
Abstract
Multiple plant pathogens cause root rot on field pea, including Aphanomyces euteiches, Rhizoctonia solani and numerous Fusarium spp. In North Dakota, root rot pathogens have been a contributing factor in the decline in area planted to field pea over the past decade. Real-time quantitative PCR (qPCR) has proven valuable in quantifying a multitude of host–pathogen interactions. However, no qPCR assays are available to effectively quantify Fusarium spp. associated with root rot in field pea. The objective of this study was to develop multiplex qPCR assays to quantify seven Fusarium spp. commonly associated with field pea (F. acuminatum, F. avenaceum, F. culmorum, F. graminearum, F. redolens, F. solani and F. sporotrichioides). Primers and associated hydrolysis probes designed for each Fusarium sp. were combined into three multiplexed assays. Assay amplification efficiencies ranged from 93 to 108 and the regression coefficient for all assays was greater than 0.97 on serial dilutions of pure fungal DNA. All three multiplex assays amplified down to 40 pg µL−1 Fusarium DNA. Multiplex qPCR assays proved sensitive and specific based on evaluations of pea plants inoculated under greenhouse conditions and plants displaying symptoms of root rot collected from commercial pea fields. Annealing temperatures and reaction components were the same for all assays, providing interchangeability and versatility in each multiplexed assay. These assays will aid researchers in advancing the information available for the Fusarium–host interactions for many important crop systems.
Résumé
De nombreux agents pathogènes causent le pourridié chez le pois potager, y compris Aphanomyces euteiches, Rhizoctonia solani et plusieurs agents du genre Fusarium. Dans le Dakota du Nord, au cours de la dernière décennie, les agents pathogènes qui causent le pourridié ont contribué au déclin de la surface plantée en pois potager. La PCR quantitative en temps réel s’est avérée utile quant à la quantification d’une multitude d’interactions hôte-agent pathogène. Toutefois, il n’existe aucun biotest faisant appel à ce type de PCR capable de quantifier efficacement les Fusarium associés au pourridié chez le pois potager. Le but de cette étude était de développer des biotests par PCR multiplexe quantitative pour quantifier sept Fusarium associés au pois potager (F. acuminatum, F. avenaceum, F. culmorum, F. graminearum, F. redolens, F. solani et F. sporotrichioides). Les amorces et les sondes d’hydrolyse conçues pour chaque Fusarium ont été combinées dans trois bitotests multiplexés. L’efficacité de l’amplification des biotests variait de 93 à 108 et le coefficient de régression pour tous les biotests était supérieur à 0.97 pour des dilutions en série d’ADN fongique pur. Les trois biotests multiplexes ont pu amplifier l’ADN de Fusarium à partir de 40 pg µL−1. En se basant sur les évaluations des plants de pois inoculés dans des conditions de culture en serre et sur des plants affichant les symptômes du pourridié, collectés sur des pois potagers cultivés commercialement, les biotests par PCR multiplexe quantitative se sont révélés sensibles et précis. Les températures de renaturation et les composants réactionnels étaient les mêmes pour tous les biotests multiplexés, chacun offrant par conséquent interchangeabilité et polyvalence. Ces biotests aideront les chercheurs à bonifier l’information disponible quant aux interactions Fusarium-hôte, et ce, pour plusieurs systèmes de production importants.
Introduction
Field pea (Pisum sativum L.) production has expanded rapidly in the USA, due in great part to production surges in North Dakota and Montana beginning around 2000 and 2005, respectively (www.nass.usda.gov). Field pea has become an economically and agriculturally valuable crop, particularly in a rotation with cereals in these two Midwestern states. North Dakota reached a high of nearly 240 000 ha planted in 2006. However, field pea production fell and remained stagnant at less than 120 000 ha from 2012 to 2015. This decrease in production was, in part, due to a wet weather cycle. However, root rot has become a substantial revenue-limiting factor in the production system. Field pea production increased in 2016 and 2017, with North Dakota producers planting over 227 000 and 190 000 ha to field peas in those 2 years, respectively (www.nass.usda.gov).
Field pea is susceptible to numerous root rotting pathogens, including Aphanomyces euteiches Drechs., Rhizoctonia solani Kuhn (teleomorph Thanatephorus cucumeris), Pythium spp. and Fusarium spp. The most frequently isolated pathogens from diseased field pea root tissues in the USA, Canada and elsewhere are Fusarium spp. (Basu et al. Citation1973; Hwang & Chang Citation1989; Fernandez Citation2007; Mathew et al. Citation2008; Chittem et al. Citation2015; Chang et al. Citation2016; Chatterton et al. Citation2016; Gossen et al. Citation2016; McLaren et al. Citation2016; Esmaeili Taheri et al. Citation2017). Yield losses have been reported to be greater than 50% (Basu et al. Citation1973); however, this can vary depending on cultivar, field history and environmental conditions (Basu et al. Citation1976). Management of fusarium root rot requires a combination of cultural practices including crop rotation, enhancing root vigour, planting quality seed and the use of seed treatment fungicides (Kraft & Pfleger Citation2001; Gossen et al. Citation2016).
Several Fusarium spp. are associated with fusarium root rot on field pea in North Dakota, and include F. acuminatum Ellis & Everhart, F. avenaceum (Fries) Sacc., F. culmorum (W.G. Smith) Sacc., F. equiseti (Corda) Sacc., F. graminearum Schwabe (teleomorph Gibberella zeae Schwein (Petch)), F. redolens Wollenweber, F. oxysporum Schlechtendahl emend. Snyder & Hansen, F. solani (Martius) Appel & Wollenweber emend. Snyder & Hansen and F. sporotrichioides Sherbakoff (Mathew et al. Citation2008; Chittem et al. Citation2015). Based on previously determined prevalence and aggressiveness of recovered Fusarium spp., all species except F. oxysporum and F. equiseti likely contribute to pea root rot in in North Dakota (Chittem et al. Citation2015). Fusarium oxysporum was frequently recovered from diseased root tissues, but displayed low relative aggressiveness as a root rot pathogen under greenhouse conditions compared with other species although it is commonly known as a wilt pathogen of field peas. Fusarium equiseti also was isolated from rotted pea roots, but at lower frequencies and aggressiveness (Chittem et al. Citation2015).
Fusarium solani is no longer considered a single species, but rather is comprised of three clades with numerous species within the F. solani species complex (FSSC) (O’Donnell Citation2000). Fusarium solani was isolated from approximately 35% of North Dakota pea fields surveyed in 2008 and 2009 (Chittem et al. Citation2015). Four F. solani isolates from diseased pea root tissues collected in North Dakota during 2014 and 2015 were classified in clade III, FSSC 5 and 11 (Chitrampalam & Nelson Citation2015). Fusarium graminearum also has been described as a species complex (O’Donnell et al. Citation2000); however, isolates collected from field peas in North Dakota have not been placed into specific lineages.
Above-ground leaf yellowing and stunting caused by fusarium root rot of field pea may not be visible until plants begin to flower. Expanding black to brown root lesions can be easily confused with roots rots caused by other soilborne pathogens (Porter et al. Citation2015). Methods for detecting and identifying plant pathogens in diseased plant tissues include plating onto solid growth medium, traditional PCR and real-time quantitative PCR (qPCR). Direct plating of diseased root tissues requires optimal incubation conditions and the appropriate growth media for the production of fungal structures. Morphological identification typically requires several weeks and accurate identification of Fusarium spp. is difficult. Because field pea roots may be colonized by multiple Fusarium spp., accurate representation of the pathogen complex can be challenging. While traditional PCR can be used for species identification, effective quantification of each species present within a sample is not possible. qPCR assays are more efficient, sensitive and specific than either direct plating or traditional PCR methods and can be used to rapidly quantify numerous targets within a sample.
The development of successful multiplex qPCR assays using DNA extracted from field pea root tissues would drastically reduce the time and expertise required to identify and quantify Fusarium spp. The objective of this study was to develop and validate multiplex hybridization probe-based qPCR assays to quantify seven Fusarium spp. (F. acuminatum, F. avenaceum, F. culmorum, F. graminearum, F. redolens, F. solani and F. sporotrichioides) directly from diseased field pea root tissue. These seven species were chosen based on frequency of detection and aggressiveness determined in previous research (Chittem et al. Citation2015).
Materials and methods
Isolation and maintenance of Fusarium species
Isolates of 12 sequenced Fusarium spp. (F. oxysporum KSU#11390, F. poae KSU#11470, F. konzum KSU#11615, F. circinatum KSU#H-10847, F. equiseti KSU#11439, F. acuminatum, KSU#11442, F. avenaceum KSU#11440, F. culmorum KSU#11427, F. graminearum KSU#15050, F. sporotrichioides KSU#11552, F. solani KSU#11420, F. redolens KSU#11616) were obtained from Kansas State University. In addition, isolates of seven Fusarium spp. (F. acuminatum, F. avenaceum, F. culmorum, F. graminearum, F. redolens, F. solani and F. sporotrichioides) were collected from diseased field pea root tissues collected in 2014. Diseased tissue was surface-sterilized in 0.8% NaOCl for 30 s and placed onto 0.5× potato dextrose agar (PDA: 19.5 g potato dextrose agar, 7.5 g agar, 1 litre distilled H2O) amended with streptomycin and neomycin, both at a concentration of 50 mg L−1. Cultures were allowed to grow for 24 h at room temperature under fluorescent lighting. Hyphal-tip segments were transferred aseptically onto 0.5× PDA with no antibiotics and allowed to grow for 6 days. Isolates were further purified by single spore isolation onto 0.5× PDA (Leslie & Summerell Citation2006). All isolates were identified to species based on morphology (Nelson et al. Citation1983; Leslie & Summerell Citation2006). Fungal tissue was scraped from the agar surface for each Fusarium spp. and lyophilized using a Labconco Freezone 6 console freeze dry system (Labconco, Kansas City, MO) for 24 h at −40°C under 0.133 mBar of vacuum pressure. Dried fungal tissue was transferred to 2 mL VWR round bottom screw cap cryovials (Cat. # 10018–746, VWR, Radnor, PA) and placed at −80°C for long-term storage.
Molecular identification of Fusarium isolates
Morphological identification of isolates of all Fusarium spp. used in qPCR development and validation experiments was verified through amplification and sequencing of the ribosomal Internal transcribed spacer (ITS) region and Translation elongation factor 1α (EF-1α) genes using the universal primer set ITS1/ITS4 (White et al. Citation1990) and primer set E1/E2 (O’Donnell et al. Citation1998), respectively. Visual confirmation of amplified PCR products was obtained via electrophoresis on a 1% agarose gel. The PCR products were treated with Exo-I (Affymetrix, OH) and rAPid phosphatase (Roche, IN) following the manufacturer’s protocol and sequenced from both directions (GenScript USA Inc., NJ). Resulting sequence data were compared with the FUSARIUM-ID v1.0 database (Geiser et al. Citation2004) for each targeted Fusarium spp. using BLAST and were aligned using MEGA 5.2 (Tamura et al. Citation2011). Identities of isolates of F. acuminatum, F. culmorum, F. graminearum, F. redolens, F. solani and F. sporotrichioides were verified with sequences available from FUSARIUM-ID v1.0. Resulting sequences were also blasted against the NCBI GenBank database. The subsequent NCBI GenBank accession number with 100% homology was recorded for each species (). No F. avenaceum sequence was available in the FUSARIUM-ID v1.0 database; therefore, American Type Culture Collection (ATCC) culture 60 644 of F. avenaceum was obtained, DNA was extracted and a section of the EF-1α gene was amplified (O’Donnell et al. Citation1998). The sequence was deposited in NCBI GenBank database as accession number KU981027. This sequence was used for isolate confirmation as F. avenaceum.
Table 1. qPCR primer and probe sequences from the targeted translation elongation factor 1α (EF-1α) gene for the amplification of seven Fusarium species and GenBank database reference sequence accessions used for primer and probe design.
Greenhouse production of infected root tissue
Infected plants for multiplex assay validation were generated by inoculating field pea seedlings with sterile water, single, or multiple Fusarium spp. under greenhouse conditions (). Seeds of ‘DS Admiral’ peas were surface-sterilized in a 0.8% NaOCl solution for 30 s and planted in ~30 g of sterile perlite. One pea seed was planted into one 3.8 × 14 cm ‘Cone-tainer’TM (Stuewe & Sons Inc. Tangent, OR). Peas were watered daily and maintained in a greenhouse under 16 h light at 22–25°C. Eleven treatments were evaluated: seven single and three multiple Fusarium spp. inoculations and one sterile water (non-inoculated) control. Purified cultures of F. acuminatum, F. avenaceum, F. culmorum, F. graminearum, F. redolens, F. solani and F. sporotrichioides were grown in liquid CarboxyMethyl-Cellulose (CMC) medium (15.0 g CMC, 1.0 g NH4NO3, 1.0 g KH2PO4 monobasic, 0.5 g MgSO4 . 7H2O, 1.0 g yeast extract, 1 litre distilled or de-ionized water) for 9 days under continuous fluorescent light for production of macroconidia (Tuite Citation1969).
Table 2. Fusarium species used to inoculate field pea plants under greenhouse conditions, average root rot rating, frequency at which the plating method and qPCR assays detected the inoculated species and quantity of fungal DNA as determined by multiplex qPCR.
Six field pea plants (replicates) were inoculated per treatment in two greenhouse trials (two biological replicates) to provide a total of 12 plants per treatment for qPCR assay validation. Isolates used to inoculate greenhouse-reared plants were purified from the field samples as described above. Inoculum contained macroconidia from six isolates of a single species. Therefore, treatments that included two or three Fusarium species were inoculated with a total of 12 or 18 isolates, respectively. Each plant was inoculated by carefully pipetting 10 mL of a solution containing 1 × 105 macroconidia mL−1 sterile water of one or multiple Fusarium spp., or sterile water, to the base of each plant 9 days after planting. Nine days after inoculation, roots were washed free of perlite, surface-sterilized in a 0.8% NaOCl solution, rinsed in sterile tap water and rated using a 0 to 5 (0 = no visible symptoms, 5 = tap root severed) scale (; Infantino et al. Citation2006; Ondrej et al. Citation2008). Roots were subsequently plated for morphological identification and molecular confirmation as described above.
Fig. 1 (Colour online) Field pea root rot rating scale for evaluation of greenhouse inoculated plants; 0 = no visible symptoms, 5 = tap root severed (adapted from Infantino et al. Citation2006; Ondrej et al. Citation2008).

Total genomic DNA extraction
Total genomic DNA was extracted from fungal cultures of all Fusarium spp. using a modified CTAB method (Doyle & Doyle Citation1987). One hundred milligrams of fungal tissue was collected from the agar surface, lyophilized and pulverized in 2 mL XXTuff reinforced microvials (Biospec Products, Bartlesville, OK) with 3–5 metal 2.3 mm diameter chrome steel beads (Biospec Products, Bartlesville, OK). Seven hundred and fifty microlitres of Carlson Lysis buffer amended with 1.5 µL of β-mercaptoethanol and 20 µg of RNase was added to each tube. Tubes were vortexed and incubated for 30–40 min in a 65°C water bath with a single inversion at 10 min intervals. A half-volume of phenol:chloroform:isoamyl alcohol (25:24:1) was added to each tube. Tubes were vigorously vortexed and centrifuged at 14 000 g for 10 min. The supernatant was transferred to a new microfuge tube and an equal volume of isopropanol was added. Tubes were inverted gently several times, and centrifuged for 2 min at 14 000 g. The DNA pellet was washed twice with 70% ethanol, air dried, and resuspended in a modified TE buffer (10 mM Tris, 0.1 mM EDTA, pH 8.0). Total genomic DNA was extracted from 150 mg of lyophilized, symptomatic field pea root tissue generated in the greenhouse and field using the FastDNA Spin Kit (MP Biomedicals) following manufacturer’s instructions. Cell Lysis Solution Y provided by the FastDNA Spin Kit was used for extractions. DNA quantity and quality were evaluated using Nanodrop and gel electrophoresis on a 1% agarose gel, respectively.
Quantitative PCR assay development and validation
The EF-1α housekeeping gene is used widely for phylogenetic analysis (Kristenson et al. Citation2005) and is available for many Fusarium spp. in NCBI and/or FUSARIUM ID v1.0. Primers and probes were designed using the Primer3 Input program Version 0.4.0 (Untergasser et al. Citation2012) targeting a 75–120 bp amplicon () from different regions within the EF-1α gene to prevent cross-hybridization and non-specific binding among each of the seven targeted Fusarium spp. Sequence alignment indicated sufficient sequence diversity existed within the gene region encompassed by the E1/E2 primers to design the Fusarium spp. specific qPCR assays. The small qPCR amplicon target size of 75–120 bp allowed multiple regions within E1/E2 sequence to be targeted for primer/probe design. In silico evaluation of the developed primer and probes indicated no homology to non-target Fusarium spp. available in the FUSARIUM-ID v1.0 database (data not shown).
Three primer pairs designed from the EF-1α gene of each species were reduced to a single primer pair with a corresponding hybridization probe based on assay efficiency, sensitivity and specificity. Probe-based simplex reactions consisted of 20 αl, with a final concentration of 1X SsoAdvanced Universal Probe Supermix (Bio-Rad Laboratories), 150 nM forward and reverse primer, 50 nM hybridization probe and 80 ng DNA. The iQ Multiplex Powermix (Bio-Rad Laboratories) replaced the SsoAdvanced Universal Probes Supermix in the F. acuminatum assay due to the interaction between the ROX in the SsoAdvanced Universal Probes Supermix and the CAL Fluor Red-610 fluorophore. This was done to limit overlapping fluorescence from adjacent adsorption channels after multiplexing. Amplification was optimal following 3 min at 95°C, followed by 39 cycles of 10 s at 95°C, 10 s at 57°C and 30 s at 72°C. All reactions were performed in triplicate. Primer pairs and probes designed for each targeted Fusarium spp. had the same cycling parameters. Therefore, the three multiplex assay combinations were based on the interaction of the fluorophores previously placed with simplex assays and assay efficiencies. Assay one includes F. graminearum and F. sporotrichioides, assay two includes F. avenaceum and F. culmorum, and assay three includes F. acuminatum, F. redolens and F. solani.
Quantitative PCR assay primer specificity
The specificity of each probe-based simplex and multiplex assay was evaluated against DNA from 19 organisms, including field pea, 12 target and non-target Fusarium spp. and common fungal, oomycete and bacterial pathogens of field peas (). Standard curves to determine efficiency and sensitivity for each simplex and multiplex assay were generated using 10-fold serial dilutions of pure fungal DNA starting at a concentration from 40 ng µL−1. Standard curves were also developed from DNA extracted from root tissues inoculated with each targeted Fusarium spp. under greenhouse conditions. In both cases, assay efficiency was calculated from the slope of the regression line (Efficiency = 10(−1/slope)−1) by plotting log DNA concentrations against crossing threshold (Ct) value using the CFX manager Software v. 2.1 (BioRad Laboratories Inc., Hercules, CA). The baseline Ct value was not adjusted. A threshold of 40 pg µL−1 was used to determine the presence of the organism in all experiments for all hybridization probe-based assays. Analysis of covariance (α = 0.05) was performed to compare the slopes and intercepts of regression lines developed from simplex and multiplex qPCR assays performed using DNA extracted from pure fungal cultures of each targeted Fusarium spp.
Table 3. Specificity of simplex and multiplex qPCR assays designed for quantification of seven Fusarium species.
Quantitative PCR assay validation
DNA extracted from roots of greenhouse-generated plants inoculated with water, a single or multiple Fusarium spp. was evaluated using the multiplex qPCR assays. DNA from greenhouse inoculated root tissue was standardized to 40 ng µL−1. Fungal DNA was quantified within plant tissues by extrapolating against the regression line generated by amplifying 10-fold serial dilutions of pure fungal DNA. Fungal DNA quantity in greenhouse inoculated root tissues from the two trials were combined based on Levene’s test for variance homogeneity. Simple t-tests were performed to evaluate significant differences between fungal DNA quantified via the simplex assays and the corresponding multiplex assays for each targeted Fusarium sp. Simple t-tests were also performed to compare Fusarium spp. DNA quantities within each multiplex assay. A Spearman correlation was performed to compare fungal DNA quantity and root rot ratings of plants inoculated with a single Fusarium sp. All qPCR reactions were run in triplicate and statistical analyses were performed using SAS v. 9.4 (α = 0.05).
Quantitative PCR of field samples
Twenty-six plants from two commercial pea fields in North Dakota displaying characteristic Fusarium root rot symptoms were used to further validate the multiplex qPCR assays. Roots were rated as previously described for greenhouse-reared plants. All field samples evaluated using the multiplex assays had a root rot rating equal to or greater than 3. Frequency of Fusarium spp. associated with root tissues was determined by plating diseased root tissues from each plant, purifying and morphologically identifying isolates as described above. All three multiplex qPCR assays were performed in duplicate on DNA extracted from the diseased root tissues from all 26 plants to determine frequency of Fusarium spp. Fungal DNA quantity was determined as described above in greenhouse-reared plants.
Results
Quantitative PCR assay development and validation
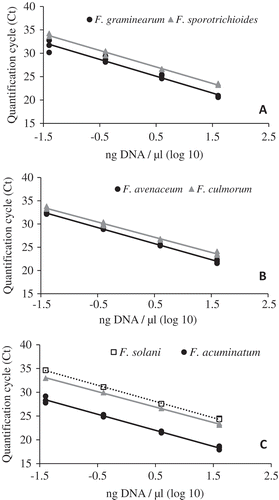
Sensitivity was similar for all simplex assays, with the limit of detection of 40 pg µL−1 of pure fungal DNA (data not shown). Each simplex assay was highly specific to the corresponding targeted Fusarium sp. No fluorescent signal was generated during the first 35 amplification cycles when evaluating non-target species (). Simplex probe-based qPCR assays developed to quantify each of the seven-targeted Fusarium sp. displayed similar assay efficiencies, ranging from 95 to 107 when evaluating pure fungal DNA, well within the recommendations for multiplexing. When comparing simplex to multiplex assays using 10-fold serial dilutions of pure fungal DNA from each target, assay sensitivity decreased, but efficiency generally was not affected (). The slope of the regression line differed significantly only when comparing simplex and multiplex assays targeting F. solani, indicating a difference in assay efficiency. Slopes of lines generated from all other assays were not significantly different when comparing simplex to multiplex assays for the same target. Intercepts of lines generated from amplification of serial dilutions of pure fungal DNA were significantly higher after multiplexing, indicating a decrease in sensitivity for assays targeting F. graminearum, F. avenaceum, F. culmorum and F. solani. Neither slopes nor intercepts of lines generated from amplification of serial dilutions of pure fungal DNA of F. sporotrichioides, F. acuminatum and F. redolens differed significantly when comparing simplex and multiplex assays targeting those species ().
Table 4. qPCR assay linear regressions obtained using the random coefficients model generated from total genomic DNA extracted from pure fungal cultures of seven Fusarium species. DNA was adjusted to 40 ng µL−1 and diluted serially over four orders of magnitude.
Assay efficiencies, calculated from the slope of the regression line generated by the three multiplexed qPCR assay amplifying DNA extracted from pure fungal cultures of seven Fusarium spp. ranged from 93 to 108 (; ). Correlation coefficients (R2) indicate that the data fit the regression lines well, with values greater than 0.97 for all assays. Multiplex PCR assay efficiencies and R2 values using the DNA extracted directly from diseased root tissues were similar to those obtained with DNA extracted from fungal tissues. Assay efficiencies remained between 87 and 106 and R2 values remained high, ≥0.97, for DNA extracted from inoculated plants (; ).
Table 5. qPCR efficiencies and coefficients of determination (R2) for the three multiplex assays to detect seven Fusarium species in pure fungal tissue and field pea roots.
Fig. 2 A standard curve of log10 DNA concentration and quantification cycle (Ct) across four, 10-fold serial DNA dilutions for multiplex qPCR. (A) Assay 1; GramF/R/Pr to quantify Fusarium graminearum (black circles) and SporF/R/Pr to quantify Fusarium sporotrichioides (grey triangles); (B) Assay 2; AveF/R/Pr to quantify Fusarium avenaceum (black circles) and CulF/R/Pr to quantify Fusarium culmorum (grey triangles); (C) Assay 3; AcuF/R/Pr to quantify Fusarium acuminatum (black circles), RedF/R/Pr to quantify Fusarium redolens (grey triangles), and SolF/R/Pr to quantify Fusarium solani (open squares). Pure fungal cultures were used to obtain DNA for the development of the standard curve.
Hybridization probe-based assays were further validated using DNA extracted from root tissues of plants inoculated with single or multiple species (). All inoculated plants had symptoms of root rot disease when harvested. All seven Fusarium spp. were quantified via the multiplex assays from plants inoculated with the corresponding single or multiple-species. qPCR detection frequencies for some species did not change when inoculated alone or co-inoculated. Frequencies decreased for the other species, including F. graminearum, F. avenaceum, F. redolens and F. acuminatum, but remained greater than 80% for all (). Spearman’s analysis indicated a significant positive correlation (P < 0.0001; r = 0.89) between fungal DNA quantity and root rot ratings from the single Fusarium spp. inoculations.
Comparison between quantitative PCR assay and traditional plating
Detection incidence ranged from zero to 100% when plating root tissues from each of the 12 plants inoculated with single or multiple Fusarium spp. under greenhouse conditions. Frequency of detection with the qPCR assays was nearly always greater than, or equal to, that of traditional plating assays. This is particularly true where two or three Fusarium spp. were inoculated. When F. graminearum and F. sporotrichioides were co-inoculated, the multiplex qPCR detection incidence of F. graminearum was 83% compared with 67% for plating, and no F. sporotrichioides was recovered via plating, but the multiplex qPCR assay detected this species in all plants. When co-inoculated, multiplex qPCR detected F. avenaceum less frequently than did plating assays, 83 vs. 92%. This was the only instance where a decrease in detection with qPCR was observed. However, this multiplex assay detected F. culmorum more frequently at 92 vs. 0% for plating. Finally, when F. solani, F. redolens and F. acuminatum were co-inoculated, qPCR detected these species at frequencies of 92, 83 and 83%, compared with the 25, 0 and 75% recovered via plating, respectively.
Fusarium spp. DNA quantification followed the same trend as recovery frequencies via plating (). When F. graminearum and F. sporotrichioides were co-inoculated, fungal DNA quantity of F. graminearum was not significantly different than when it was inoculated alone. In contrast, F. sporotrichioides quantity was significantly lower than when inoculated alone. Additionally, when co-inoculated, F. graminearum DNA quantity was significantly higher than F. sporotrichioides. When co-inoculating F. avenaceum and F. culmorum, roots inoculated with both species had significantly lower fungal DNA quantity than when either species was inoculated alone. However, F. culmorum had significantly lower fungal DNA quantity than F. avenaceum when co-inoculated. Quantities of DNA of F. acuminatum and F. solani were not significantly different between the single and co-inoculations. However, F. redolens DNA quantity was significantly different between the single and co-inoculations. Where three species were co-inoculated, differences in fungal DNA quantity were only significant when comparing F. redolens to F. acuminatum and F. solani ().
Field samples were collected to allow a side-by-side comparison of the plating method and multiplex qPCR method. The multiplex qPCR assays detected each targeted Fusarium sp. in as many, or more field-sampled plants as the plating method (). Fusarium culmorum and F. solani were not identified by plating, but were detected at frequencies of 8 and 27%, respectively, by the multiplex qPCR assays. Fusarium graminearum was not detected by either plating or PCR. While it was more common for the qPCR assays to detect a targeted species in a plant, there were a few single plants where the target was isolated in plating assays, but not amplified by qPCR (data not shown). This is likely due to differences in tissues sampled when Fusarium was present at very low levels. Fungal DNA quantities of each species from individual plant samples was determined and did not correlate with detection frequency.
Table 6. Frequency at which the plating method and qPCR assays detected the seven targeted Fusarium species in field peas displaying symptoms of root rot collected from commercial fields and quantity of fungal DNA.
Discussion
Fusarium spp. cause economically important diseases of cereal grains, corn, soybean, canola, dry edible beans and cool season pulse crops such as field pea, lentil and chickpea (Leslie & Summerell Citation2006). Previous research has indicated that in North Dakota, and other regions in the Northern Great Plains, numerous Fusarium spp. are associated with root rot of field pea (Basu et al. Citation1973; Clarkson Citation1978; Hwang and Chang Citation1989; Fernandez Citation2007; Mathew et al. Citation2008; Chittem et al. Citation2015; Chang et al. Citation2016; Chatterton et al. Citation2016; Gossen et al. Citation2016; McLaren et al. Citation2016; Esmaeili Taheri et al. Citation2017). However, it can be difficult to discern which of these species are most damaging using non-quantitative methods. Morphological identification of Fusarium spp. can be useful when conducting survey research; however, the resources required to evaluate a large number of samples are large. While previous PCR assays have been developed to detect Fusarium spp. in various host systems (Strausbaugh et al. Citation2005; Waalwijk et al. Citation2008; Ward et al. Citation2008; Yli-Mattila et al. Citation2008; Brunner et al. Citation2009; Nicolaisen et al. Citation2009; Demeke et al. Citation2010; Atoui et al. Citation2012; Nazari et al. Citation2014; Preiser et al. Citation2015), they do not amplify all seven targeted Fusarium spp. associated with the root rot complex of field pea. Additionally, attempts to multiplex previously developed assays targeting Fusarium spp. associated with pea root rot were unsuccessful based on assay parameters, efficiency, sensitivity or specificity (data not shown).
Three novel multiplex hybridization probe-based qPCR assays specific for detecting and quantifying F. acuminatum, F. avenaceum, F. culmorum, F. graminearum, F. solani, F. sporotrichioides and F. redolens directly from diseased field pea root tissues were developed in this study. These assays were specific for the target species, with no amplification of other Fusarium spp. or other pea pathogens evaluated. Sequences of the EF1-α gene of each Fusarium sp. used for assay design were compiled from isolates associated with root rot in field pea and were identical to sequences in publicly available databases, thereby making these assays potentially transferrable to other host-pathosystems. All assays use the same protocol and cycling parameters, including annealing temperature. This provides the flexibility to interchange the simplex assays, creating new multiplex assays for the specific needs of the researcher. This is particularly beneficial in other host crops where combinations established in this research are not required. Additionally, our results indicated that the simplex assays had similar efficiencies, ranging from 95 to 107% across four, 10-fold serial dilutions for each target Fusarium sp., further supporting the versatility of the primers and probes designed in this research. We are not aware of other qPCR assays developed to date with this versatility. Specificity evaluations may need to be conducted against isolates of target Fusarium spp. collected from regions outside the Northern Great Plains or from different hosts, due to interspecific genetic variation.
The reduced sensitivity observed when simplex assays were combined into multiplex assays, as indicated by significantly higher regression line intercepts, may be due to hybridization competition between primers and probes in the multiplex reactions. This is unlikely to be a limiting factor for the effectiveness of these assays. Detecting minute amounts of DNA is likely not biologically significant, as many Fusarium spp. inhabit soil and root tissues but are not aggressive pathogens (Chittem et al. Citation2015). Additionally, the multiplex qPCR assays developed here have proven more sensitive than traditional plating.
Assay efficiency was not compromised when comparing pure fungal DNA and DNA extracted from root tissues infected under greenhouse conditions. All greenhouse inoculations utilizing a single Fusarium sp. resulted in detection and quantification of each targeted species by the corresponding assay. When pea plants were inoculated with multiple Fusarium spp., all targeted species were detected, but not with 100% frequency. This may be due to interspecific competition between or among Fusarium spp. As the preliminary results reported here, and previous results indicate, one species may outcompete another during infection and colonization (Velluti et al. Citation2000; Nazari et al. Citation2014). In previous studies, when co-inoculated onto irradiated maize kernels, F. graminearum inhibited the growth of F. moniliforme and F. proliferatum (Velluti et al. Citation2000). Additionally, a higher level of F. sporotrichioides biomass was detected than F. langsethiae when these two species were co-inoculated (Nazari et al. Citation2014). Although the assays developed in this study were not conducted to compare all species combinations, results from co-inoculations evaluated here indicate that F. graminearum colonized field pea root tissues to a greater extent than F. sporotrichioides; F. avenaceum colonized tissues to a greater extent than F. culmorum, and F. solani and F. acuminatum colonized pea root tissues more than F. redolens. These indications are further supported by the average root rot ratings for greenhouse plants inoculated with a single Fusarium sp. The Spearman’s correlation analysis also indicated a positive correlation between fungal DNA quantities and root rot ratings in plants inoculated with a single Fusarium sp., evidence of the assays’ ability to quantify the targeted Fusarium spp. causing disease. These results are indirectly supported by a survey of North Dakota field peas, which indicated that F. avenaceum, F. solani and F. acuminatum were among the most commonly isolated Fusarium spp. (Chittem et al. Citation2015). Further research in the area of co-inoculations is needed in this pathosystem, and these qPCR assay will provide a platform to do so efficiently and effectively.
In the studies reported here, qPCR detected the target Fusarium spp. at equal or greater frequency than did traditional plating. Plating assays failed to recover some species when co-inoculations were conducted and when evaluating the field samples, limiting the ability of this technique to adequately evaluate species interactions in planta. Plating assays did not detect one of the two or three species co-inoculated in greenhouse assays, and these species were detected at significantly lower levels in the multiplex reaction. While environmental conditions could have contributed to differences in in vitro growth, the qPCR assays were more sensitive in detecting lower fungal quantities than the plating method. This further highlights the utility of these qPCR assays in in planta studies.
Acknowledgements
The authors thank C. Doetkott for assistance with statistical analyses, Olivia Vaadeland for assistance in sample collection and processing, and Dr John Leslie, Kansas State University, for providing Fusarium isolates.
Additional information
Funding
References
- Atoui A, El Khoury A, Kallassy M, Lebrihi A. 2012. Quantification of Fusarium graminearum and Fusarium culmorum by real-time PCR system and zearalenone assessment in maize. Int J Food Microbiol. 154:59–65.
- Basu PK, Brown NJ, Crête R, Gourley CO, Johnston JW, Pepin HS, Seaman WL. 1976. Yield loss conversion factors for Fusarium root rot of pea. Can Plant Dis Surv. 56:25–32.
- Basu PK, Crête R, Donaldson AG, Gourley CO, Haas JH, Harper FR, Lawrence CH, Seaman WL, Toms HNW, Wong SI, et al. 1973. Prevalence and severity of diseases of processing peas in Canada, 1970-71. Can Plant Dis Surv. 53:49–57.
- Brunner K, Paris MPK, Paolino G, Bürstmayr H, Lemmens M, Berthiller F, Schuhmacher R, Krska R, Mach RL. 2009. A reference-gene-based quantitative PCR method as a tool to determine Fusarium resistance in wheat. Anal Bioanal Chem. 395:1385–1394.
- Chang KF, Wu LF, Fu H, Yang Y, Hwang SF, Turnbull GD, Strelkov SE. 2016. Microorganisms and the occurrence of root rot of field pea in Alberta in 2015. Can Plant Dis Surv. 96:186–187.
- Chatterton S, Harding MW, Bowness R, Vucurevich C, Storozynsky Q, Dubitz T, Nielsen J, Olson M. 2016. Survey of root rot in Alberta field pea in 2015. Can Plant Dis Surv. 96:188–190.
- Chitrampalam P, Nelson JB. 2015. Multilocus phylogeny reveals an association of agriculturally important Fusarium solani species complex (FSSC) 11, and clinically important FSSC 5 and FSSC 3+ 4 with soybean roots in the north central United States. A Van Leeuw J Microb. 109:335–347.
- Chittem K, Mathew FM, Gregoire M, Lamppa RS, Chang YW, Markell SG, Bradley CA, Barasubiye T, Goswami RS. 2015. Identification and characterization of Fusarium spp. associated with root rots of field pea in North Dakota. Eur J Plant Pathol. 143:641–649.
- Clarkson JDS. 1978. Pathogenicity of Fusarium spp. associated with foot‐rots of peas and beans. Plant Pathol. 27:110–117.
- Demeke T, Grӓfenhan T, Clear RM, Phan A, Ratnayaka I, Chapados J, Patrick SK, Gaba D, Levesque CA, Seifert KA. 2010. Development of a specific TaqMan® real-time PCR assay for quantification of Fusarium graminearum clade 7 and comparison of fungal biomass determined by PCR with deoxynivalenol content in wheat and barley. Int J Food Microbiol. 141:45–50.
- Doyle J, Doyle JL. 1987. Genomic plant DNA preparation from fresh tissue-CTAB method. Phytochem Bull. 19.11:11–15.
- Esmaeili Taheri A, Chatterton S, Foroud NA, Gossen BD, McLaren DL. 2017. Identification and community dynamics of fungi associated with root, crown, and foot rot of field pea in western Canada. Eur J Plant Pathol. 147:489–500.
- Fernandez MR. 2007. Fusarium populations in roots of oilseed and pulse crops grown in eastern Saskatchewan. Can J Plant Pathol. 87:945–952.
- Geiser DM, Del Mar Jiménez-Gasco M, Kang S, Makalowska I, Veeraraghavan N, Ward TJ, Zhang N, Kuldau GA, O’Donnell K. 2004. FUSARIUM-ID v. 1.0: A DNA sequence database for identifying Fusarium. In: Mulè G, Bailey JA, Cooke BM, Logrieco A, editors. Molecular diversity and PCR-detection of toxigenic Fusarium species and ochratoxigenic fungi. Springer, Dordrecht p. 473–479.
- Gossen B, Conner R, Chang K, Pasche J, McLaren D, Henriquez M, Chatterton S, Hwang S. 2016. Identifying and managing root rot of pulses on the Northern Great Plains. Plant Dis. 100:1965–1978.
- Hwang SF, Chang KF. 1989. Incidence and severity of root rot disease complex of field pea in northeastern Alberta in 1988. Can Plant Dis Surv. 69:139–141.
- Infantino A, Kharrat M, Riccioni L, Coyne CJ, McPhee KE, Grünwald NJ. 2006. Screening techniques and sources of resistance to root diseases in cool season food legumes. Euphytica. 147:201–221.
- Kraft JM, Pfleger FL. 2001. Compendium of pea diseases and pests. 2nd ed. St. Paul: The American Phytopathological Society.
- Kristenson R, Torp M, Kosiak B, Holst-Jensen A. 2005. Phylogeny and toxigenic potential is correlated in Fusarium species as revealed by partial translation elongation factor 1 alpha gene sequences. Mycol Res. 109:173–186.
- Leslie JF, Summerell BA. 2006. The Fusarium laboratory manual. Vol. 2. No. 10. Ames (IA): Blackwell Publishing.
- Mathew FM, Barasubiye T, Markell SG, Goswami RS. 2008. Detection and identification of Fusarium species in field pea roots. Phytopathology. 98:S100 (abstr.).
- McLaren DL, Henderson TL, Kim YM, Chang KF, Kerley TJ. 2016. Field pea disease in Manitoba in 2015. Can Plant Dis Surv. 96:191–192.
- Nazari LE, Pattori EL, Terzi VA, Morcia CA, Rossi I. 2014. Influence of temperature on infection, growth, and mycotoxin production by Fusarium langsethiae and F. sporotrichioides in durum wheat. Food Microbiol. 39:19–26.
- Nelson PE, Toussoun TA, Marasas WFO. 1983. Fusarium species—An illustrated manual for identification. University Park (PA): Pennsylvania State University Press.
- Nicolaisen M, Supronienė S, Nielsen LK, Lazzaro I, Spliid NH, Justesen AF. 2009. Real-time PCR for quantification of eleven individual Fusarium species in cereals. J Microbiol Meth. 76:234–240.
- O’Donnell K. 2000. Molecular phylogeny of the Nectria haematococca-Fusarium solani species complex. Mycologia. 92:919–938.
- O’Donnell K, Kistler HC, Cigelnik E, Ploetz RC. 1998. Multiple evolutionary origins of the fungus causing Panama disease of banana: concordant evidence from nuclear and mitochondrial gene genealogies. Proc Natl Acad Sci USA. 95:2044–2049.
- O’Donnell KH, Kistler C, Tacke BK, Casper HH. 2000. Gene genealogies reveal global phylogeographic structure and reproductive isolation among lineages of Fusarium graminearum, the fungus causing wheat scab. Proc Natl Acad Sci USA. 97:7905–7910.
- Ondrej M, Dostalova R, Trojan R. 2008. Evaluation of virulence of Fusarium solani isolates on pea. Plant Protect Sci. 44:9–18.
- Porter LD, Pasche JS, Chen W, Harveson RM. 2015. Isolation, identification, storage, pathogenicity tests, hosts, and geographic range of Fusaruim solani f. sp. pisi causing Fusarium root rot of pea. Plant Health Prog. 15:136–145.
- Preiser VD, Goetsch M, Sulyok R, Krska RL, Mach A, Farnleitner A, Brunner K. 2015. The development of a multiplex real-time PCR to quantify Fusarium DNA of trichothecene and funmonisin producing strains in maize. Anal Meth. 7:1358–1365.
- Strausbaugh CA, Overturf K, Koehn AC. 2005. Pathogenicity and real-time PCR detection of Fusarium spp. in wheat and barley roots. Can J Plant Pathol. 27:430–438.
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 28:2731–2739.
- Tuite J. 1969. Plant pathological methods, Fungi and bacteria. Minneapolis (MN): Burgess Publishing Company.
- Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, Rozen SG. 2012. Primer3—New capabilities and interfaces. Nucleic Acids Res. 40:e115.
- Velluti A, Marin S, Bettucci L, Ramos AJ, Sanchis V. 2000. The effect of fungal competition on colonization of maize grain by Fusarium moniliforme, F. proliferatum, and F. graminearum and on fumonisin B1 and zearalenone formation. J Food Microbiol. 59:59–66.
- Waalwijk C, Koch SH, Ncube E, Allwood J, Flett B, De Vries I, Kema GHJ. 2008. Quantitative detection of Fusarium spp. and its correlation with fumonisin content in maize from South African subsistence farmers. World Mycotox J. 1:39–47.
- Ward TJ, Clear RM, Rooney AP, O’Donnell K, Gaba D, Patrick S, Starkey DE, Gilbert J, Geiser DM, Nowicki TW. 2008. An adaptive evolutionary shift in Fusarium head blight pathogen populations is driving the rapid spread of more toxigenic Fusarium graminearum in North America. Fungal Genet Biol. 45:473–484.
- White TJ, Bruns T, Lee S, Taylor JW. 1990. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, Gelfand DH, Sninsky JJ, White TJ, editors. PCR protocols: a guide to methods and applications. New York (NY): Academic Press, Inc.
- Yli-Mattila T, Paavanen-Huhtala S, Jestoi M, Parikka P, Hietaniemi V, Gagkaeva T, Sarlin T, Haikara A, Laaksonen S, Rizzo A. 2008. Real-time PCR detection and quantification of Fusarium poae, F. graminearum, F. sporotrichioides, and F. langsethiae in cereal grains in Finland and Russia. Arch Phyto Plant Protect. 41:243–260.