
Abstract
The ascomycete fungus Ophiostoma novo-ulmi is the highly aggressive pathogen responsible for the current, highly destructive, pandemic of Dutch elm disease (DED). In spite of its economic and ecological impact, O. novo-ulmi is not considered a model species, even though this fungus is easily grown in the laboratory and amenable to standard genetic and molecular investigations. The nuclear genomes of O. novo-ulmi and related species O. ulmi were recently sequenced and annotated, thus providing new opportunities for deciphering the complex basis of parasitic fitness in these pathogens. Comparative in silico analyses with genomes from other, well-characterized fungal and bacterial genomes have confirmed that the DED pathogens possess several hundred orthologues of genes encoding putative pathogenicity factors. Physiological, molecular, genomic and transcriptomic approaches are currently being used for studying yeast-mycelium dimorphism, a trait that the DED fungi share with several other pathogens of plants and mammals. The response of O. novo-ulmi to specific external stimuli suggests that oxylipins may be involved in yeast-mycelium transition. Orthologues of genes encoding enzymes involved in the biosynthesis of oxylipins are present in the nuclear genome of O. novo-ulmi. Genome-wide analyses of gene expression in the yeast and mycelial phases have revealed that over 10% of the nuclear genes are differentially expressed between these conditions. Comparisons with transcriptomes of the human pathogens Candida albicans and Histoplasma capsulatum showed that regulation patterns differed among the three species, thus highlighting the importance of developing resources for non-model organisms such as the DED fungi.
Résumé
Le champignon ascomycète Ophiostoma novo-ulmi est l’agent pathogène très agressif qui est responsable d’une pandémie de maladie hollandaise de l’orme (MHO) dévastatrice. En dépit de son impact économique et écologique, O. novo-ulmi n’est pas considéré comme une espèce modèle, bien que ce champignon soit facilement cultivé en laboratoire et qu’il se prête aux analyses génétiques et moléculaires. Les génomes nucléaires d’O. novo-ulmi et de l’espèce apparentée O. ulmi sont depuis peu séquencés et annotés, ce qui offre de nouvelles opportunités pour élucider la nature complexe du parasitisme chez ces agents pathogènes. Des comparaisons in silico avec des génomes bien caractérisés chez des espèces bactériennes ou fongiques ont confirmé que les agents de la MHO possèdent plusieurs centaines de séquences orthologues de gènes présumés encoder des facteurs de pathogénie. Des approches physiologiques, moléculaires, génomiques et transcriptomiques sont mises à contribution pour l’analyse du dimorphisme levure-mycélium, un trait que les agents de la MHO partagent avec plusieurs autres agents pathogènes des plantes et des mammifères. La réponse d’O. novo-ulmi à certains stimuli externes spécifiques suggère l’implication d’oxylipines dans la transition levure-mycélium. Des orthologues de gènes encodant des enzymes impliquées dans la biosynthèse des oxylipines sont présents dans le génome nucléaire d’O. novo-ulmi. Des analyses d’expression génique à l’échelle du génome ont révélé que plus de 10% des gènes nucléaires étaient différentiellement exprimés entre les phases levuriforme et mycélienne. Les comparaisons avec les transcriptomes des agents anthropopathogènes Candida albicans et Histoplasma capsulatum ont montré que les patrons de régulation différaient entre les trois espèces étudiées, illustrant ainsi l’importance de développer des ressources pour des organismes non modèles tels les agents de la MHO.
Introduction
The onset of Dutch elm disease (DED) in the early 1900s has been a catastrophic event for populations of elm species native to Europe and North America (). It has been estimated that over one billion adult trees were killed by two successive pandemics of DED caused by the ascomycete fungi Ophiostoma ulmi (Buisman) Nannf. and O. novo-ulmi Brasier (Paoletti et al. Citation2006). While elms have not been eradicated from these continents, the post-DED landscape certainly looks different from what it used to be. Prior to DED, elms were planted extensively in cities because they were considered highly resistant to disease and to abiotic stresses such as soil compaction, salt and desiccation. Large elms were also a common sight in the countryside. This is no longer the case. Many cities lost large numbers of elm trees and decided to replace them with other species. In North America, this led to extensive planting of white ash (Fraxinus americana L.) and Norway maple (Acer platanoides L.) which were considered highly resistant to diseases and pests. Nowadays, the former is being decimated by the emerald ash borer (Agrilus planipennis Fairmaire), whereas the latter is facing epidemics of tar spot since Rhytisma acerinum (Pers.) Fr., a native pathogen of A. platanoides in Eurasia, was accidentally introduced in North America (Hudler et al. Citation1987).
Fig. 1 (Colour online) Symptoms of Dutch elm disease (DED): (A) extensive crown dieback in a mature American elm (Ulmus americana) naturally infected by DED; note the healthy U. americana individual in the background; (B) close-up of external symptoms developing within 2 weeks on a U. americana sapling inoculated with Ophiostoma novo-ulmi under greenhouse conditions; (C) typical internal symptoms characterized by brown streaks in the xylem of a DED-infected U. americana individual. Streaks result from the oxidation of phenolics produced in response to infection (photo courtesy of G. Bussières, Université Laval).

Given the impact of DED, one might expect the causal agents to be considered as model tree pathogens. As can be seen from the title chosen for this paper, I will discuss genome-wide studies of O. novo-ulmi from the perspective that it is not a model species. The rationale for this position is simple: although O. novo-ulmi possesses several biological and genetic attributes that are desirable in a model organism (see Bernier et al. Citation2015 for a recent review), the small number of research teams currently working on the DED pathosystem throughout the world and the limited financial resources available mean that DED research is less ‘competitive’ than research on current model plant pathogenic fungi. It is my belief, however, that omics approaches can rapidly bring much new knowledge of the traits that contribute to parasitic fitness (sensu Andrivon Citation1993) in the DED fungi.
Things we knew about the DED fungi prior to next generation sequencing
It was 25 years ago that Brasier (Citation1991) proposed that two distinct species of Ophiostoma caused DED. Until then, the disease was thought to be caused by a single taxon, known as O. ulmi by the end of the 1980s (), which included two biotypes, the so-called non-aggressive and aggressive strains (Gibbs & Brasier Citation1973). Apart from obvious differences in virulence (used here in a quantitative way), colony morphology, growth temperature optima and production of a hydrophobin known as cerato-ulmin (reviewed in Brasier Citation1991), the strains could also be unequivocally distinguished by their electrophoretic soluble protein (Jeng & Hubbes Citation1983) and isozyme (Bernier et al. Citation1983) patterns. The reason, however, for conferring the status of species to each of the two strains was their strong reproductive isolation in nature. Eventually, a third species (O. himal-ulmi Brasier & M.D. Mehrotra) was described (Brasier & Mehrotra Citation1995), whereas O. novo-ulmi was split into two subspecies, novo-ulmi and americana (Brasier & Kirk Citation2001). Molecular markers based on restriction fragment length polymorphisms (RFLPs) and random amplified polymorphic DNA (RAPDs) were used to further characterize genetic variation in natural populations of DED fungi (e.g. Hintz et al. Citation1991; Hoegger et al. Citation1996). Comparisons of whole genomes by pulsed-field gel electrophoresis (PFGE) showed the occurrence of polymorphisms in chromosome size and number within O. ulmi and O. novo-ulmi (Dewar & Bernier Citation1993, Citation1995; Dewar et al. Citation1997; Et-Touil et al. Citation1999). Robust estimates of genome size in DED fungi, however, could not be obtained by PFGE-based approaches.
Table 1. Current nomenclature of the Dutch elm disease fungi.
While O. himal-ulmi is apparently confined to the western portion of the Himalayas where it behaves as an endophyte on Ulmus wallichiana Planch. (Brasier & Mehrotra Citation1995), O. ulmi and O. novo-ulmi have been overlapping for several decades in Europe and North America. This coexistence has led to documented cases of interspecific hybridization in several European locations (Brasier et al. Citation1998; Solla et al. Citation2008). In particular, an isolate of O. novo-ulmi subsp. novo-ulmi with unusually low virulence was found to be an introgressant carrying an O. ulmi allele at the Pat1 pathogenicity locus (Et-Touil et al. Citation1999). There is also strong molecular evidence that the MAT1-1 mating-type allele found in O. novo-ulmi populations was acquired though interspecific hybridization with O. ulmi (Paoletti et al. Citation2006). The impact of this event was major since it allowed O. novo-ulmi to take advantage of sexual reproduction. This, in turn, allows for hybridization between the two subspecies within O. novo-ulmi and there is evidence that it is a driver for the emergence of highly fit individuals (Brasier & Kirk Citation2010).
Mendelian analysis of progeny recovered from controlled O. ulmi × O. novo-ulmi interspecific crosses has shown that pathogenicity is under polygenic control (Brasier & Gibbs Citation1976). However, the actual number of pathogenicity genes and the nature of the proteins they encode remain elusive. The hydrophobin cerato-ulmin, secreted abundantly by O. novo-ulmi and O. himal-ulmi, was shown to affect components of fitness other than pathogenicity in experiments carried out with null mutants (Bowden et al. Citation1996) or with over-expressing mutants (Temple et al. Citation1997) obtained by genetic transformation (Royer et al. Citation1991). In spite of experimental evidence for elm cell wall degradation during pathogenesis (Svaldi & Elgersma Citation1982) and production of cell-wall degrading enzymes in vitro by the DED pathogens (e.g. Binz & Canevascini Citation1996, Citation1997), the role of these enzymes remains an open question. Knockout mutants of O. novo-ulmi that did not produce endopolygalacturonase were still pathogenic to elm saplings (Temple et al. Citation2009). Suggestions that toxins might be involved in pathogenesis (e.g. Scheffer et al. Citation1987) have not been verified by genetic or molecular analyses. Pathogenicity of O. novo-ulmi does not appear to be linked to its relative tolerance to elm phytoalexins since Proctor et al. (Citation1994) reported that mutants with increased sensitivity to mansonone E remained pathogenic to U. americana L.
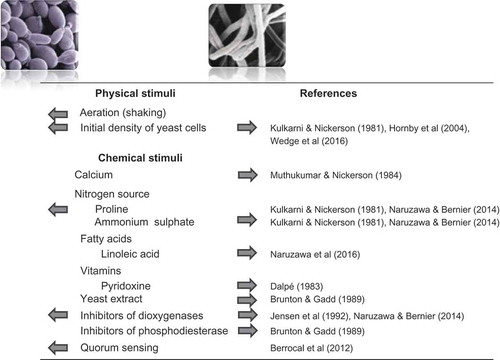
An intriguing feature of the DED fungi is their ability to switch between yeast-like and mycelial growth (). This trait is frequently encountered in Ophiostoma species, whether they are pathogens (such as the DED fungi) or saprobes (e.g. O. piceae (Münch) Syd. & P. Syd.). Yeast-mycelium dimorphism is also observed in several fungi that are pathogenic to humans (e.g. Candida albicans (C.P. Robin) Berkhout and the Ophiostomatoid fungus Sporothrix schenckii Hektoen & C.F. Perkins) or plants (e.g. Verticillium dahliae Kleb. and Ustilago maydis (DC.) Corda). In C. albicans, S. schenckii and U. maydis, a clear link has been established between pathogenicity and the ability to transition between the two growth forms (see Gauthier Citation2015 for a recent review). No such hard evidence exists for the DED fungi except for one report by Richards (Citation1994) that a spontaneous mutant of O. novo-ulmi deficient for the production of yeast-like spores and synnematal spores was also impaired for pathogenicity. Nevertheless, several physical and chemical factors affecting yeast-mycelium dimorphism have been documented in O. novo-ulmi (). It must be noted, however, that most of this work was conducted on a single isolate (NRRL6404) of O. novo-ulmi subsp. americana and that several experiments pre-dated the subdivision of the DED fungi into different species and subspecies. When the effect of external factors on yeast-mycelium was assessed on a subset of isolates representing the three DED-causing Ophiostoma species, important differences were observed among isolates irrespective of their taxonomic position (Naruzawa & Bernier Citation2014).
Fig. 2 (Colour online) Summary of the effect of various physical and chemical factors on the yeast-mycelium transition in the Dutch elm disease fungi (photos of yeast-like spores and mycelium courtesy of M. Aoun and K.V. Plourde, Université Laval). Arrows indicate the effect of a given stimulus. Initial cell density can have opposite effects: inoculation with a low yeast cell density stimulates mycelial growth, whereas a high concentration inoculum favours yeast growth.
Next generation sequencing of the O. ulmi and O. novo-ulmi genomes: the dawn of a new era?
The advent of next generation sequencing (NGS) technologies has made the sequencing of most fungal genomes an endeavour that is no longer restricted to a happy few research groups or consortia with large financial resources. As a result, the genomes of O. ulmi and O. novo-ulmi subsp. novo-ulmi were sequenced independently by two teams which both published results of their work in 2013. The genome of O. ulmi W9 (OuW9) was sequenced with Illumina GAIIx as paired-end reads and mate-paired reads, with an average coverage of 200X (Khoshraftar et al. Citation2013). The final 31.5 Mb assembly contained 3415 contigs. Annotation of the genome was facilitated by RNA sequencing (RNA-Seq) of a sample grown in the yeast phase, which provided transcriptomic confirmation for 80% of the 8639 predicted protein-coding gene models. A different strategy was used for sequencing the genome of O. novo-ulmi H327 (OnuH327), as the original goal of the project was to compare the performance of five different genomics core facilities using the same technology, namely the Roche/454 GS-FLX Titanium System, with an average coverage slightly over 60X×. Additionally, a single 8-kb, paired-end library was prepared and sequenced at one site. The final assembly was 31.8 Mb and included eight chromosome-length linear scaffolds, one circular mitochondrial contig of 66.4 kb, and one putative 4.2-kb linear plasmid. Based on ab initio predictions, the nuclear genome harboured 8613 protein coding genes, whereas the mitochondrion encoded 15 genes and 26 tRNAs (Forgetta et al. Citation2013). The number of predicted nuclear genes was subsequently increased to 8640 following further annotation including RNA-Seq data from the yeast-like and mycelial growth phases which provided coverage for 99% of the predicted gene models (Comeau et al. Citation2015). Repetitive elements account for only 3% of the nuclear genome of OnuH327 and include 1600 retrotransposons and 400 DNA transposons. Analysis of NGS data showed the occurrence of 22 regions of excessively high AT (excluding telomeres) distributed over all 8 chromosomes. These regions tended to harbour OPHIO DNA transposons (Bouvet et al. Citation2007) and SWING retrotransposons (Comeau et al. Citation2015). Most transposons found in the OnuH327 genome tended to be heavily methylated by repeat-induced point mutation (RIP) and are thus presumed to be inactive, with the exception of copies of OPHIO1 which have retained transposase activity (Bouvet et al. Citation2008).
Genome statistics for O. ulmi and O. novo-ulmi subsp. novo-ulmi genomes are summarized in , along with data for the closely related saprobe O. piceae (a sapstaining fungus frequently encountered in conifers) and the more distant Grosmannia clavigera (Rob.-Jeffr. & R.W. Davidson) Zipfel, Z.W. de Beer & M.J. Wingf. (a pathogen vectored by the mountain pine beetle, Dendroctonus ponderosae Hopkins). As is evident from this table, general features of the O. ulmi and O. novo-ulmi genomes are highly similar, including the number of predicted nuclear genes. Most (n = 8220; 95.1%) nuclear genes are shared by the two species (Nigg et al. Citation2015): less than 200 O. novo-ulmi open reading frames have no orthologs in O. ulmi or any other sequence in databases (Comeau et al. Citation2015). The number of predicted genes in the saprobe (O. piceae) is higher than in the three pathogens. Whether this observation is anecdotal or has a biological significance will require comparative analyses of a larger set of genomes from Ophiostomatoid saprobes and pathogens.
Table 2. Summary of genome statistics for Dutch elm disease pathogens and two other Ophiostomatales.
As mentioned previously, molecular evidence had been presented by Paoletti et al. (Citation2006) that O. novo-ulmi had acquired the MAT1-1 allele through interspecific hybridization with O. ulmi. This was further confirmed when the genomes of O. ulmi W9 (Mat1-2) and O. novo-ulmi H327 (Mat1-1) were aligned, as a large (~360 kb) portion of higher-than-average identity (>95%) sequence was observed around the MAT1 locus. Shorter regions that may have been introgressed from O. ulmi were also observed on all chromosomes of OnuH327 (Comeau et al. Citation2015). Moreover, the availability of whole-genome sequences for O. ulmi and O. novo-ulmi revealed that mating-type organization was more complex than initially thought. The typical architecture of the mating-type locus of heterothallic Sordariomycetes consists of three genes (MAT1-1-1, MAT1-1-2 and MAT1-1-3) in Mat1-1 isolates, and one gene (MAT1-2) in Mat1-2 isolates. The genomes of both OuW9 and OnuH327 were found to contain one additional, but divergent copy of MAT1-2, termed MAT1-2-1H327. This particular sequence is also present in several other Sordariomycetes, although it was not observed in the one isolate of O. himal-ulmi analysed (Comeau et al. Citation2015). The function of this gene is unknown. Although the OnuH327 genome also contains a gene encoding an orthologue of DNA excision repair protein ERCC-1 involved in mating-type switching, no evidence was obtained for mating-type switching in isolate H327 or other isolates tested by Comeau et al. (Citation2015).
Orthologues of genes that are known to encode pathogenicity and/or virulence factors are obvious candidates for further studies in the DED fungi. Comeau et al. (Citation2015) identified a subset of 1731 O. novo-ulmi genes which had orthologues in the Pathogen-Host Interactions Database (PHI-base; Urban et al. Citation2015), based on a Blast search (E value cutoff = e−20). This is almost three times the figure (n = 610) reported previously for O. ulmi by Khoshraftar et al. (Citation2013) who, however, had queried an earlier version of PHI-base which contained fewer reference sequences.
Sequences found in PHI-base include several genes encoding secreted proteins and genes encoding carbohydrate-active enzymes (CAZymes; Cantarel et al. Citation2009). As reported by Comeau et al. (Citation2015), O. novo-ulmi harbours relatively few CAZyme genes (n = 311) compared with other Sordariomycetes for which genomes are available, including the wilt fungi Fusarium oxysporum f.sp. lycopersici W.C. Snyder & H.N. Hanse and V. dahliae which have 694 and 559 CAZyme-encoding genes, respectively. One possible explanation for this large difference is that species such as O. novo-ulmi and G. clavigera (269 CAZyme-encoding genes) benefit from the help of bark beetles to get access to the host xylem, whereas F. oxysporum and V. dahliae do not (Comeau et al. Citation2015).
The arsenal of genes contributing to parasitic fitness also includes genes encoding enzymes that are able to detoxify and/or metabolize defence-related molecules produced by the host. A total of 48 cytochrome P450 oxidase (CYP450)-encoding genes were found in the OnuH327 genome (Comeau et al. Citation2015). Based on the nomenclature of Nelson (Citation2009), at least 16 of these genes may be involved in the degradation of plant defence compounds. Functional validation of one O. novo-ulmi gene orthologous to a gene encoding a CYP52P6 protein involved in defence against plant terpenes was attempted using quantitative PCR (qPCR). Results were deemed inconclusive since the accumulation of O. novo-ulmi Cyp52p6 transcripts did not differ significantly from that of Cyp51f1, a gene encoding membrane ergosterol which was considered a housekeeping gene and thus selected as control. However, Dai et al. (Citation2015) recently reported that transcription of an orthologue of Cyp51F1 in the Ophiostomatoid fungus Leptographium qinlingensis Tang & Chen increased significantly when cultures were exposed to monoterpenes. Therefore, the potential contribution of Cyp51f1 (and other CYP450 genes) to parasitic fitness of the DED fungi remains unknown and awaits further investigation.
Cytochrome P450 oxidases also play a role in the biosynthesis of secondary metabolites. The latter are generally encoded by batteries of genes arranged in clusters. It is noteworthy that the OnuH327 genome contains only seven clustered secondary metabolite pathway genes and that most clusters are incomplete. The exception is a cluster including genes involved in the biosynthesis of sterigmatocystin, a precursor of aflatoxin (Comeau et al. Citation2015).
As observed by Khoshraftar et al. (Citation2013), the fatty acid biosynthesis pathway is strikingly reduced in the O. ulmi W9 genome compared with Saccharomyces cerevisiae Meyen ex E.C. Hansen. The same is true for the O. novo-ulmi H327 genome which contains orthologues for S. cerevisiae genes encoding cytosolic acetyl-coA carboxylase (Acc1) and fatty acid synthases (Fas1 and Fas2) but no orthologues for the mitochondrial counterparts of these enzymes (Comeau et al. Citation2015). Furthermore, the OnuH327 genome harbours only one (Elo2) of the three elongase-encoding genes found in S. cerevisiae. This seemingly incomplete fatty acid biosynthesis pathway, however, does not mean that the DED fungi are incapable of synthesizing fatty acids de novo. Rather, Jensen et al. (Citation1992) reported that O. novo-ulmi NRRL6404 produced mostly linoleic acid (C18:2; 50% of all phospholipids extracted from yeast-like cells), followed by oleic acid (C18:1), palmitic acid (C16:0) and linolenic acid (C18:3), and low (less than 3%) concentrations of stearic acid (C18:0) and palmitoleic acid (C16:1). In contrast, the four major fatty acids produced by S. cerevisiae are palmitoleic acid and oleic acid, followed by palmitic acid and stearic acid (Klug & Daum Citation2014).
Unsaturated fatty acids (UFAs) are building blocks for a wide variety of complex lipids, including oxylipins. The latter result from the oxidation of UFAs by dioxygenase enzymes that include lipoxygenases (LOX) and cyclooxygenases (COX). Jensen et al. (Citation1992) had reported earlier that the presence of dioxygenase inhibitors in liquid cultures of O. novo-ulmi NRRL6404 converted mycelial growth to yeast growth. More specifically, propyl gallate (a LOX inhibitor) did have a significant effect, whereas COX inhibitors tested by these authors had no effect. In their study of yeast-mycelium dimorphism in the DED fungi, Naruzawa and Bernier (Citation2014) confirmed the effect of propyl gallate and reported that salicylic acid, a COX inhibitor that Jensen et al. (Citation1992) had not included in their study, also favoured yeast growth. Based on the above results, the DED fungi were expected to harbour Lox and Cox genes. A search for these genes in the OuW9 and OnuH327 genomes revealed the occurrence of two Cox genes but no Lox gene (Naruzawa Citation2015; Naruzawa et al. Citation2016). Since oxylipins have not been characterized in the DED fungi, it is not known whether they have a direct effect on yeast-mycelium dimorphism in these species. On the other hand, Naruzawa et al. (Citation2016) recently showed that exposure of O. ulmi and O. novo-ulmi to exogenous linoleic acid stimulated the formation of mycelium in liquid shake culture. Since linoleic acid is a substrate for oxylipin synthesis by cyclooxygenases, it is therefore possible that fatty acid metabolism is important for yeast-mycelium dimorphism in the DED fungi. Further experimental proof must await the recovery of mutants impaired for COX activity.
Genomic and biochemical data further suggest that the regulation of yeast-mycelium transition in the DED fungi extends beyond oxylipin and fatty acid metabolism. A query of the OuW9 and OnuH327 genomes shows that they harbour orthologues for genes belonging to the three pathways that have been characterized in model dimorphic fungi, namely the cyclic AMP-dependent protein kinase A (cAMP-PKA), the mitogen-activated protein kinase (MAPK) and the Pal/Rim cascades. Furthermore, yeast-mycelium dimorphism in the DED fungi is also influenced by cell density employed in the initial inoculum (Kulkarni & Nickerson Citation1981; Hornby et al. Citation2004; Wedge et al. Citation2016) and this, in turn, is a result of quorum sensing (Hornby et al. Citation2004). A fusel alcohol derived from isoleucine, 2-methyl-1-butanol, has been identified as the principal quorum sensing molecule in O. novo-ulmi NRRL6404 (Berrocal et al. Citation2012) and other molecules may be involved. Orthologues of genes regulated by quorum sensing in C. albicans (Bandara et al. Citation2012) are present in the OnuH327 genome and are obvious targets for molecular analyses of this phenomenon in the DED fungi.
Transcriptomic analyses of important biological traits of the DED fungi
The first genome-wide gene expression studies of DED fungi were based on expressed sequenced tags (ESTs). Resources developed for O. novo-ulmi included a collection of ESTs representing over 2000 different genes (Hintz et al. Citation2011) expressed in axenic yeast-like cultures and smaller sets of ESTs associated with asexual and sexual reproduction (Jacobi et al. Citation2010) and growth at 15 or 31°C (GenBank accession EG355849–EG356125). An attempt was made to recover large sets of fungal ESTs expressed during colonization of U. americana calli by O. novo-ulmi. To this end, expression libraries were enriched in upregulated genes by suppression subtractive hybridization (SSH). However, only four confirmed fungal sequences were recovered among the 314 unisequences that were annotated (Aoun et al. Citation2010).
Khoshraftar et al. (Citation2013) produced the first comprehensive transcriptome for a DED fungus. In this case, the transcriptome of a single yeast-phase culture of O. ulmi W9 was obtained by RNA-Seq, as a tool for validating gene models predicted in silico. This resource is useful since it provides a catalogue of the genes expressed during yeast growth and allows for comparisons with other fungal species with a yeast phase. It does not, however, provide information on the molecular bases of yeast-mycelium transition in DED fungi. As a first step towards this goal, the transcriptomes of the yeast and mycelial growth phases of O. novo-ulmi were obtained and compared (Nigg et al. Citation2015). Comparison of RNA-Seq data (n = 7341 genes retained after trimming and filtering) from both phases revealed that close to 12% of the gene content was differentially expressed between the two phases. In particular, several transcription factors and 11 CAZyme genes were overexpressed during yeast growth, whereas genes coding for serine/threonine kinases and mitogen-activated kinases (MAPK), 16 CAZyme genes and one COX gene were upregulated in mycelia. It is noteworthy that comparative genome analyses by Nigg et al. (Citation2015) showed that O. novo-ulmi shared many orthologues with the dimorphic human pathogens C. albicans (n = 2774) and Histoplasma capsulatum Darling (n = 5292). Yet, when the O. novo-ulmi yeast- and mycelial transcriptome profiles were compared with those of C. albicans and H. capsulatum, few orthologues showed similar regulation of expression between the two growth phases. Therefore, it is important to develop specific tools and resources for exploring regulatory pathways in non-model species such as O. novo-ulmi that are distantly related to the classical model fungal species.
Mating-type genes MAT1-1-1, MAT1-1-2 and MAT1-1-3 were part of the 899 predicted genes that were not detected in the O. novo-ulmi RNA-Seq dataset (Nigg et al. Citation2015). This observation is not surprising, as Jacobi et al. (Citation2010) had previously shown that MAT1-1-3 transcripts were significantly more abundant in perithecia and synnemata than mycelium. However, transcripts of gene g2571 encoding the MAT1-2-1H327 divergent mating-type 2 allele (Comeau et al. Citation2015) were abundant in both yeast and mycelium RNA-Seq samples. Moreover, there was no significant difference in the numbers of transcripts of the cerato-ulmin (CU) gene between yeast and mycelial cultures grown in liquid medium. This contradicted results from a previous study (Tadesse et al. Citation2003) in which the CU gene was shown to be upregulated in mycelial cells, based on qPCR analyses. Apart from the fact that two different analytical approaches were used, a likely explanation for this discrepancy is that all experiments described by Nigg et al. (Citation2015) were conducted in Ophiostoma complete medium (OCM), whereas Tadesse et al. (Citation2003) grew mycelia in OCM and yeasts in Ophiostoma minimal medium (OMM).
Comparisons between transcriptomes obtained from stable yeast and mycelial cell populations, while informative, provide a static assessment of differences in gene expression associated with these conditions. In a complementary, ongoing experiment, the dynamics of gene expression throughout transition from yeast to mycelium in liquid culture are being investigated in a time course RNA-Seq analysis. Preliminary results, based on the profiling of over 7500 genes, indicate that significant transcriptional shifts happen early (within 2 h) during the transition (Nigg et al. unpublished results).
The next steps
To the best of the author’s knowledge, genome-wide analyses of the DED fungi have not yet proceeded beyond what is described in the previous sections. Significant progress has been accomplished within the last few years, starting with the public release of the full genome sequences of O. ulmi and O. novo-ulmi. At the same time, NGS-based studies of the DED fungi have been carried out by a handful of research groups. Many more exciting developments are to be expected once additional groups enter this field, either directly or indirectly. Here are a few thoughts on some of the anticipated developments.
Data mining of the current genomic resources available for O. ulmi and O. novo-ulmi should help interested scientists identify gene sequences of interest. This approach has already been used by de Salas et al. (Citation2015) in their study of two quorum sensing-related genes in the saprobe O. piceae. Putative pathogenicity genes can also be readily identified among genes with orthologues in the PHI-base database. The occurrence, within Ophiostomatoid fungi, of several species complexes that include both pathogens and saprobes makes this group of organisms particularly attractive for comparative genomics of these different fungal lifestyles. These studies will be further facilitated once a larger set of genomes from saprophytic and pathogenic Ophiostomatoid fungi is available. As of January 2016, the genomes of 18 Ophiostomatoid species belonging to the Ophiostomatales (eight species) and the Microascales (10 species) had been deposited in the National Center for Biotechnology Information (NCBI) genome database (http://www.ncbi.nlm.nih.gov/genome; accessed on 18.1.2016) but several more are expected to become publicly available in 2016.
The advent of RNA-Seq, a quantitative sequencing-based method for mapping transcribed regions of the genome (Nagalakshmi et al. Citation2008), is another landmark in the development of NGS technologies. In addition to its advantages over older technologies (reviewed by Wang et al. Citation2009), RNA-Seq is particularly well suited for studying non-model organisms since it can detect transcripts for which there is no genomic sequence available. As mentioned previously, RNA-Seq data were used in the annotation of the genomes of O. ulmi (Khoshraftar et al. Citation2013) and O. novo-ulmi (Comeau et al. Citation2015), as well as in analyses of the yeast-mycelium dimorphism in O. novo-ulmi (Nigg et al. Citation2015). The relatively low cost of RNA-Seq will facilitate a more systematic analysis of gene expression associated with relevant biological traits of the DED pathogens. Several of these traits (e.g. response to physical or chemical stimuli, development of reproductive structures, and exposure to known plant defence metabolites) can be assessed in vitro under a variety of conditions. Transcriptomic analysis of pathogenesis will require obtaining RNA-Seq data from elms inoculated with the DED fungi. This is technically feasible, as shown recently by Perdiguero et al. (Citation2015), although methodological improvements are required for improving the yield of fungal sequences in experiments conducted in planta.
The efficiency of gene knockout protocols for O. novo-ulmi also needs to be substantially improved in order to fully exploit the new knowledge gained from NGS approaches. A few O. novo-ulmi mutants with altered phenotypes for mycelial growth (Pereira et al. Citation2000; Plourde et al. Citation2008) and virulence (Plourde & Bernier Citation2014) have been obtained by random insertional mutagenesis, whereas genes encoding cerato-ulmin (Bowden et al. Citation1996) and endopolygalacturonase (Temple et al. Citation2009) were selectively inactivated by targeted disruption. The latter, however, is difficult to achieve in O. novo-ulmi due to the action of a very efficient DNA repair system, based on non-homologous end joining (NHEJ), which is commonly encountered in filamentous ascomycetes. In Neurospora crassa Shear & B.O. Dodge, this problem was circumvented by inactivating the Mus52 gene involved in NHEJ and using a Δmus52 mutant in further gene replacement experiments (Ninomiya et al. Citation2004). This procedure, however, did not improve the efficiency of targeted gene replacement in O. novo-ulmi (Naruzawa Citation2015). Therefore, new approaches, including the CRISPR-Cas9 system of RNA-guided mutagenesis (Jinek et al. Citation2012; Nødvig et al. Citation2015), need to be examined in order to meet the challenge of producing mutants of DED fungi for large-scale functional validation of targeted genes. In the meantime, knockdown by RNA interference (Fire et al. Citation1998) provides an alternative procedure for investigating gene expression in O. novo-ulmi (Carneiro et al. Citation2010, Citation2013).
Functional analysis of mutants requires that efficient phenotyping assays are available. These may include phenotype microarray (PM) approaches similar to those used in the characterization of wild isolates of O. ulmi, O. novo-ulmi and fungal endophytes of elms (Blumenstein et al. Citation2015). Large-scale testing of pathogenicity, however, remains a challenge. Until a pathogenicity testing platform becomes available for DED fungi, an apple-based assay developed originally for Cryphonectria parasitica (Murrill) M.E. Barr (Fulbright Citation1984) can be used successfully for identifying O. novo-ulmi insertional mutants with altered virulence (Plourde & Bernier Citation2014).
Concluding remarks
The last decade has seen the emergence of technologies that hold great potential for accelerating the rate of discovery in non-model organisms, including the DED pathogen O. novo-ulmi. While omics approaches are often associated with large-scale initiatives that benefit from impressive research budgets, it is important to keep in mind that the democratization of NGS-based technologies also benefits small research teams with more modest funding. One of the challenges, in this case, is to plan biological experiments as efficiently as possible, in order to maximize the ‘return on the investment’ (in particular, fees for services from NGS sequencing- and bioinformatics platforms). Once good quality raw data files from genomic or transcriptomic analyses are received from the platforms, it is then mostly a matter of extracting as much as possible of biologically relevant information from the data, through appropriate statistical analyses and thorough searches of databases and scientific literature. Another challenge, which remains to be met successfully in O. novo-ulmi and several other important plant pathogens, is to design efficient procedures for the validation in vitro and in planta of genes identified in silico. This is, in my view, the next frontier which will hopefully be crossed within the next decade.
Acknowledgements
Obtaining a high-quality assembly of the O. novo-ulmi H327 genome was made possible by Ken Dewar (McGill University) who supervised this collaborative sequencing project through the Association of Biomolecular Resource Facilities (ABRF). I am also indebted to past and present members of my research team for their contributions to DED research over 25 years.
Additional information
Funding
References
- Andrivon D. 1993. Nomenclature for pathogenicity and virulence: the need for precision. Phytopathology. 83:889–890.
- Aoun M, Jacobi V, Boyle B, Bernier L. 2010. Identification and monitoring of Ulmus americana transcripts during in vitro interactions with the Dutch elm disease pathogen Ophiostoma novo-ulmi. Physiol Mol Plant Pathol. 74:254–266.
- Bandara HMHN, Lam OLT, Jin LJ, Samaranayake L. 2012. Microbial chemical signaling: a current perspective. Crit Rev Microbiol. 38:217–249.
- Bernier L, Aoun M, Bouvet GF, Comeau A, Dufour J, Naruzawa ES, Nigg M, Plourde KV. 2015. Genomics of the Dutch elm disease pathosystem: are we there yet? iForest. 8:149–157.
- Bernier L, Jeng RS, Hubbes M. 1983. Differentiation of aggressive and non-aggressive strains of Ceratocystis ulmi by polyacrylamide gel electrophoresis of intramycelial enzymes. Mycotaxon. 17:456–472.
- Berrocal A, Navarrete J, Oviedo C, Nickerson KW. 2012. Quorum sensing activity in Ophiostoma ulmi: effects of fusel oils and branched chain amino acids on yeast-mycelial dimorphism. J Appl Microbiol. 113:126–134.
- Binz T, Canevascini G. 1996. Xylanases from the Dutch elm disease pathogens Ophiostoma ulmi and Ophiostoma novo-ulmi. Physiol Mol Plant Pathol. 49:159–175.
- Binz T, Canevascini G. 1997. Purification and partial characterization of the extracellular laccase from Ophiostoma novo-ulmi. Curr Microbiol. 35:278–281.
- Blumenstein K, Macaya-Sanz D, Martín JA, Albrectsen BR, Witzell J. 2015. Phenotype MicroArrays as a complementary tool to next generation sequencing for characterization of tree endophytes. Front Microbiol. 6:1033.
- Bouvet GF, Jacobi V, Bernier L. 2007. Characterization of three DNA transposons in the Dutch elm disease fungi and evidence of repeat-induced point (RIP) mutations. Fungal Genet Biol. 44:430–443.
- Bouvet GF, Jacobi V, Plourde KV, Bernier L. 2008. Stress-induced mobility of OPHIO1 and OPHIO2, DNA transposons of the Dutch elm disease fungi. Fungal Genet Biol. 45:565–578.
- Bowden CG, Smalley E, Guries RP, Hubbes M, Temple B, Horgen PA. 1996. Lack of association between cerato-ulmin production and virulence in Ophiostoma novo-ulmi. Mol Plant-Microbe Interact. 9:556–564.
- Brasier CM. 1991. Ophiostoma novo-ulmi sp. nov., causative agent of current Dutch elm disease pandemics. Mycopathologia. 115:151–161.
- Brasier CM, Gibbs JN. 1976. Inheritance of pathogenicity and cultural characters in Ceratocystis ulmi: hybridization of aggressive and non-aggressive strains. Ann Appl Biol. 83:31–37.
- Brasier CM, Kirk SA. 2001. Designation of the EAN and NAN races of Ophiostoma novo-ulmi as subspecies. Mycol Res. 105:547–554.
- Brasier CM, Kirk SA. 2010. Rapid emergence of hybrids between the two subspecies of Ophiostoma novo-ulmi with a high level of pathogenic fitness. Plant Pathol. 59:186–199.
- Brasier CM, Kirk SA, Pipe ND, Buck KW. 1998. Rare interspecific hybrids in natural populations of the Dutch elm disease pathogens Ophiostoma ulmi and O. novo-ulmi. Mycol Res. 102:45–57.
- Brasier CM, Mehrotra MD. 1995. Ophiostoma himal-ulmi sp. nov., a new species of Dutch elm disease fungus endemic to the Himalayas. Mycol Res. 99:205–215.
- Brunton AH, Gadd GM. 1989. The effect of exogenously-supplied nucleosides and nucleotides and the involvement of adenosine 3ʹ:5ʹ-cyclic monophosphate (cyclic AMP) in the yeast mycelium transition of Ceratocystis (= Ophiostoma) ulmi. FEMS Microbiol Lett. 60:49–53.
- Buisman CJ. 1932. Ceratostomella ulmi, de geslachtelijke vorm van Graphium ulmi Schwarz [Ceratostomella ulmi, the sexual form of Graphium ulmi Schwarz]. Tijdschr PlZiekt. 38:1–8. Dutch.
- Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B. 2009. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res. 37:D233–D238.
- Carneiro JS, de la Bastide PY, Chabot M, Lerch L, Hintz WE. 2010. Suppression of polygalacturonasegene expression in the phytopathogenic fungus Ophiostoma novo-ulmi by RNA interference. Fungal Genet Biol. 47:399–405.
- Carneiro JS, de la Bastide PY, Hintz WE. 2013. Regulated gene silencing in the fungal pathogen Ophiostoma novo-ulmi. Physiol Mol Plant Pathol. 82:28–34.
- Comeau AM, Dufour J, Bouvet GF, Nigg M, Jacobi V, Henrissat B, Laroche J, Levesque RC, Bernier L. 2015. Functional annotation of the Ophiostoma novo-ulmi genome: insights into the phytopathogenicity of the fungal agent of Dutch elm disease. Genome Biol Evol. 7:410–430.
- Dai L, Li Z, Yu J, Ma M, Zhang R, Chen H, Pham T. 2015. The CYP51F1 gene of Leptographium qinlingensis: sequence characteristic, phylogeny and transcript levels. Int J Mol Sci. 16:12014–12034.
- Dalpé Y. 1983. L’influence de la carence en pyridoxine sur la morphologie et l’ultrastructure cellulaire de Ceratocystis ulmi [Influence of pyridoxine deficiency on the morphology and cellular ultrastructure of Ceratocystis ulmi]. Can J Bot. 61:2079–2084. French.
- de Hoog GS, Scheffer RJ. 1984. Ceratocystis versus Ophiostoma: a reappraisal. Mycologia. 76:292–299.
- de Salas F, Martínez MJ, Barriuso J. 2015. Quorum-sensing mechanisms mediated by farnesol in Ophiostoma piceae: effect on secretion of sterol esterase. Appl Environ Microbiol. 81:4351–4357.
- Dewar K, Bernier L. 1993. Electrophoretic karyotypes of the elm tree pathogen Ophiostoma ulmi (sensu lato). Mol Gen Genet. 238:43–48.
- Dewar K, Bernier L. 1995. Inheritance patterns of nonhomologous chromosomes in Ophiostoma ulmi sensu lato. Curr Genet. 27:541–549.
- Dewar K, Bousquet J, Dufour J, Bernier L. 1997. A meiotically reproducible chromosome length polymorphism in the ascomycete fungus Ophiostoma ulmi (sensu lato). Mol Gen Genet. 255:38–44.
- DiGuistini S, Wang Y, Liao NY, Taylor G, Tanguay P, Feau N, Henrissat B, Chan SK, Hesse-Orce U, Massoumi Alamouti S, et al. 2011. Genome and transcriptome analyses of the mountain pine beetle-fungal symbiont Grosmannia clavigera, a lodgepole pine pathogen. Proc Natl Acad Sci USA. 108:2504–2509.
- Et-Touil A, Brasier CM, Bernier L. 1999. Localization of a pathogenicity gene in Ophiostoma novo-ulmi and evidence that it may be introgressed from O. ulmi. Mol Plant-Microbe Inter. 12:6–15.
- Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. 1998. Potent and specific genetic interference by double-stranded RNA in Caernorhabditis elegans. Nature. 391:806–811.
- Forgetta V, Leveque G, Dias J, Grove D, Lyons Jr R, Genik S, Wright C, Singh S, Peterson N, Zianni M, et al. 2013. Comparison of multiple genome sequencing centers and analysis of the Dutch elm disease fungus genome using the Roche/454 GS-FLX titanium system. J Biomol Tech. 24:39–49.
- Fulbright DW. 1984. Effect of eliminating ds-RNA in hypovirulent Endothia parasitica. Phytopathology. 74:722–724.
- Gauthier GM. 2015. Dimorphism in fungal pathogens of mammals, plants, and insects. PLoS Pathog. 11:e1004608.
- Gibbs JN, Brasier CM. 1973. Correlation between cultural characters and pathogenicity in Ceratocystis ulmi from Britain, Europe and America. Nature. 241:381–383.
- Haridas S, Wang Y, Lim L, Massoumi Alamouti S, Jackman S, Docking R, Robertson G, Birol I, Bohlmann J, Breuil C. 2013. The genome and transcriptome of the pine saprophyte Ophiostoma piceae, and a comparison with the bark beetle-associated pine pathogen Grosmannia clavigera. BMC Genomics. 14:373.
- Hintz W, Pinchback M, De La Bastide P, Burgess S, Jacobi V, Hamelin R, Breuil C, Bernier L. 2011. Functional categorization of unique expressed sequence tags obtained from the yeast-like growth phase of the elm pathogen Ophiostoma novo-ulmi. BMC Genomics. 12:431.
- Hintz WE, Jeng RS, Hubbes M, Horgen PA. 1991. Identification of three populations of Ophiostoma ulmi (aggressive subgroup) by mitochondrial DNA restriction-site mapping and nuclear DNA fingerprinting. Exp Mycol. 15:316–325.
- Hoegger PJ, Binz T, Heiniger U. 1996. Detection of genetic variation between Ophiostoma ulmi and the NAN and EAN races of O. novo-ulmi in Switzerland using RAPD markers. Eur J For Pathol. 26:57–68.
- Hornby JM, Jacobitz-Kizzier SM, McNeel DJ, Jensen EC, Treves DS, Nickerson KW. 2004. Inoculum size effect in dimorphic fungi: extracellular control of yeast-mycelium dimorphism in Ceratocystis ulmi. Appl Environ Microbiol. 70:1356–1359.
- Hudler GW, Banik MT, Miller SG. 1987. Unusual epidemic of tar spot on Norway maple in upstate New York. Plant Dis. 71:65–68.
- Jacobi V, Dufour J, Bouvet GF, Aoun M, Bernier L. 2010. Identification of transcripts upregulated in asexual and sexual fruiting bodies of the Dutch elm disease pathogen Ophiostoma novo-ulmi. Can J Microbiol. 56:697–705.
- Jeng RS, Hubbes M. 1983. Identification of aggressive and non-aggressive strains of Ceratocystis ulmi by polyacrylamide gradient gel electrophoresis of intramycelial proteins. Mycotaxon. 17:445–455.
- Jensen EC, Ogg C, Nickerson KW. 1992. Lipoxygenase inhibitors shift the yeast/mycelium dimorphism in Ceratocystis ulmi. Appl Environ Microbiol. 58:2505–2508.
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. 2012. A programmable dual-RNA guided DNA endonuclease in adaptive bacterial immunity. Science. 337:816–821.
- Khoshraftar S, Hung S, Khan S, Gong Y, Tyagi V, Parkinson J, Sain M, Moses AM, Christendat D. 2013. Sequencing and annotation of the Ophiostoma ulmi genome. BMC Genomics. 14:162.
- Klug L, Daum G. 2014. Yeast lipid metabolism at a glance. FEMS Yeast Res. 14:369–388.
- Kulkarni RK, Nickerson KW. 1981. Nutritional control of dimorphism in Ceratocystis ulmi. Exp Mycol. 5:148–154.
- Muthukumar G, Nickerson KW. 1984. Ca(II)-calmodulin regulation of fungal dimorphism in Ceratocystis ulmi. J Bacteriol. 159:390–392.
- Nagalakshmi U, Wang Z, Waern K, Shou C, Raha D, Gerstein M, Snyder M. 2008. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science. 320:1344–1349.
- Naruzawa ES 2015. Bases moléculaires du dimorphisme levure-mycélium chez le champignon phytopathogène Ophiostoma novo-ulmi [Molecular bases of yeast-mycelium dimorphism in the plant pathogenic fungus Ophiostoma novo-ulmi] [dissertation]. Quebec City (QC): Université Laval. French.
- Naruzawa ES, Bernier L. 2014. Control of yeast-mycelium dimorphism in vitro in Dutch elm disease fungi by manipulation of specific external stimuli. Fungal Biol. 118:872–884.
- Naruzawa ES, Malagnac F, Bernier L. 2016. Effect of linoleic acid on reproduction and yeast-mycelium dimorphism in the Dutch elm disease pathogens. Botany. 94:31–39.
- Nelson DR. 2009. The cytochrome P450 homepage. Hum Genomics. 4:59–65.
- Nigg M, Laroche J, Landry CR, Bernier L. 2015. RNAseq analysis highlights specific transcriptome signatures of yeast and mycelial growth phases in the Dutch elm disease fungus Ophiostoma novo-ulmi. G3 (Bethesda). 5:2487–2495.
- Ninomiya Y, Suzuki K, Ishii C, Inoue H. 2004. Highly efficient gene replacements in Neurospora strains deficient for nonhomologous end-joining. Proc Natl Acad Sci USA. 101:12248–12253.
- Nødvig CS, Nielsen JB, Kogle ME, Mortensen UH. 2015. A CRISPR-Cas9 system for genetic engineering of filamentous Fungi. PLoS One. 10:e0133085.
- Paoletti M, Buck KW, Brasier CM. 2006. Selective acquisition of novel mating type and vegetative incompatibility genes via interspecies gene transfer in the globally invading eukaryote Ophiostoma novo-ulmi. Mol Ecol. 15:249–262.
- Perdiguero P, Venturas M, Cervera MT, Gil L, Collada C. 2015. Massive sequencing of Ulmus minor’s transcriptome provides new molecular tools for a genus under the constant threat of Dutch elm disease. Front Plant Sci. 6:541.
- Pereira V, Royer JC, Hintz WE, Field D, Bowden C, Kokurewicz K, Hubbes M, Horgen PA. 2000. A gene associated with filamentous growth in Ophiostoma novo-ulmi has RNA-binding motifs and is similar to a yeast gene involved in mRNA splicing. Curr Genet. 37:94–103.
- Plourde KV, Bernier L. 2014. Inoculation of apple (Malus x domestica ‘Golden Delicious’) fruits provides a rapid virulence assay for the Dutch elm disease fungus Ophiostoma novo-ulmi. Plant Pathol. 63:1078–1085.
- Plourde KV, Jacobi V, Bernier L. 2008. Use of insertional mutagenesis to tag putative parasitic fitness genes in the Dutch elm disease fungus Ophiostoma novo-ulmi subsp. novo-ulmi. Can J Microbiol. 54:797–802.
- Proctor RH, Guries RP, Smalley EB. 1994. Lack of association between tolerance to the elm phytoalexin mansonone E and virulence in Ophiostoma novo-ulmi. Can J Bot. 72:1355–1364.
- Richards WC. 1994. Nonsporulation in the Dutch elm disease fungus Ophiostoma ulmi: evidence for control by a single nuclear gene. Can J Bot. 72:461–467.
- Royer JC, Dewar K, Hubbes M, Horgen PA. 1991. Analysis of a high frequency transformation system for Ophiostoma ulmi, the causal agent of Dutch elm disease. Mol Gen Genet. 225:168–176.
- Scheffer RJ, Liem JI, Elgersma DM. 1987. Production in vitro of phytotoxic compounds by non-aggressive and aggressive isolates of Ophiostoma ulmi, the Dutch elm disease pathogen. Physiol Mol Plant Pathol. 30:321–335.
- Schwarz MB. 1922. Das zweigensterben der olmen, trauerweiden und pfirschbäume [The twig dying of the elms, willows, and peach trees]. Meded phytopath Lab Willie Commelin Scholten. 5:1–73. Dutch.
- Solla A, Dacasa MC, Nasmith C, Hubbes M, Gil L. 2008. Analysis of Spanish populations of Ophiostoma ulmi and O. novo-ulmi using phenotypic characteristics and RAPD markers. Plant Pathol. 57:33–44.
- Svaldi R, Elgersma DM. 1982. Further studies on the activity of cell wall degrading enzymes of aggressive and non-aggressive isolates of Ophiostoma ulmi. Eur J For Pathol. 12:29–36.
- Tadesse Y, Bernier L, Hintz WE, Horgen PA. 2003. Real time RT-PCR quantification and Northern analysis of Cerato ulmin (CU) gene transcription in different strains of the phytopathogens Ophiostoma ulmi and O. novo ulmi. Mol Genet Gen. 269:789–796.
- Temple B, Bernier L, Hintz WE. 2009. Characterization of the polygalacturonase gene of the Dutch elm disease pathogen Ophiostoma novo-ulmi. N Z J For Sci. 39:29–37.
- Temple B, Horgen PA, Bernier L, Hintz WE. 1997. Cerato-ulmin, a hydrophobin secreted by the causal agents of Dutch elm disease, is a parasitic fitness factor. Fungal Genet Biol. 22:39–53.
- Urban M, Pant R, Raghunath A, Irvine AG, Pedro H, Hammond-Kosack K. 2015. The pathogen-host interactions database: additons and future developments. Nucl Acids Res. 43:D645–D655.
- Wang Z, Gerstein M, Snyder M. 2009. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 10:57–63.
- Wedge ME, Naruzawa ES, Nigg M, Bernier L. 2016. Diversity in yeast-mycelium dimorphism response of the Dutch elm disease pathogens: the inoculum size effect. Can J Microbiol. (In Press). doi:10.1139/cjm-2015-0795