst interactions and to examine the expression profiles of wheat miRNAs under Pst stress. Furthermore, we discussed how these miRNAs modulate defence mechanisms in wheat during Pst infection.
Materials and methods
Plant materials and treatments
For the biotic stress treatment in wheat cultivar ‘Suwon 11’, Pst races of Chinese yellow rust 23 (CYR23) and 31 (CYR31) were used for the incompatible and compatible interactions, respectively. The plants were grown in a growth chamber with a 16-h photoperiod (photosynthetic photon flux density, 36 μmol m−2 s−1) at 16°C. Seven-day-old seedlings at the primary leaf stage were separately inoculated with fresh urediniospores of CYR23 and CYR31 using a paintbrush; control plants were inoculated with sterile water. The inoculated and control plants were kept for 24 h at 100% humidity and then maintained in the growth chamber. Wheat leaves were excised at 0, 12, 24, 48, 72 and 120 hours post-inoculation (hpi), quickly frozen in liquid nitrogen and stored at −80°C. Three independent biological replications were performed for each treatment.
microRNA array and RNA gel blot analysis
The microRNA array platform was designed and constructed by Iyer et al. (Citation2012). Following this principle and based on the known sequence information of miRNAs from the miRNA database (http://microrna.sanger.ac.uk/sequences), a total of 188 miRNAs for known miRNAs was selected, which covers the available conserved and unique plant miRNAs from 95 miRNA families of eight different plant species. Antisense probes for each miRNA (20 μM) were printed in duplicate using the Genetix Qpix2 robot on a Hybond-N+ membrane (Supplementary Table 1). The array platform included four external controls (MAC2, MAC3, MAC4 and MAC5) (Supplementary Table 2), which help to measure the efficiency of the microRNA array system.
The RNA sample extracted from wheat leaves was used for hybridization. Total RNA was extracted using the TrizolTM reagent (Life Technologies, Carlsbad, CA) following the manufacturer’s instructions. One hundred µg of total RNA was separated on a 15% denaturing PAGE, and the small RNAs (14–28 nt) were extracted from the gel and used for the array. The small RNAs were dephosphorylated using antarctic phosphatase (New England Biolabs, Ipswich, MA) and then radiolabelled with γ-32P-ATP and PNK. The radiolabelled small RNAs were hybridized to the miRNA array membrane at 37°C for 12 h in hybridization buffer (50% formamide, 5× SSPE and 5× Denhart’s solution). The membrane was washed three times (20 min each) with washing buffer (2 × SSC and 0.1% SDS) at 42°C. After hybridization, detection was performed using Phosphor-Imager screens (FLA-7000, Fujifilm).
For the RNA gel blot assay, 80 µg of total RNA isolated from wheat leaves was separated on a denaturing 15% urea–PAGE gel and electro-transferred to a Hybond-N+ membrane using a Semi-Dry Transfer Cell (Bio-Rad, Hercules, CA). The DNA oligonucleotide probes were labelled with γ-32P-ATP and PNK. The hybridization method was the same as that described above for the array.
miRNAs sequence analysis
As the array platform was constructed from several conserved and non-conserved miRNAs of different plants, we analysed the wheat conserved and non-conserved miRNAs identified in this study, respectively. If the sequences of miRNAs that were positive in array were identical to the sequences of known wheat miRNAs with the mismatch of 3 bp, we identified them as the same miRNA. If the sequences were unknown in wheat, they were mapped to reference sequences using SOAP (http://soap.genomics.org.cn) and selected genomes (wheat transcript assemblies from TIGR and our lab) with mismatches of 3 bp to obtain the candidate precursor sequences. If the candidate precursor sequences could be folded into a stable hairpin structure (http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi), we defined it as a new miRNA. Otherwise, they were abandoned.
Primer design and cDNA synthesis
The mature miRNAs primers were designed according to the method described by Feng et al. (Citation2012). The specific primer of each miRNA was used as the reverse transcription primer for testing the miRNAs containing a stem-loop structure, consisting of 44 conserved and 6 variable nucleotides that are specific for the 3′ ends of the miRNA sequence. The forward primers for the RT-qPCR were designed based on the mature miRNA sequence, whereas approximately 6 bp was removed at the 3′ end and approximately 6 bp was added at 5′ end according to the Tm value. A common primer originated from the reverse transcription primer was used as reverse primers for the RT-qPCR of miRNAs. All primers are listed in Supplemental Table 3.
The total RNA extracted from each sample of wheat leaves was treated with DNase I to remove any contaminating genomic DNA. The RNA concentration was detected before and after DNase I treatment using a NanoDropTM 1000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA), and formamide denaturing gel electrophoresis was conducted to visualize the integrity of the RNA. Before the reverse transcription of the mature miRNAs, 3 μg total RNA and 4.0 μL primer mix (0.5 μL each miRNA RT primer and 0.5 μL inner reference gene reverse transcription primer) were combined in a total volume of 10 μL with RNase-free water and incubated at 85°C for 5 min followed by cooling on ice. Then, 10 μL mix (6 μL 5× RT-Buffer (Invitrogen), 2 μL 2.5 mM dNTPs (Invitrogen), 1 μL RNase inhibitor and 1 μL M-MLV RT Enzyme 200U (Life Technologies) were added to a final volume of 30 μL. The reaction was performed at 42°C for 60 min and 72°C for 10 min. All of the cDNA samples were 20-fold diluted with sterile water before being used as the template in the RT-qPCR analysis.
Real-time quantitative PCR
All of the quantitative PCR amplifications were performed using a CFX96TM Real-Time System (Bio-Rad) with SYBR Green I (Life Technologies) to detect double-stranded cDNA synthesis. The translation elongation factor 1 alpha-subunit (EF) gene (GenBank accession no. M90077) was used as a control (Kong & Yang Citation2010). The real-time PCR amplifications were performed in a reaction mix including 12.5 μL 2× SYBR Premix Ex TaqTM (Takara, Dalian, China), 2.0 μL 20× first-strand cDNA, 0.2 μM each primer and 10.1 μL sterile water. The PCR procedures were as follows: 95°C for 3 min, 40 cycles of 95°C for 10 s and 60°C for 10 s, then 72°C for 30 s; melting curves were generated immediately after the completion of the RT-qPCR cycle to detect primer dimerization and other artifacts of the amplification. The relative expression level of the miRNAs in the Pst-inoculated plants at each time point was calculated as the fold of the mock-inoculated plants at that time point using the comparative 2−ΔΔCT method. The experiments were repeated in triplicate as independent biological replicates using newly extracted RNA and synthesized cDNA samples.
Results
Identification of miRNAs in wheat leaves by microRNA array analysis and RNA gel blot assay
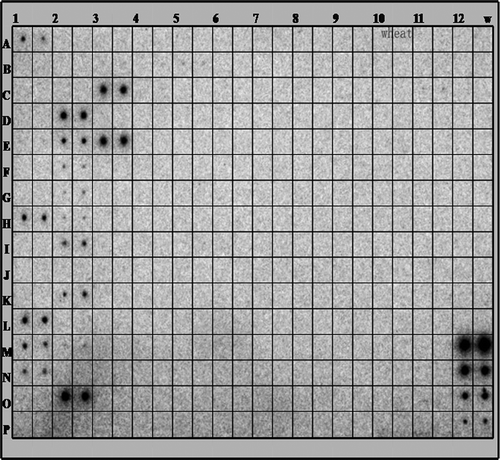
To date, the miRBase Sequence Database (Release 18.0) contains 18 226 mature miRNA products from 168 species, whereas only 41 miRNAs of 36 families have been found in wheat (http://www.mirbase.org). To identify more wheat miRNAs, we selected 188 miRNAs of 95 miRNA families from five plant species to employ a miRNA array. As shown in , the hybridization of the external controls (MC2-5) was obvious and stable, demonstrating that the hybridization system was feasible. We identified 18 candidate miRNAs of 11 families (miR156, miR159, miR160, miR164, miR165, miR166, miR167, miR168, miR169, miR390 and miR528) in wheat ().
Fig. 1 Identification of miRNAs by microarray. The identification of miRNAs was screened by miRNA array analysis. Total RNA was extracted from the leaves of 10-day-old wheat seedlings. One hundred micrograms of total RNA was used for the array. The extracted small RNAs were 5′ end labelled and probed against the antisense DNA oligonucleotides spotted onto the membrane.
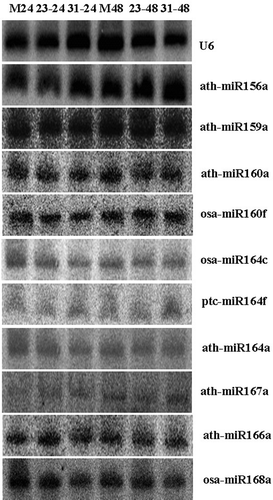
To confirm the candidate miRNAs identified by array, 18 candidate miRNAs were further validated by RNA gel blot analysis. Being different to the results of array, only 14 candidate miRNAs were hybridized to DNA probes, and the hybridization bands of four miRNAs were approximately 100–150 bp (data not shown), which were speculated to be the size of the precursor or non-specific hybridization. The hybridization bands of another 10 miRNAs (ath-miR156a, ath-miR159a, ath-miR160a, osa-miR160f, ath-miR164a, osa-miR164c, ptc-mir 164f, ath-miR167a, ath-miR166a and osa-miR168a) were approximately 21 bp (), which represented the specific hybridization of mature miRNAs.
Fig. 2 Validation of miRNAs using northern blot. Total RNA (80 μg) was used for the RNA gel blot analysis for the mock and wheat leaves inoculated with CYR23 and CYR31. The wheat leaves were sampled for the analysis at 24 and 48 hpi. M means MOCK, 23 means CYR23, and 31 means CYR31. U6 was used as the loading control. The sizes of the hybridization bands of ath-miR156a, ath-miR159a, ath-miR160a, osa-miR164c, ptc-mir164f, ath-miR164a, ath-miR167a, osa-miR160f, ath-miR166a and osa-miR168a were approximately 21 bp.
Identification of known and new members of miRNAs in wheat
According to miRBase, several identical or homologous sequences for these 10 miRNAs were identified in T. aestivum, with the exception of ath-miR166a and osa-miR168a. We analysed the sequence homology of all of the wheat known mature miRNAs (). Specifically, the mature miRNA sequence of tae-miR167 is fully identical to ath-miR167a. The last nucleotide of tae-miR156 and tae-miR159ab is different than ath-miR156a and ath-miR159a, respectively. tae-miR160 can be aligned with ath-miR160a, whereas there is one nucleotide change compared with osa-miR160f and ptc-miR160h. tae-miR164 can be aligned with ath-miR164a, whereas there is one nucleotide change compared with osa-miR164c and two nucleotide changes compared with ptc-miR164f and sbi-miR164c. According to the criteria presented above, these mature sequences of known wheat miRNAs show high similarity among different plants with the mismatches less than 3 bp; therefore, we identified them as the same miRNA.
Table 1. Sequence analysis of the array-identified miRNAs in wheat.
To identify new miRNAs in wheat, the sequences of ath-miR166a and osa-miR168a identified in the microarray analysis, which were unknown in wheat, were mapped to reference sequences using SOAP (http://soap.genomics.org.cn) and selected genomes (wheat transcript assemblies from TIGR and our lab). Fortunately, we found that they could be mapped to genomic sequences with no mismatch and we named them as PN-tae-166a and PN-tae-168. The candidate precursors of these miRNAs were in silico cloned from the wheat genome information. The sequenced candidate precursors could be folded into standard stem-loop structures using RNAfold WebServer (http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi) (, ).
Table 2. Pre-miRNA sequence of potentially new members of conserved miRNAs in wheata.
Fig. 3 (Colour online) The stem-loop structures of new miRNAs. The sequences were mapped to reference sequences using SOAP (http://soap.genomics.org.cn) and selected genomes (wheat transcript assemblies from TIGR and our lab) and were folded into standard stem-loop structures using RNAfold WebServer (http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi). (A, PN-tae-miR166a; B, PN-tae-miR168a).

According to the results of the RNA gel blot analysis and based on microarray and sequence analysis, we detected five known wheat miRNAs (tae-miR156, tae-miR159, tae-miR160, tae-miR164 and tae-miR167) and two new miRNAs (PN-tae-miR166a and PN-tae-miR168a) in wheat cultivar ‘Suwon 11’ ().
Expression level of wheat miRNAs in response to Pst
To verify whether miRNAs participate in the interactions between wheat and Pst, we chose tae-miR156, tae-miR159, tae-miR160, tae-miR164, tae-miR167, PN-tae-miR166a and PN-tae-168a for further characterization using quantitative real-time PCR (). The transcriptional accumulation of tae-miR156 was approximately 2 times higher at 12 hpi than the level at 0 hpi in the incompatible interaction, whereas it was 5 times higher at 48 hpi in the compatible interaction (). The transcriptional accumulation of tae-miR159 and tae-miR160 were upregulated at 12 hpi and then declined from 18 to 120 hpi to the control level in both the compatible and incompatible interactions (). In the incompatible interaction, the transcription of tae-miR164 increased at 12 hpi and then decreased slightly to the control level from 18 to 120 hpi. In the compatible interaction, the transcription of tae-miR164 in the wheat leaves decreased dramatically from 12 to 24 hpi and then increased to the control level from 48 to 120 hpi (). The relative expression of tae-miR167 was upregulated at 12 hpi and then began to decline from 18 to 48 hpi, after which it sharply decreased to the control level from 72 to 120 hpi (). The transcriptional accumulation of PN-tae-miR166a was only upregulated at 24 hpi, to approximately eight times the level of that at 0 hpi in the incompatible interaction, whereas there was no induction in the compatible interaction (). In contrast, the relative expression of PN-tae-miR168a was downregulated at 24 hpi in the compatible interaction. Although the expression also showed a downregulation in the incompatible interaction, it was not significant when compared with the compatible interaction ().
Fig. 4 Relative expression analysis of miRNAs by RT-qPCR. Seven mature miRNAs were confirmed and selected for the transcript accumulation analysis in wheat after challenge with CYR23 and CYR31. (A, tae-miR156; B, tae-miR159; C, tae-miR160; D, tae-miR164; E, tae-miR167; F, PN-tae-miR166a; G, PN-tae-miR168a). The data were normalized to the expression level of wheat translation elongation factor 1 alpha-subunit (EF). The vertical bars represent the standard deviations. The relative expression level of the miRNAs in the Pst-inoculated plants at each time point was calculated as the fold of the mock-inoculated plants at that time point using the comparative 2−ΔΔCT method. The vertical bars represent the standard deviations. The experiments were repeated in triplicates of independent biological replicates using newly extracted RNA and synthesized cDNA samples.
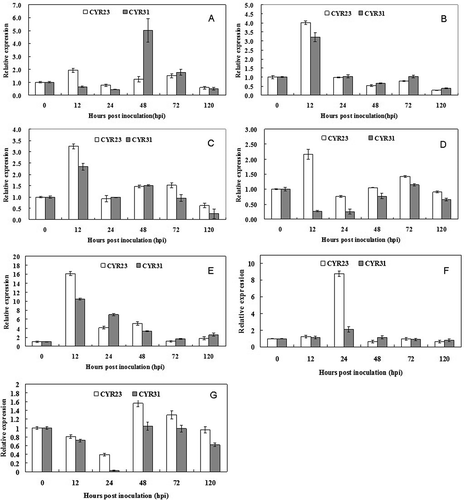
In conclusion, all of these miRNAs were involved in the interaction between wheat and Pst, and nearly all of them were regulated at 12 hpi or 24 hpi. We speculated that the conserved miRNAs might play a more important role in the resistance of wheat to Pst by regulating the various types of genes involved in the different signalling pathways.
Target genes prediction for miRNAs
To explore the miRNAs involved in the interaction between wheat and Pst, we analysed the potential targets of conserved Pst-inducible miRNAs in wheat based on the potential target genes predicted by Yao et al. (Citation2007). We predicted the target genes for the known and new conserved miRNAs using psRNATarget (http://plantgrn.noble.org/psRNATarget) and compared the results with the data of Yao et al. (Citation2007) and data for Arabidopsis thaliana (). Accurate target genes for the known miRNAs were predicted, and most of them were transcription factors and auxin response factors. Similar to ath-miR166a, the targets of PN-tae-miR166a were predicted to be Class III HD-Zip proteins, also a type of transcription factor. The target genes of ath-miR168a were histone-lysine N-methyltransferases, but we did not find target genes for PN-tae-miR168a using psRNATarget.
Table 3. The information of the selected miRNAs and their targets in wheat and Arabidopsis thalianaa.
Discussion
miRNAs are a class of small RNAs that play important roles in post-transcriptional gene regulation by degrading target mRNA or inhibiting gene translation. Several studies have indicated that miRNAs are essential for plant development and for responding to hormone signalling and environmental stresses (Aukerman & Sakai Citation2003; Jin Citation2008; Padmanabhan et al. Citation2009; Lv et al. Citation2010). Wheat stripe rust is one of the most destructive diseases of wheat worldwide. It has been proven that breeding efforts and the rational utilization of disease-resistant varieties are the safest, most economical and most effective methods to control this disease (Chen Citation2005). Understanding the interactions between wheat and the stripe rust pathogen provides essential clues to the implementation of resistance genes in wheat breeding. Numerous studies have investigated the mechanisms of wheat resistance to Pst (Coram et al. Citation2008a, Citation2008b, Citation2010). Recently, miRNAs emerged as key components in plant responses and adaptation to pathogen stress (Jin Citation2008; Padmanabhan et al. Citation2009). Although many miRNAs have been discovered, there is no evidence confirming the involvement of miRNAs in the interaction between wheat and Pst.
Most miRNAs are conserved between different plants, enabling the analysis of their expression using a set of selected array probes. Microarray analysis, a high-throughput tool to mine miRNA, was applied in this study to clarify whether the currently known conserved miRNAs could be induced by Pst. We first prepared a miRNA microarray containing 188 probes that were complementary to known miRNAs in 95 miRNA families from eight plant species. Although many plant miRNAs are highly conserved (Axtell & Bartel Citation2005; Zhang et al. Citation2006), only 18 miRNAs from 11 miRNA families were detected using this microarray. To confirm the accuracy of the array further, 18 miRNAs were validated using RNA gel blot analysis, and the hybridization bands of only 10 miRNAs were approximately 21 nt. To confirm them as new miRNAs, it is essential to find the sequences of the pre-miRNAs, which can be folded into a standard stem-loop structure. In this study, we found only two standard stem-loop structures for the miRNAs identified using microarray, belonging to new miRNA families. For those undetected conserved miRNAs, their low expression caused by their tissue-specific, developmental stage-specific or disease-specific patterns (Yin et al. Citation2008) requires further exploration. Recently, deep sequencing of plant small RNA libraries also demonstrated that plants express more non-conserved miRNAs than conserved miRNAs (Ilegems et al. Citation2010).
Previous reports demonstrated that miRNAs were involved in plant resistance to pathogens (Jin Citation2008; Padmanabhan et al. Citation2009). By using RT-qPCR, five wheat miRNAs were found to be induced upon Pst infection. It is interesting to note that the most significant changes of the miRNA expression levels occurred at 12 hpi or 24 hpi, the key time points of successful infection for Pst. At 12 hpi, the germ tube of Pst is penetrating into the stoma and forming a substomatal vesicle; at 24 hpi, the haustoria forms and begins to absorb nutrition from the host cells (Wang et al. Citation2007a). As important regulators, miRNAs respond rapidly and regulate target genes to help the plants adapt to external environment changes. Consistently, the expression of tae-miR156, tae-miR164 and tae-miR167 were also induced by the powdery mildew pathogen at 12 hpi (Xin et al. Citation2010). When challenged by Pst, tae-miR164 was downregulated in the compatible interaction and upregulated in the incompatible interaction; conversely, when challenged with the powdery mildew pathogen, tae-miR164 was downregulated in the incompatible interaction and showed no significant change in the compatible interaction (Xin et al. Citation2010).
The target gene of miR156 has been validated as a member of the squamosa promoter-binding protein-like (SPL) transcription factors, which mainly regulate floral and reproductive growth (Schwarz et al. Citation2008). As tae-miR156 was found induced in wheat challenged with Pst and has also been demonstrated to be involved in the response to pathogen stress (Xin et al. Citation2010), we speculated that tae-miR156 regulates growth progress and can also respond to external stress. Previous studies revealed that tae-miR164 was involved in the auxin signalling pathway by regulating the NAC transcription factor family (Gupta et al. Citation2012). It has been reported that the NAC transcription factor was a transcriptional activator for plants in response to abiotic and biotic stresses, and NAC4 and NAC8 showed different expression profiles between the incompatible and compatible interactions of wheat and Pst (Nakashima et al. Citation2007; Xia et al. Citation2010a, Citation2010b). We also found that tae-miR164 showed different expression levels for the incompatible and compatible interactions, whereas further investigation is necessary to determine which NAC genes were regulated. miR159, representing one of the most ancient miRNAs in plants, is a conserved basal regulator and is predicted to have 20 or more target genes in Arabidopsis (Rajagopalan et al. Citation2006). It was predicted that tae-miR159 regulates MYB transcription factors in wheat and was induced in both the incompatible and compatible interactions of wheat to Pst in our study. We had proven it was stably expressed in samples after different treatments (Feng et al. Citation2012), thus we speculated that it is involved in the basal resistance of wheat. In our study, tae-miR160 and tae-miR167 showed a similar expression trend in wheat inoculated with CYR23 and CYR31. tae-miR167 showed a dramatic induction in wheat after challenge with Pst, demonstrating that tae-miR167 had an ample transcript accumulation for regulating genes involved in biological progresses. The target genes of tae-miR160 and tae-miR167 were predicted to be ARF family genes. ARF10 is negatively targeted by miR160 in A. thaliana, playing a critical role in ARF10-dependent auxin and ABA pathways (Liu et al. Citation2007). The ARF6 and ARF8 transcripts are the cleavage targets of miR167 in A. thaliana (Wu et al. Citation2006). So we speculated that the auxin signal transduction pathway regulated by tae-miR160 and tae-miR167 is involved in the basal resistance of wheat to Pst. In our study, PN-tae-miR166a was highly expressed at 24 hpi in the incompatible interaction, which resulted in the repression of Class III HD-Zip proteins. It has been demonstrated that the absence of Class III HD-ZIP expression in procambium cells leads to a wider distribution of auxin in the internal tissues (Ilegems et al. Citation2010). The auxin-stimulated SCF ubiquitination pathway contributes to limiting biotrophic pathogen colonization once plant-pathogen compatibility is established, and the repression of the auxin response pathway can increase Arabidopsis susceptibility to necrotrophic fungi (Llorente et al. Citation2008). A series of reports have also described the negative effect of auxin signalling on plant resistance to biotrophic pathogens (Navarro et al. Citation2006; Wang et al. Citation2007b). So we speculate that PN-tae-miR166a is not only involved in plant growth development, but also played roles in the resistance of wheat to Pst through the auxin signal transduction pathway. miRNAs play a very important role in most cell processes via two main pathways: target gene cleavage and translation inhibition at the post-transcriptional level. In addition, miRNAs can also regulate gene expression at the transcriptional level via histone modification (Wu et al. Citation2010). The target gene of ath-miR168a was identified as histone-lysine N-methyltransferase, which is a histone-modifying enzyme. Unfortunately, we were unable to identify the target gene using psRNATarget, which might be due to the limited genome information of wheat.
In conclusion, two new members of conserved miRNAs families and five known miRNAs were identified in this study; all were regulated during the challenge of wheat by Pst. These Pst-inducible miRNAs may function in the interactions between wheat and Pst by regulating the candidate target genes; however, the complicated molecular mechanism remains to be explored.
Supplemental data
Supplemental data for this article can be accessed here: http://dx.doi.org/10.1080/07060661.2014.999124.
Supplemental Material
Download MS Word (82.5 KB)Acknowledgements
We thank Dr Guiliang Tang (Department of Biological Sciences, Michigan Technological University, USA) for the protocols for the miRNA array platform and primer design. This work was supported by grants from the National Basic Research Program of China (2013CB127700), the National Science & Technology Pillar Program during the Twelfth Five-year Plan Period (2012BAD19B04), the National Natural Science Foundation of China (31271990), the Youth Science & technology star of Shaanxi (2012KJXX-15) and the 111 Project from the Ministry of Education of China (B07049).
References
- Aukerman MJ, Sakai H. 2003. Regulation of flowering time and floral organ identity by a microRNA and its APETALA2-like target genes. Plant Cell. 15:2730–2741.
- Axtell MJ, Bartel DP. 2005. Antiquity of microRNAs and their targets in land plants. Plant Cell. 17:1658–1673.
- Bartel DP. 2004. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 116:281–297.
- Chen C, Ridzon DA, Broomer AJ, Zhou Z, Lee DH, Nguyen JT, Barbisin M, Xu NL, Mahuvakar VR, Andersen MR, et al. 2005. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 33:e179.
- Chen X. 2005. Epidemiology and control of stripe rust [Puccinia striiformis f. sp. tritici] on wheat. Can J Plant Pathol. 27:314–337.
- Coram TE, Huang X, Zhan G, Settles ML, Chen X. 2010. Meta-analysis of transcripts associated with race-specific resistance to stripe rust in wheat demonstrates common induction of blue copper-binding protein, heat-stress transcription factor, pathogen-induced WIR1A protein, and ent-kaurene synthase transcripts. Funct Integr Genomic. 10:383–392.
- Coram TE, Settles ML, Chen X. 2008a. Transcriptome analysis of high-temperature adult-plant resistance conditioned by Yr39 during the wheat-Puccinia striiformis f. sp. tritici interaction. Mol Plant Pathol. 9:479–493.
- Coram TE, Wang M, Chen X. 2008b. Transcriptome analysis of the wheat-Puccinia triiformis f. sp. tritici interaction. Mol Plant Pathol. 9:157–169.
- Ekins R, Chu FW. 1999. Microarrays: their origins and applications. Trends Biotechnol. 17:217–218.
- Feng H, Huang X, Zhang Q, Wei G, Wang X, Kang Z. 2012. Selection of suitable inner reference genes for relative quantification expression of microRNA in wheat. Plant Physiol Biochem. 51:116–122.
- Galiveti CR, Rozhdestvensky TS, Brosius J, Lehrach H, Konthur Z. 2010. Application of housekeeping npcRNAs for quantitative expression analysis of human transcriptome by real-time PCR. RNA. 16:450–461.
- Gupta OP, Permar V, Koundal V, Singh UD, Praveen S. 2012. MicroRNA regulated defense responses in Triticum aestivum L. during Puccinia graminis f. sp. tritici infection. Mol Biol Rep. 39:817–824.
- Ilegems M, Douet V, Meylan-Bettex M, Uyttewaal M, Brand L, Bowman JL, Stieger PA. 2010. Interplay of auxin, KANADI and Class III HD-ZIP transcription factors in vascular tissue formation. Development. 137:975–984.
- Iyer NJ, Jia X, Sunkar R, Tang G, Mahalingam R. 2012. microRNAs responsive to ozone-induced oxidative stress in Arabidopsis thaliana. Plant Signal Behav. 7:484–491.
- Jin H. 2008. Endogenous small RNAs and antibacterial immunity in plants. FEBS Lett. 582:2679–2684.
- Katagiri F, Glazebrook J 2009. Overview of mRNA expression profiling using DNA microarrays. Curr Protoc Mol Biol. Chapter 22, Unit 22 24.
- Kong WW, Yang ZM. 2010. Identification of iron-deficiency responsive microRNA genes and cis-elements in Arabidopsis. Plant Physiol Biochem. 48:153–159.
- Li H, Zhang Z, Huang F, Chang L, Ma Y. 2009. MicroRNA expression profiles in conventional and micropropagated strawberry (Fragaria × ananassa Duch.) plants. Plant Cell Rep. 28:891–902.
- Liu H-H, Tian X, Li Y-J, Wu C-A, Zheng C-C. 2008. Microarray-based analysis of stress-regulated microRNAs in Arabidopsis thaliana. RNA. 14:836–843.
- Liu P-P, Montgomery TA, Fahlgren N, Kasschau KD, Nonogaki H, Carrington JC. 2007. Repression of AUXIN RESPONSE FACTOR10 by microRNA160 is critical for seed germination and post-germination stages. Plant J. 52:133–146.
- Llorente F, Muskett P, Sánchez-Vallet A, López G, Ramos B, Sánchez-Rodríguez C, Jordá L, Parker J, Molina A. 2008. Repression of the auxin response pathway increases Arabidopsis susceptibility to necrotrophic fungi. Mol Plant Pathol. 1:496–509.
- Lv D-K, Bai X, Li Y, Ding X-D, Ge Y, Cai H, Ji W, Wu N, Zhu Y-M. 2010. Profiling of cold-stress-responsive miRNAs in rice by microarrays. Gene. 459:39–47.
- Ma J, Huang X, Wang X, Chen X, Qu Z, Huang L, Kang Z. 2009. Identification of expressed genes during compatible interaction between stripe rust (Puccinia striiformis) and wheat using a cDNA library. BMC Genom. 10:586.
- Mica E, Gianfranceschi L, Pè ME. 2006. Characterization of five microRNA families in maize. J Exp Bot. 57:2601–2612.
- Nakashima K, Tran LSP, Van Nguyen D, Fujita M, Maruyama K, Todaka D, Ito Y, Hayashi N, Shinozaki K, Yamaguchi-Shinozaki K. 2007. Functional analysis of a NAC-type transcription factor OsNAC6 involved in abiotic and biotic stress-responsive gene expression in rice. Plant J. 51:617–630.
- Navarro L, Dunoyer P, Jay F, Arnold B, Dharmasiri N, Estelle M, Voinnet O, Jones JDG. 2006. A plant miRNA contributes to antibacterial resistance by repressing auxin signaling. Science. 312:436–439.
- Padmanabhan C, Zhang X, Jin H. 2009. Host small RNAs are big contributors to plant innate immunity. Curr Opin Plant Biol. 12:465–472.
- Peltier HJ, Latham GJ. 2008. Normalization of microRNA expression levels in quantitative RT-PCR assays: identification of suitable reference RNA targets in normal and cancerous human solid tissues. RNA. 14:844–852.
- Rajagopalan R, Vaucheret H, Trejo J, Bartel DP. 2006. A diverse and evolutionarily fluid set of microRNAs in Arabidopsis thaliana. Gene Dev. 20:3407–3425.
- Rhoades MW, Reinhart BJ, Lim LP, Burge CB, Bartel B, Bartel DP. 2002. Prediction of plant microRNA targets. Cell. 110:513–520.
- Schwarz S, Grande AV, Bujdoso N, Saedler H, Huijser P. 2008. The microRNA regulated SBP-box genes SPL9 and SPL15 control shoot maturation in Arabidopsis. Plant Mol Biol. 67:183–195.
- Shukla LI, Chinnusamy V, Sunkar R. 2008. The role of microRNAs and other endogenous small RNAs in plant stress responses. BBA - Gene Regul Mech. 1779:743–748.
- Sunkar R, Girke T, Jain PK, Zhu JK. 2005. Cloning and characterization of microRNAs from rice. Plant Cell. 17:1397–1411.
- Wang C-F, Huang L-L, Buchenauer H, Han Q-M, Zhang H-C, Kang Z-S. 2007a. Histochemical studies on the accumulation of reactive oxygen species (O2- and H2O2) in the incompatible and compatible interaction of wheat-Puccinia striiformis f. sp. tritici. Physiol Mol Plant Pathol. 71:230–239.
- Wang D, Pajerowska-Mukhtar K, Culler AH, Dong X. 2007b. Salicylic acid inhibits pathogen growth in plants through repression of the auxin signaling pathway. Curr Biol. 17:1784–1790.
- Wang X, Tang C, Zhang G, Li Y, Wang C, Liu B, Qu Z, Zhao J, Han Q, Huang L, et al. 2009. cDNA-AFLP analysis reveals differential gene expression in compatible interaction of wheat challenged with Puccinia striiformis f. sp. tritici. BMC Genom. 10:289.
- Wu L, Zhou H, Zhang Q, Zhang J, Ni F, Liu C, Qi Y. 2010. DNA methylation mediated by a microRNA pathway. Mol Cell. 38:465–475.
- Wu M-F, Tian Q, Reed JW. 2006. Arabidopsis microRNA167 controls patterns of ARF6 and ARF8 expression, and regulates both female and male reproduction. Development. 133:4211–4218.
- Xia N, Zhang G, Liu X-Y, Deng L, Cai G-L, Zhang Y, Wang X-J, Zhao J, Huang L-L, Kang Z-S. 2010a. Characterization of a novel wheat NAC transcription factor gene involved in defense response against stripe rust pathogen infection and abiotic stresses. Mol Biol Rep. 37:3703–3712.
- Xia N, Zhang G, Sun Y-F, Zhu L, Xu L-S, Chen X-M, Liu B, Yu Y-T, Wang X-J, Huang L-L, Kang Z-S. 2010b. TaNAC8, a novel NAC transcription factor gene in wheat, responds to stripe rust pathogen infection and abiotic stresses. Physiol Mol Plant Pathol. 74:394–402.
- Xin M, Wang Y, Yao Y, Xie C, Peng H, Ni Z, Sun Q. 2010. Diverse set of microRNAs are responsive to powdery mildew infection and heat stress in wheat (Triticum aestivum L.). BMC Plant Biol. 10:23.
- Xu T, Wang B, Liu X, Feng R, Dong M, Chen J. 2012. Microarray-based identification of conserved microRNAs from Pinellia ternata. Gene. 493:267–272.
- Yao Y, Guo G, Ni Z, Sunkar R, Du J, Zhu JK, Sun Q. 2007. Cloning and characterization of microRNAs from wheat (Triticum aestivum L.). Genome Biol. 8:96.
- Yin JQ, Zhao RC. 2007. Identifying expression of new small RNAs by microarrays. Methods. 43:123–130.
- Yin JQ, Zhao RC, Morris KV. 2008. Profiling microRNA expression with microarrays. Trends Biotechnol. 26:70–76.
- Zeng HQ, Zhu YY, Huang SQ, Yang ZM. 2010. Analysis of phosphorus-deficient responsive miRNAs and cis-elements from soybean (Glycine max L.). J Plant Physiol. 167:1289–1297.
- Zhang B, Pan X, Cannon CH, Cobb GP, Anderson TA. 2006. Conservation and divergence of plant microRNA genes. Plant J. 46:243–259.