
Abstract
Zn(II)2Cys6 transcription factor genes encode transcription regulators that manage the infection potential and production of secondary metabolites such as toxins in fungi. In this study, a gene named rolP that encodes a putative Zn(II)2Cys6 transcription factor regulating leucinostatin production in the filamentous fungus Paecilomyces lilacinus was characterized by a gene knockout approach. The deduced proprotein consists of 705 amino acids and is highly homologous to the Zn(II)2Cys6 transcription factor of the entomopathogenic fungus Metarhizium brunneum. Predictive analysis of the secondary structure of the proportion showed that it has two domains. Paecilomyces lilacinus can produce nematotoxins leucinostatins A and B. Deletion of rolP from the P. lilacinus wild-type strain Pl36-1 leads to the absence of leucinostatin A, while a large increase in the leucinostatin A level was achieved in the rolP overexpression strain Ov-Pl36-1. During the process of nematode infection, rolP showed high expression levels at 48–72 h and peaked at 48 h. Bioassay tests confirmed the requirement of rolP for leucinostatin production. However, the root-knot second-stage juvenile-toxicity was minimized to 35.3% in ΔrolP; toxicity was 97.2% with Ov-Pl36-1 compared with 68.4% with the wild type strain Pl36-1. Interestingly, neither knockout nor overexpression of the rolP gene affected the growth or sporulation of P. lilacinus. Additionally, fungal nutrition and acidic media might stimulate the rolP activity of the wild type strain against nematodes. These findings suggested that the rolP gene is required for the induction and production of leucinostatins and is considered a leucinostatin regulatory gene in P. lilacinus.
Résumé
Les gènes du facteur de transcription Zn(II)2Cys6 encodent les régulateurs de transcription qui gèrent le potentiel d’infection et la production de métabolites secondaires, comme des toxines, chez les champignons. Dans cette étude, un gène nommé rolP, qui encode un facteur de transcription putatif Zn(II)2Cys6 régulant la production de leucinostatine chez le champignon filamenteux Paecilomyces lilacinus, a été caractérisé grâce à un gène inactivé. La proprotéine qui en a été déduite consiste en 705 acides aminés et est fortement homologue au facteur de transcription Zn(II)2Cys6 du champignon entomopathogène Metarhizium brunneum. L’analyse prévisionnelle de la structure secondaire de la proportion a montré qu’elle possède deux domaines. P. lilacinus peut produire les leucinostatines A et B, des nématotoxines. La délétion du gène rolP de la souche sauvage Pl36-1 de P. lilacinus provoque l’absence de leucinostatine A, tandis que l’on a noté une forte augmentation du taux de leucinostatine A lors de la surexpression du gène rolP de la souche OvPl36-1. Durant l’infection causée par le nématode, le gène rolP a affiché des taux élevés d’expression de 48 à 72 h, et a atteint son plus haut taux à 48 h. Des biotests ont confirmé que le gène rolP était nécessaire à la production de leucinostatine. Toutefois, la toxicité induite par le gène ∆rolP chez les larves de deuxième stade du nématode cécidogène était réduite à 35.3%; la toxicité chez la souche Ov-Pl36-1 était de 97.2%, comparativement à 68.4% chez la souche sauvage Pl36-1. Chose intéressante, ni l’inactivation ni la surexpression du gène rolP n’ont influencé la croissance ou la sporulation de P. lilacinus. De plus, chez la souche sauvage, le mode d’alimentation du champignon et l’acidité du milieu peuvent stimuler l’activité du gène rolP contre les nématodes. Ces résultats suggèrent que le gène rolP est nécessaire à l’induction et à la production des leucinostatines et qu’il est considéré comme un gène de régulation de la leucinostatine chez P. lilacinus.
Introduction
Root-knot nematodes (Meloidogyne incognita) are plant parasites with worldwide distribution causing economically important losses in a wide variety of crops (Luc et al. Citation2005). The fungal species Paecilomyces lilacinus (Thom) Samson has been reported to infect nematodes including M. incognita (Khan et al. Citation2004; Yang et al. Citation2015). The fungus employs diverse strategies to infect its host nematodes, including adherence, penetration and digestion (Tunlid & Jansson Citation1991). Currently, several pathogenicity-related enzymes in P. lilacinus, such as serine proteases and chitinases, which interact with the nematode cuticle and egg shell, have been identified (Khan et al. Citation2003; Morton et al. Citation2004; Yang et al. Citation2007).
The production of paecilotoxins (leucinostatins) in P. lilacinus is an indicator of nematicidal activity (Cutler et al. Citation2000; Park et al. Citation2004). Leucinostatins are a large and diverse family of secondary metabolites produced by bacteria, fungi and plants (Schneider Citation2005; Das et al. Citation2006; Tang et al. Citation2006). Leucinostatin A and B are the most abundant components of this family. Leucinostatin A causes inhibition of mitochondrial ATP synthesis (Shima et al. Citation1990) and various phosphorylation pathways (Lucero et al. Citation1978). Leucinostatins from P. lilacinus are reported to kill nematodes (Park et al. Citation2004) and have been widely used for their biocontrol (Kiewnick & Sikora Citation2006; Anastasiadis et al. Citation2008; Siddiqui & Futai Citation2009). However, limited information on the molecular pathogenic mechanisms of leucinostatins against nematodes is available.
The Zn(II)2Cys6 family of transcriptional regulator proteins has been reported to be a characteristic class of fungal transcription factors (Todd & Andrianopoulos Citation1997), in which the N-terminus carries a DNA-binding domain with a six-cysteine zinc atom binuclear cluster (Baum et al. Citation1987; Marmorstein et al. Citation1992). Members of this family are involved in diverse functions and act as regulators in different metabolic pathways, ranging from carbon and nitrogen metabolism and stress responsiveness to the synthesis of secondary metabolites (Todd & Andrianopoulos Citation1997). To date, little is known regarding the regulators involved in the secondary metabolism of P. lilacinus. Like many other aspects of eukaryotic transcriptional control, many Zn(II)2Cys6 proteins can change fungal physiology. However, the relationship between the Zn(II)2Cys6 protein action mechanism and loss-of-function phenotype remains unclear.
We have obtained a random-insertional mutant of P. lilacinus (named R147) that is compromised with regard to leucinostatin production. We speculated that a gene important in the leucinostatin pathway was disrupted in R147. We had cloned a 1-kb fragment from R147 by TAIL-PCR. Sequence analyses indicated that the fragment is a part of a putative Zn(II)2Cys6 transcription factor gene. In the present study, we cloned the full-length of the corresponding gene (rolP) by TAIL-PCR and conducted functional analyses of the encoded protein. The objectives of this study were to explore the relationship between rolP and leucinostatin production and to investigate rolP expression in P. lilacinus. Toward these objectives, Agrobacterium tumefaciens-mediated transformation (ATMT) was used in the present study to delete the Zn(II)2Cys6 coding gene and the loss-of-function in the gene knockout strains was studied.
Materials and methods
Strains and culture conditions
The P. lilacinus wild-type strain Pl36-1 (WT) was isolated from nematode eggs collected from Hubei Province, China. It was grown on potato dextrose agar (PDA) medium at 28°C. The weak virulent P. lilacinus mutant R147 was obtained using the ATMT-based random insertional mutagenesis method (Xiao et al. unpublished). The absence of leucinostatins A and B in this mutant strain was confirmed using matrix-assisted laser desorption/ionization-time of flight mass spectrometry (MALDI-TOF MS) to analyse its cell-free filtrate when cultured in medium B (Mikami et al. Citation1989). Pl36-1 was transformed using the Agrobacterium tumefaciens stain AGL1. The AGL1 was cultured on Luria-Bertani (LB) (Sambrook et al. Citation1989) plates containing 50 μg mL−1 rifampicin and 100 μg mL−1 streptomycin at 25°C. The Escherichia coli strain DH5α was used in all DNA manipulations and was typically grown in LB medium at 37°C. All other media used for culturing fungi and bacteria have been described previously (Mikami et al. Citation1989; Sambrook et al. Citation1989; Bundock et al. Citation1995) (Supplementary Table 1). The eggs and juveniles of the root-knot nematode M. incognita were separated and washed from cucumber roots using water and sodium phosphate buffer (pH 7).
Sequencing of genomic DNA flanking T-DNA
Thermal asymmetric interlaced PCR (TAIL-PCR) was used to clone the DNA fragments flanking the inserted T-DNA from the R147 strain. Genomic DNA from R147 was used as template in successive reactions along with the following primers: (a) T-DNA left border-specific primers L1: 5′-ACACTCCGTTGGGAACTTAGCAGGT-3′, L2: 5′-CTTAGTAAACTGATGTAGAAATAGG-3′, L3: 5′-GACGCATTCGGAAGAATTGGTAGAC-3′; (b) right border-specific primers R1: 5′-TGTGAGTTGGGATAGAGCCCGATAA-3′, R2: 5′-TACCTTGCGAACGATGTTGAGA-3′, R3: 5′-TCAGCAGGCGTTACACAATACTTCA-3′; and (c) arbitrary degenerate primers AD1, AD2, AD3 and AD4 (Wang et al. Citation2010). The PCR procedures and programs followed Wang et al. (Citation2010) with minor modifications (Supplementary Table 2). The final TAIL-PCR products were purified using a DNA Gel-Extraction Kit (Axygen, Union City, CA), cloned into the pMD18-T vector (Takara, Dalian, China), and sequenced by Invitrogen Trading Co. Ltd. (Shanghai, China). The flanking sequences at both ends of the T-DNA were combined and assembled with the previously obtained 1-kb fragment by DNAStar software (Madison, WI). The assembled sequence was analysed using the Blastn and Blastx algorithms available at NCBI’s GenBank (http://blast.ncbi.nlm.nih.gov/Blast.cgi). The putative open reading frame based on the sequence analyses was regarded as a gene and named rolP.
Construction of knockout and overexpression vectors
The knockout vector pCAMBIA1301B () was constructed based on the pCAMBIA1301 (GenBank accession number: AF234297) vector (Hajdukiewicz et al. Citation1994) containing a selectable marker gene coding for hygromycin resistance (HYG). First, two selectable marker gene fragments (‘KpnI-PtrpC-bar-TtrpC-BamHI’ and ‘HindIII-PtrpC-CBX-TtrpC-AhdI’; ) were individually amplified by PCR from the vector pEGAD (GenBank accession number: AF218816) and pDonrCbx (Genbank accession number: EU360889) under the control of trpC promoter (GenBank accession number: X02390), respectively. The amplification primers are listed in . Second, the ‘HindIII-PtrpC-CBX-TtrpC-AhdI’ fragment was digested with HindIII and AhdI, purified and then substituted for the HindIII/AhdI region in pCAMBIA1301, yielding p1301. Third, two 1000-bp arms (left arm and right arm) homologous to the 5ʹ region and 3ʹ region of rolP, respectively, were amplified from the wild type strain Pl36-1. The left arm was flanked with EcoRI and KpnI, while the right arm was flanked with BamHI and XbaI. Fourth, the left arm was digested with KpnI, the ‘KpnI-PtrpC-bar-TtrpC-BamHI’ fragment was digested with KpnI and BamHI, and the right arm was digested with BamHI. These fragments were purified and then ligated to each other by T4 ligase. The resultant fragment was referred to as rolPA. Fifth, the rolPA fragment was digested with EcoRI and XbaI and ligated into the same sites of the vector p1301, yielding pCAMBIA1301B. This recombinant plasmid, carrying both ‘HindIII-PtrpC-CBX-TtrpC-AhdI’ and ‘EcoRI-leftarm-KpnI-PtrpC-bar-TtrpC-BamHI–rightarm-XbaI’ fragments, was transferred into A. tumefaciens strain AGL-1 and selected for kanamycin resistance. The resultant strain was used for knockout of the rolP gene from the P. lilacinus wild type strain Pl36-1.
Fig. 1 Construction of the knockout and overexpression vectors. a, The pCAMBIA1301B knockout vector. b, The fragment of the EcoRI-left arm-KpnI-PtrpC-bar-TtrpC-BamHI-right arm-XbaI containing the selectable marker bar gene coding for the resistance of glufosinate and four restriction sites (EcoRI, KpnI, BamHI and XbaI). c, The fragment of the HindIII-PtrpC-CBX-TtrpC-AhdI containing a selectable marker CBX gene coding for carboxin resistance and containing two restriction sites (HindIII and AhdI). d, The fragment of the Zn(II)2Cys6 transcription factor gene (rolP) containing two restriction sites (EcoRI and XbaI).
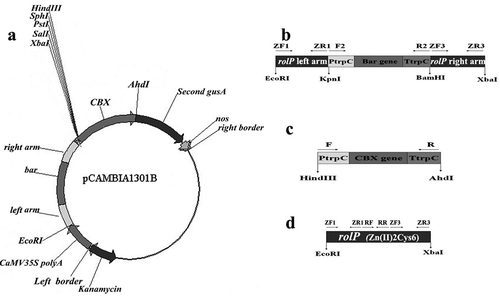
Table 1. Primers used for the reconstruction of the binary vector.
The overexpression vector pCAMBIA1301C was constructed based on the vector p1301. Briefly, the fragment of rolP was amplified by PCR using two primers (ZF1 and ZR3) tailed with EcoRI and XbaI restriction sites, respectively (). Afterwards, this fragment was double-digested with EcoRI/XbaI and inserted into the p1301 after digestion with the same enzymes. The recombinant vector was named pCAMBIA1301C, containing HindIII-PtrpC-CBX-TtrpC-AhdI and EcoRI-rolP-XbaI fragments, and was transferred into A. tumefaciens strain AGL-1.
Verification of rolP mutants by PCR and southern blot analysis
To confirm the success of gene knockout, genomic DNA was extracted from the transformants. PCR was carried out to amplify rolP using the primers ZF1 and ZR3 (). The PCR conditions were 95°C for 5 min; 30 cycles of 94°C for 30 s, 65.2°C for 1 min 30 s, 72°C for 2 min; and a final cycle of 72°C for 10 min. DNA from the wild-type strain P36-1 was used as the positive control.
For Southern blot analysis, 20 ug of genomic DNA from the P. lilacinus wild type P36-1 or transformants was digested with EcoRI/XbaI (for rolP gene) or HindIII/AhdI (for CBX) and fractionated on a 0.75% agarose gel. Depurination, capillary blotting to nylon membrane and UV-crosslinking were performed according to Brown (Citation2001). Blots were prehybridized for 30 min at 65°C in hybridization solution (250 mM sodium phosphate, 1 mM EDTA, 7% SDS, pH 7.2), hybridized overnight in the same solution with digoxigenin-labelled rolP fragments amplified by gene-specific primer pair RF/RR or CBX-A/CBX-B () and digoxigenin (DIG)-dUTP (Roche, Germany). After hybridization, the blot was washed at 65°C twice for 15 min with 5 × SSC plus 0.1% SDS, once for 15 min with 1 × SSC plus 0.1% SDS, and finally for 15 min in 0.1 × SSC plus 0.1% SDS.
Growth and sporulation test
The wild type and confirmed gene knockout strains (ΔrolP) were cultured on PDA for 9 days at 28°C in the dark. One mycelial disc (5 mm) from the edge of each colony was transferred to fresh medium (15 mL of 4% PDA in 9-cm diameter Petri dish). All Petri dishes were incubated in the dark at 28°C. After 48 h, the diameter of each colony was measured daily in two perpendicular directions until 9 days. For sporulation, the spores on 9-day-old plates were collected in sterilized distilled water and counted using a hemocytometer. The experiment was repeated twice with three replicates each.
Extraction and detection of mycotoxin
The mycotoxins ‘leucinostatins’ were extracted from the WT and the ΔrolP strains following Mikami et al. (Citation1989). Briefly, mycelia from PDA plates were inoculated into a 250-mL Erlenmeyer flask containing 100 mL of medium B and incubated on a shaker at 200 rpm in 28°C for 15 days. One hundred millilitres of the culture filtrate was adjusted to pH 3.0 with 1 M HCl and extracted with ethyl acetate of the same volume. The extract was washed with 5% NaHCO3 and concentrated in vacuo. Crude paecilotoxin (leucinostatins) was dissolved in a small amount of methanol and analysed using MALDI-TOF MS. The experiment was repeated twice with three replicates.
Nematode bioassay
The pathogenicity of WT and ΔrolP strains was examined on M. incognita. A total of 0.5 mL of conidial suspension (106 conidia mL−1) or cell-free filtrate (collected from 20-day medium B culture of P. lilacinus by centrifugation and filtered through a sterile 0.22-μm filter) was mixed with 0.5 mL of suspension containing 100 M. incognita eggs or juvenile. The control treatment consisted of 0.5 mL of medium B mixed with 0.5 mL of the M. incognita (eggs or juvenile) suspension. All treatments were incubated at 28°C. The corrected egg parasitism ratio and corrected nematode mortality rates were recorded. Each isolate was examined at 48-h intervals. The infection process of eggs and second-stage juvenile (J2) mortality were investigated under a light microscope. The experiment was repeated twice with three replications for each treatment. The corrected mortality rates were estimated according to the formula of Abbott (Citation1925): {(Ca-Ta)/Ca}, where Ca = number of live J2 or unparasitized eggs in the control after treatment, and Ta = number of live J2 or unparasitized eggs after treatment.
Semi-quantitative RT-PCR analysis
Mixtures of M. incognita (100 juveniles + 100 eggs per 0.5 mL) were prepared, and then infected with 0.5 mL of a conidial suspension (106 conidia mL−1) of the WT or ΔrolP strains of P. lilacinus. Total RNA was extracted from infected nematodes at different time points (12, 36, 48, 60, 72 and 96 h after infection) using the Trizol Total RNA Isolation Kit (Invitrogen, Shanghai, China). The residual genomic DNA was removed by treating the RNA sample with RNase-free DNase (Invitrogen). Five micrograms of total RNA was used and cDNA synthesis was performed using the oligo dT primer with the AMV First Strand cDNA Synthesis Kit (Invitrogen) following the manufacturer’s instructions. For semi-quantitative reverse transcription PCR (SqRT-PCR), the primer pair ZF1/ZR1 was used to amplify a partial sequence of the rolP gene. The β-tubulin gene was used as an endogenous control with the primers TubF (5′ACAACCGCCAACATGCGTGAGATTGTGA3′) and TubR (5′CTCCATCTCGTCCATTCCTTTCGCCTG). SqRT-PCR reactions were performed in 10 µL solution containing 2 µL of cDNA template (19.2 ng µL−1), 3 µL of specific primers (100 nM concentration for each) and 5 µL of the PCR Mix (CWbio, Beijing, China). The PCR reactions were performed at 95°C for 10 min and 40 cycles of 30 s at 95°C, 1 min at 65.2°C and 1 min at 72°C. The amplified PCR products were detected by 1.5% agarose gel electrophoresis, and the images were analysed using TotalLab v11 software (www.trtallab.com). The experiment was repeated twice (three replications for each experiment).
Effect of carbon and nitrogen sources on rolP gene expression and larvicidal activity
The WT and ΔrolP strains were cultured at 28°C on a rotary shaker at 200 rpm in 250-mL flasks containing 100 mL of medium B with different carbon or nitrogen sources. The medium B contained sucrose (19.98 g L−1 carbon) and peptone (0.65 g L−1 nitrogen) as a carbon and nitrogen source, respectively. For the carbon test, sucrose was replaced by D-glucose, D-fructose, maltose, sorbitol or lactose, and for the nitrogen test, peptone was replaced by (NH4)2SO4, (NH4)2CO3, NH4NO3, KNO3 or L-alanine. The unchanged medium B was applied as a control. To ensure the same conditions for all samples, the amount of carbon or nitrogen was adjusted to be constantly fixed at specific sets of conditions. The initial pH was adjusted to 7.0 prior to sterilization using 1 M HCl. After 15 days of culturing, the mycelia were collected by centrifugation and subjected to total RNA extraction. The cell-free filtrates were mixed with juvenile water suspension (100 J2 per 0.5 mL) of M. incognita (0.5 mL cell-free filtrate + 0.5 mL nematode suspension) to test the nematode mortality rate. The control contained 0.5 mL of cell-free filtrate in medium B (unchanged) mixed with 0.5 mL of M. incognita juvenile suspension (100 J2 per 0.5 mL). The experiment was repeated twice with three replicates for each treatment.
Effect of pH on P. lilacinus rolP gene expression and virulence against nematodes
The initial pH of medium B was adjusted from 3.0 to 8.0 by adding 1 M HCl or 1 M NaOH. Thereafter, 1 mL of WT conidial suspension (106 conidia mL−1) was transferred into medium B and cultured at 28°C. After 15 days, the conidia and mycelia were collected for total RNA extraction. Next, 0.5 mL of cell-free filtrates and 0.5 mL of juvenile water suspension (100 J2 of M. incognita per 0.5 mL) were mixed to test the nematode mortality rate. The control consisted of 0.5 mL of cell-free filtrate in medium B mixed with 0.5 mL of M. incognita juvenile suspension (100 J2 per 0.5 mL). The experiment was repeated twice with three replicates per treatment.
Statistical analysis
Statistical analysis was carried out using SPSS 12.0.1. The data were presented as the means ± SD. All of the data were subjected to one-way analysis of variance and, when appropriate, a multiple-range test was conducted using Duncan’s multiple range test at P ≤ 0.05.
Results
Cloning of rolP and sequence analysis
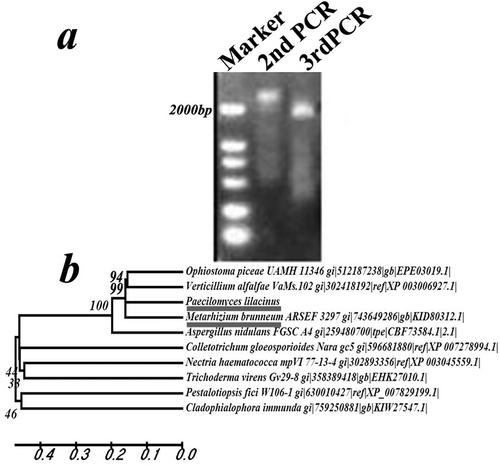
A 2005-bp fragment was amplified from the P. lilacinus mutant strain R147 by Tail-PCR (). The rolP gene sequence was deposited in GenBank under the accession number KR023984. The 2005-bp fragment was cloned using the primers R1/R2/R3 and AD and the previously obtained 1000-bp fragment was cloned using the primers L1/L2/L3 and AD. The two fragments were spliced and assembled in one 2897-bp fragment. Based on the homologous analysis, we found that the fragment part (2338-bp) is the full Zn(II)2Cys6 transcription factor commonly present in other fungal species. Therefore, the full-length gene was cloned and detected using the primers ZF1 and ZR3 (). The cDNA fragment was 2118 bp, encoding 705 amino acids. The DNA sequences contained three introns compared with the cDNA sequences (Supplementary Fig. 2a). Homologous analysis of the rolP gene sequence with other fungal species indicated that rolP was highly similar to its orthologues in Metarhizium brunneum, Verticillium alfalfae and Ophiostoma piceae (). Predictive analysis of the secondary structure of the rolP protein identified two conserved domains: a Zn(II)2Cys6 fungal-type DNA-binding domain (23–52 aa) and a fungal-specific transcription factor domain (269–523 aa) (Supplementary Fig. 2b).
Fig. 2 Molecular analysis of rolP. a, PCR products of the rolP gene of the R147 strain by the Tail-PCR method. b, Phylogenetic analysis of rolP and its homologues from other fungal species.
Validation and stability analysis of transformants
The amplification product of the knocked-out strains was 3468 bp of the EcoRI-left arm- KpnI-PtrpC-bar-TtrpC-BamHI-right arm-XbaI fragment using the primers ZF1 and ZR3, while it was 2338 bp for each of the overexpression and WT strains. The CBX marker gene fragment was 1652 bp in overexpression strains, and was not detected in knockout or WT strains (). This indicated that the plasmid had been integrated into the P. lilacinus strains through A. tumefaciens.
Fig. 3 Validation of the rolP knockout and overexpression strains. a, PCR analyses of WT and transformants. Lane 1, Takara 250 bp DNA ladder; Lanes 2–4, amplification of rolP. 2, the knockout strain ΔrolP, 3, the overexpression strain Ov-Pl36-1, 4, the wild type strain Pl36-1. Lanes 5–6, amplification of the CBX resistance gene. 5, the Ov-Pl36-1 strain and 6, the WT strain. b, Southern blot analysis of the rolP gene in the colonies of WT and transformants. Lane 1: The pCAMBIA1301B vector; Lane 2, the ΔrolP strain; Lane 3, the Ov-Pl36-1 strain; Lane 4, the WT strain. c, Southern blot analysis of the CBX gene in the colonies of WT and transformants. Lane 1, the pCAMBIA1301B vector; Lane 2, the Δrolp strain; Lanes 3, the Ov-Pl36-1 strain; Lane 4, the WT strain.
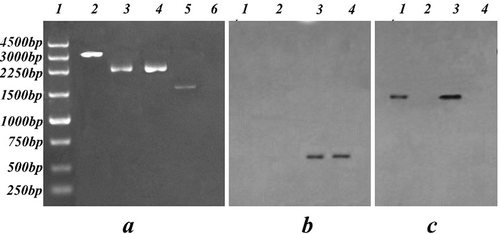
In Southern blot analysis, the overexpression and WT strains each produced a single band, while no bands were detected from ΔrolP when using the rolP gene middle-region (from RF to RR) as a probe (). To differentiate between the WT and the overexpression strain, the CBX gene was applied as a probe. The results showed a single band in the pCAMBIA1301B and overexpression vectors (lanes 1 and 3, respectively), while neither ΔrolP nor the WT strain showed any bands (lanes 2 and 4, respectively) (). The results confirmed that the ΔrolP and overexpression Ov-Pl36-1 strains were successfully obtained.
Effects of rolP gene knock-out and overexpression on growth and sporulation
The P. lilacinus 36-1, ΔrolP and Ov-Pl36-1 strains were cultured on PDA plates. The three strains appeared similar both in morphology and growth rate. The WT strain achieved a growth rate of 0.59 cm day−1 with a conidial concentration of 3.84 × 107 conidia mL−1, while the ΔrolP strain grew 0.50 cm day−1 with a conidial concentration of 3.40 × 107 conidia mL−1. The Ov-Pl36-1 strain achieved a growth rate of 0.55 cm day−1 with a conidial concentration of 3.56 × 107 conidia mL−1 ( and Supplementary Fig. 1).
Table 2. Radial growth and sporulation yield of P. lilacinus strains.
Metabolites and nematode bioassay tests
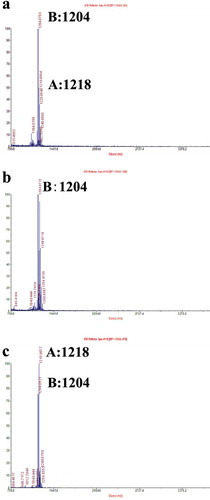
MALDI-TOF MS was used to detect and determine the crude production of leucinostatins for P. lilacinus 36-1 and its transformants. Both leucinostatins A and B were detected in the extracts of the WT strain (). In the ΔrolP strain, leucinostatin A was undetectable, while leucinostatin B was recorded at a high intensity (). In the Ov-Pl36-1 strain, both leucinostatins A and B were highly detectable, and leucinostatin A was recorded at a higher intensity rate, almost double the standard level, and leucinostatin B was recorded at a lower rate than WT ().
Fig. 4 (Colour online) Determination of leucinostatins from P. lilacinus strains. Leucinostatin production of P. lilacinus 36-1 and its transformants was determined by MALDI-TOF MS. Leucinostatin A molecular weight: 1218. Leucinostatin B molecular weight: 1204. a, The metabolites of the WT strain P. lilacinus 36-1. b, The metabolites of the knockout strain ΔrolP. c, The metabolites of the overexpressed strain Ov-Pl36-1.
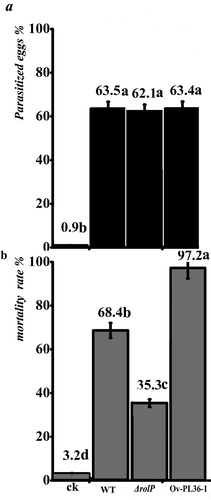
The pathogenicity test was carried out using the WT strain, the weakly virulent strain R147, ΔrolP and Ov-Pl36-1 against eggs or larvae of M. incognita. No significant difference could be detected among the strains regarding their ability to parasitize eggs, with parasitism rates ranging from 62.1% to 63.5% (). By contrast, significance was detected in the juvenile-corrected mortality rates among the concerned strains compared with the rate (68.4%) of the WT. J2-toxicity was minimized with ΔrolP strain recording a J2-corrected mortality of 35.3%, while the J2-corrected mortality was maximized to 97.2% with Ov-Pl36-1 ().
Fig. 5 Pathogenicity test of P. lilacinus strains against M. incognita. The pathogenicity of the P. lilacinus strains was determined by examining the effectiveness of each strain against eggs and second-stage juveniles (J2) of M. incognita. a, The results of corrected-parasitized eggs. b, The results of the J2-corrected mortality rate.
Expression analysis of the rolP gene at early stages of infection
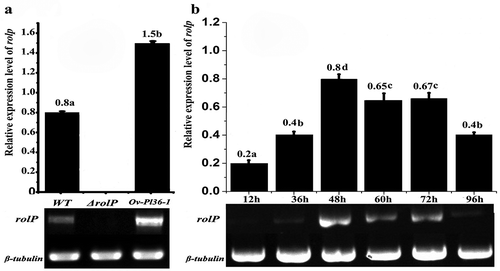
The expression levels of rolP in the WT and transformants during the nematode infection process were analysed by SqRT-PCR. Expression of rolP was detected in the WT and over-expressed strains but not in the ΔrolP strain. Moreover, the relative expression level of rolP in over-expressed strain was almost double that of the WT strain (). At different infection periods using the WT strain, the expression levels of the rolP gene were variable, while the expression levels of the β-tubulin gene were stable during different culturing periods. The rolP expression levels were highly detected (0.65–0.8) at infection periods between 48 and 72 h and peaked at 48 h. On the other hand, the expression levels were lower (0.2–0.4) at 12, 36 and 96 h ().
Fig. 6 Semi-quantitative reverse transcription PCR analysis of rolP genes in P. lilacinus. a, The expression levels of rolP in the WT, ΔrolP and Ov-pl36-1 strains at 48 h. b, The expression levels of rolP in the WT strain at different infection periods.
Influence of carbon and nitrogen sources in media on rolP gene expression and fungal virulence against root-knot juveniles
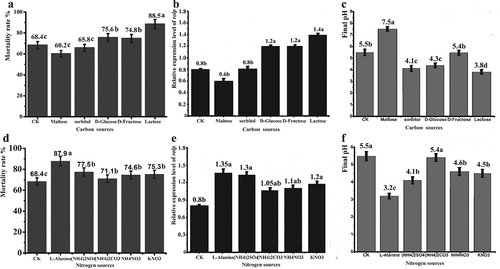
Because the components of media play an important role in the activity and toxin production of biocontrol agents, different carbon and nitrogen sources were applied to medium B to study their effect on WT rolP gene expression and toxicity of its cell-free culture against the root-knot nematode M. incognita. Among the five carbon sources, cell-free cultures with lactose, D-glucose or D-fructose achieved a significant increase in the J2-corrected mortality rates, recording values of 88.5%, 75.6% and 74.8%, respectively. Interestingly, the same carbon source treatments achieved the highest rolP expression in the WT strain, recording values of 1.4, 1.2 and 1.2, respectively. Conversely, maltose achieved the lowest corrected mortality rates and the lowest rolP expression (0.6) (). Regarding nitrogen sources, cell-free cultures with all tested nitrogen sources achieved significant J2-corrected mortality rates. The highest corrected mortality rate (87.9%) was achieved with L-alanine (3.30 g L−1), followed by (NH4)2SO4 (2.75 g L−1), (NH4)2CO3 (3.71 g L−1), NH4NO3 (0.9 g L−1) and KNO3 (2.27 g L−1). The same trend was achieved with the rolP expression level because all nitrogen sources recorded a significant increase in the WT rolP expression level. The highest expression level (1.35) was achieved with L-alanine (3.30 g L−1) (). The control (standard medium B) recorded a corrected mortality rate of 68.4% and a rolP gene expression level of 0.8. Regarding the cell-free culture of ΔrolP, its toxicity against J2 was very low (35.3%) and was not influenced by changing the carbon or nitrogen source (data not shown). As shown in and , the final pH values of cell-free culture indicated that an acidic environment might be more suitable for nematode toxicity and rolP gene expression.
Fig. 7 Effect of carbon and nitrogen sources on WT toxicity and rolP gene expression. The WT strain was cultured in medium B supplemented with various carbon and nitrogen sources. The toxicity of the cell-free culture was examined against root-knot J2. SqRT-PCR was used to examine the mRNA level of the rolP gene. a, b and c show medium B with carbon sources. d, e and f show medium B with nitrogen sources. The mortality rates of J2 are shown in (a) and (d). The relative expression levels of the rolP gene are shown in (b) and (e). The initial and final pH values are shown in (c) and (f). The data shown here are the means ± SD (n = 3). *, P ≤ 0.05.
Influence of an acidic environment on rolP gene expression and fungal virulence against root-knot juveniles
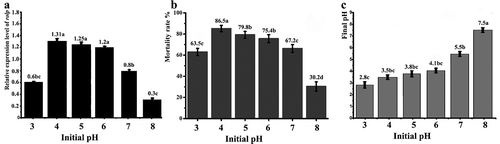
To confirm that relatively low pH values are favourable for rolP expression, the WT strain was cultured in medium B with different pH values ranging from 3 to 8. The rolP expression level was significantly higher in acidic media (pH 4–6) than in the control media (pH 7.0). The rolP expression value was 0.8 in the control, increased steadily with medium acidity and peaked at an initial pH of 4, with an expression level of 1.31. The expression levels were significantly reduced to less than those of the control with over-acidic (pH 3.0) media, resulting in an expression level of 0.6. Similarly, the expression level was decreased (0.8) with alkaline medium at pH 8 (). Bioassay tests using the obtained cell-free filtrates from the WT strain confirmed that the acidic media with pH values ranging from 4 to 6 achieved the highest M. incognita-corrected mortality rates (75.4–86.5%), while the corrected mortality rates were sharply decreased (30.2%) when the pH was 8 (). The final pH values of WT cell-free filtrates are presented in .
Fig. 8 Effect of pH on WT toxicity and the rolP gene. The WT strain was cultured in medium B with different pH values ranging from3.0 to 8.0. a, The mRNA level of the rolP gene. b, The mortality rates of root-knot J2 in cell-free culture. c, Final pH values of cell-free culture. The data shown here are the means ± SD (n = 3). *, P ≤ 0.05.
Discussion
The zinc cluster binuclear protein family is exclusively identified from fungal species and has the conserved motif CysX2CysX6CysX5-12CysX2CysX6-8Cys (MacPherson et al. Citation2006). This family has been well characterized in Saccharomyces cerevisiae (Akache et al. Citation2001). Zinc cluster proteins are also found in various other fungi, such as Kluyveromyces lactis (Witte & Dickson Citation1988), Schizosaccharomyces pombe (Goldar et al. Citation2005), Colletotrichum lagenarium (Tsuji et al. Citation2000) and Aspergillus nidulans (Gomez et al. 2002).
Zn(II)2Cys6 transcription factor genes have been shown to encode transcription regulators that control infection capability (Idnurm & Howlett Citation2001) and produce certain secondary metabolites, such as toxins, in fungi (Bok et al. Citation2006). One important characteristic of filamentous fungi is their production of a wide range of secondary metabolites. Many of these natural compounds are important. For instance, aflatoxins are secondary metabolites produced by some Aspergillus species (Sanchez et al. Citation2012). Interestingly, aflatoxin induction is regulated by the zinc cluster protein AflR. AflR positively regulates the expression of these genes by binding to the consensus motif TCGSWNNSCGR found in the promoters of the target genes (Ehrlich et al. Citation1999). Cmr1p is another example of a gene encoding a zinc cluster protein that contributes to secondary metabolite induction. Colletotrichum lagenarium is a plant pathogen that causes anthracnose of cucumbers. Plant infection by this fungus requires the production of melanin for effective appressorium penetration (Kubo & Furusawa Citation1991). Cmr1p is a positive regulator of the expression of melanin biosynthetic structural genes SCD1 and THR1 in this species (Tsuji et al. Citation2000).
The present study is the first attempt to characterize the putative Zn(II)2Cys6 transcription factor rolP of the filamentous fungus P. lilacinus. rolP was found to be a regulator of the production of the nematotoxic leucinostatins and is required for fungal pathogenicity. MALDI-TOF MS detected both leucinostatins A and B from the extracts of the WT strain (). In the ΔrolP strain, leucinostatin A was undetectable while in the Ov-Pl36-1 strain, both leucinostatins A and B were highly detectable. In addition, the results of the pathogenicity test were in harmony with those of leucinostatin detection. Although no significant differences were detected among all tested strains in parasitizing eggs, the toxicity against root-knot juveniles was minimized with the knockout strain ΔrolP, while the toxicity was maximized with the Ov-Pl36-1 strain (). Moreover, the toxicity of the cell-free culture of the WT strain was significantly improved when the fungus was cultured in acidic media or amended with specific sources of carbon or nitrogen (, ). Interestingly, rolP gene expression was increased under these specific conditions, indicating that these factors might stimulate rolP gene activity and increase leucinostatin production.
Several lines of evidence suggested that rolP is required for the pathogenicity of P. lilacinus against nematode juveniles. First, the zinc cluster binuclear family member Zn(II)2Cys6 is functional in fungal physiology, particularly primary and secondary metabolism (MacPherson et al. Citation2006), directly reflecting toxin production and the pathogenicity of certain fungi. Second, the expression level of the rolP gene increased when root-knot nematodes were infected by the WT strain. Third, rolP deletion resulted in the absence of leucinostatin A accompanied by a strong depletion of fungal toxicity against nematode juveniles, while rolP overexpression resulted in a high increase in leucinostatin A as well as fungal toxicity against nematode juveniles. Fourth, media supplemented with sole carbon or nitrogen sources significantly influenced the toxicity of the WT strain against nematode juveniles but had no effect on the ΔrolP. Thus it could be concluded that rolP is required for the production of leucinostatins and is a leucinostatin regulatory gene in P. lilacinus. Although the present study did not investigate the induction mechanism of leucinostatins, it paves the way to analyse the functions of these transcription factor genes in relation to the pathway-specific regulation of secondary metabolite biosynthesis of the rolP gene. Further studies are needed to investigate the induction mechanism of leucinostatins, the structural and conformation changes that occur upon leucinostatin binding to its targets, and the effects of such binding.
Supplemental data
Supplemental data for this article can be accessed here:http://dx.doi.org/10.1080/07060661.2015.1065437
Supplemental Figure 3
Download JPEG Image (146.6 KB){kind=link}
Supplemental Figure 2
Download JPEG Image (4.2 MB){kind=link}
Supplemental Figure 1
Download JPEG Image (3.2 MB){kind=link}
Supplemental Table 2
Download MS Word (42.5 KB)Supplemental Table 1
Download MS Word (35.5 KB)Acknowledgements
The authors would like to thank Dr Yanping Fu for providing the AGL-1 strain. In addition, we acknowledge the Talented Young Scientists Programme of Benha University, Egypt.
Additional information
Funding
References
- Abbott WS. 1925. A method of computing the effectiveness of an insecticide. J Econ Entomol. 18:265–267.
- Akache B, Wu KQ, Turcotte B. 2001. Phenotypic analysis of genes encoding yeast zinc cluster proteins. Nucleic Acids Res. 29:2181–2190.
- Anastasiadis IA, Giannakou IO, Prophetou-Athanasiadou DA. 2008. The combined effect of the application of a biocontrol agent Paecilomyces lilacinus, with various practices for the control of root-knot nematodes. Crop Prot. 27:352–361.
- Baum JA, Geever R, Giles NH. 1987. Expression of qa-1F activator protein: identification of upstream binding sites in the qa gene cluster and localization of the DNA-binding domain. Mol Cell Biol. 7:1256–1266.
- Bok JW, Chung D, Balajee SA, Marr KA, Andes D, Nielsen KF, Frisvad JC, Kirby KA, Keller NP. 2006. GliZ, a transcriptional regulator of gliotoxin biosynthesis, contributes to Aspergillus fumigatus virulence. Infect Immun. 74:6761–6768.
- Brown T. 2001. Southern blotting. Current protocols in molecular biology, unit 2.9A. Hoboken (NJ): John Wiley & Sons.
- Bundock P, Dulk-Ras A, Beijersbergen A. 1995. Transkingdom T-DNA transfer from Agrobacterium tumefaciens to Saccharomyces cerevisiae. EMBO J. 14:3206–3214.
- Cutler SR, Ehrhardt DW, Griffitts JS. 2000. Random GFP: cDNA fusions enable visualization of subcellular structures in cells of Arabidopsis at a high frequency. Proc Natl Acad Sci USA. 97:3718–3723.
- Das S, Lyla PS, Khan A. 2006. Marine microbial diversity and ecology: importance and future perspectives. Curr Sci. 90:1325–1334.
- Ehrlich KC, Montalbano BG, Cary JW. 1999. Binding of the C6-zinc cluster protein, AFLR, to the promoters of aflatoxin pathway biosynthesis genes in Aspergillus parasiticus. Gene. 230:249–257.
- Goldar MM, Jeong HT, Tanaka K, Matsuda H, Kawamukai M. 2005. Moc3, a novel Zn finger type protein involved in sexual development, ascus formation, and stress response of Schizosaccharomyces pombe. Curr Genet. 48:345–355.
- Hajdukiewicz P, Svab Z, Maliga P. 1994. The small, versatile pPZP family of Agrobacterium binary vectors for plant transformation. Plant Mol Biol. 25:989–994.
- Idnurm A, Howlett BJ. 2001. Pathogenicity genes of phytopathogenic fungi. Mol Plant Pathol. 2:241–255.
- Khan A, Williams K, Molloy MP. 2003. Purification and characterization of a serine protease and chitinases from Paecilomyces lilacinus and detection of chitinase activity on 2D gels. Prot Expr Purif. 32:210–220.
- Khan A, Williams K, Nevalainen H. 2004. Effects of Paecilomyces lilacinus protease and chitinase on the eggshell structures and hatching of Meloidogyne javanica juveniles. Biol Control. 31:346–352.
- Kiewnick S, Sikora RA. 2006. Biological control of the root-knot nematode Meloidogyne incognita by Paecilomyces lilacinus strain 251. Biol Control. 38:179–187.
- Kubo Y, Furusawa I. 1991. Melanin biosynthesis: prerequisite for successful invasion of the plant host by appressoria of Colletotrichum and Pyricularia. In: Cole GT, Hoch HC, editors. The fungal spore and disease initiation in plants and animals. New York: Plenum Publishing; p. 205–217.
- Lucero HA, Lescano WIM, Vallejos RH. 1978. Inhibition of energy conservation reactions in chromatophores of Rhodospirillum rubrum by antibiotics. Arch Biochem Biophys. 186:9–14.
- Luc M, Sikora RA, Bridge J. 2005. Plant parasitic nematodes in subtropical and tropical agriculture. Wallingford, Oxford: CAB International.
- MacPherson S, Larochelle M, Turcotte B. 2006. A fungal family of transcriptional regulators: the zinc cluster proteins. Microbiol Mol Biol Rev. 70:583–604.
- Marmorstein R, Carey M, Ptashne M, Harrison SC. 1992. DNA recognition by GAL4: Structure of a protein-DNA complex. Nature. 356:408–414.
- Mikami Y, Yazawa K, Fukushima K, Arai T, Udagawa S, Samson RA. 1989. Paecilotoxin production in clinical or terrestrial isolates of Paecilomyces lilacinus strains. Mycopathologia. 108:195–199.
- Morton CO, Kerry B, Hirsch P. 2004. Infection of plant-parasitic nematodes by nematophagous fungi – a review of the application of molecular biology to understand infection processes and to improve biological control. Nematology. 6:161–170.
- Park JO, Hargreaves JR, McConville EJ. 2004. Production of leucinostatins and nematicidal activity of Australian isolates of Paecilomyces lilacinus (Thom) Samson. Lett App Microbiol. 38:271–276.
- Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual. New York: Cold Spring Harbor Laboratory Press. Cold Spring Harbor; 1626 pp.
- Sanchez JF, Somoza AD, Keller NP, Wang CC. 2012. Advances in Aspergillus secondary metabolite research in the post-genomic era. Nat Prod Rep. 29:351–371.
- Schneider G. 2005. Enzymes in the biosynthesis of aromatic polyketide antibiotics. Curr Opin Struct Biol. 15:629–636.
- Shima A, Fukushima K, Arai T, Terada H. 1990. Dual inhibitory effects of the peptide antibiotics leucinostatins on oxidative phosphorylation in mitochondria. Cell Struct Funct. 15:53–58.
- Siddiqui ZA, Futai K. 2009. Biocontrol of Meloidogyne incognita on tomato using antagonistic fungi, plant growth-promoting rhizobacteria and cattle manure. Pest Manag Sci. 65:943–948.
- Tang G-L, Cheng Y-Q, Shen B. 2006. Polyketide chain skipping mechanism in the biosynthesis of the hybrid nonribosomal peptide-polyketide antitumor antibiotic leinamycin in Streptomyces atroolivaceus S-140. J Nat Prod. 69:387–393.
- Todd RB, Andrianopoulos A. 1997. Evolution of a fungal regulatory gene family: the Zn(II)2Cys6 binuclear cluster DNA binding motif. Fungal Genet Biol. 21:388–405.
- Tsuji G, Kenmochi Y, Takano Y, Sweigard J, Farrall L, Furusawa I, Horino O, Kubo Y. 2000. Novel fungal transcriptional activators, Cmr1p of Colletotrichum lagenarium and Pig1p of Magnaporthe grisea, contain Cys2His2 zinc finger and Zn(II)2Cys6 binuclear cluster DNA-binding motifs and regulate transcription of melanin biosynthesis genes in a developmentally specific manner. Mol Microbiol. 38:940–954.
- Tunlid A, Jansson S. 1991. Proteases and their involvement in the infection and immobilization of nematodes by nematophagous fungus Arthrobotrys oligospora. Appl Environ Microbiol. 57:2868–2872.
- Wang JP, Wang JX, Liu F, Pan C. 2010. Enhancing the virulence of Paecilomyces lilacinus against Meloidogyne incognita eggs by overexpression of a serine protease. Biotechnol Lett. 32:1159–1166.
- Witte MM, Dickson RC. 1988. Cysteine residues in the zinc finger and amino acids adjacent to the finger are necessary for DNA binding by the LAC9 regulatory protein of Kluyveromyces lactis. Mol Cell Biol. 8:3726–3733.
- Yang F, Abdelnabby H, Xiao Y 2015. Novel point mutations in β-tubulin gene for carbendazim resistance maintaining nematode pathogenicity of Paecilomyces lilacinus. Eur J Plant Pathol. (in press). doi: 10.1007/s10658-015-0665-0.
- Yang JK, Tian BY, Liang LM, Zhang K-Q. 2007. Extracellular enzymes and the pathogenesis of nematophagous fungi. Appl Microbiol Biotech. 75:21–31.