
Abstract
The interactions in the tertiary structure of a ribosomal RNA fragment in the GTPase Associating Center (GAC) have been experimentally studied, but the roles of the bound and diffuse cations in its folding pathway have not yet been fully elucidated. Melting experiments have shown that the temperature of the first of the two distinguishable transitions in the unfolding pathway of the GAC RNA can be regulated by altering the magnesium concentration, yet the physical interpretation of such ion-dependent effects on folding have not been clearly understood in spite of the availability of crystal structures that depict many GAC RNA–ion interactions. Here, we use umbrella sampling and molecular dynamics (MD) simulations to provide a physical description for the first transition in this unfolding pathway, with a focus on the role of a chelated magnesium ion. Our results indicate that the presence of cations mediating the local interaction of two loops stabilizes the folded state relative to the unfolded or partially folded states. Also, our findings suggest that a bridging magnesium ion between the two loops improves the stabilizing effect. This is consistent with the multistep unfolding pathway proposed for the GAC RNA and highlights the importance of ions in the first unfolding step. The results suggest how MD simulations can provide insight into RNA unfolding pathways as a complementary approach to experiments.
Introduction
The folding of RNA molecules into their functional structures depends on their interaction with diffuse, associated, and bound cations that neutralize the RNA phosphate groups and trigger the formation of complex tertiary structures (Draper, Citation2004; Woodson, Citation2010). In bulk solvent, diffuse ‘hydrated’ cations can transiently interact with RNA atoms. In contrast, bound or chelated ions bind to the RNA atoms directly, a process that involves displacement of one or more water residues in the first hydration shell of the ion and RNA. This partial dehydration requires overcoming a considerable energetic barrier (~12 kcal/mol for removing one water molecule from the first shell of magnesium (Allnér, Nilsson, & Villa, Citation2012)) in the process of ion binding, especially for magnesium (Draper, Citation2004).
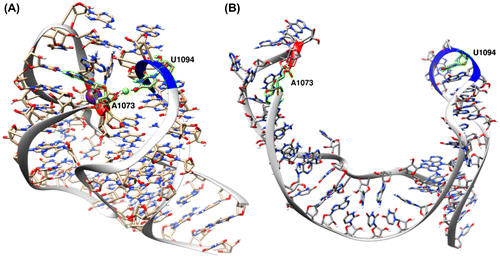
The 58-nucleotide RNA in the GTPase-Associating Center (GAC RNA) has been extensively investigated to better understand RNA–ion interactions (Bukhman & Draper, Citation1997; Conn, Draper, Lattman, & Gittis, Citation1999; Conn, Gittis, Lattman, Misra, & Draper, Citation2002; Leipply & Draper, Citation2011; Misra & Draper, Citation2001; Shiman & Draper, Citation2000; Wang, Lu, & Draper, Citation1993; Wimberly, Guymon, McCutcheon, White, & Ramakrishnan, Citation1999). This highly conserved RNA structure interacts with the L11 protein in the 23S ribosomal subunit to facilitate GTP hydrolysis during transcription (Sergiev, Bogdanov, & Dontsova, Citation2005). Thermal unfolding experiments have shown that a bound monovalent ion (with preference for and K+ over other monovalent cations such as Li+, Na+, Rb+, and Cs+) is crucial in stabilizing the tertiary structure of the GAC RNA (Shiman & Draper, Citation2000; Wang et al., Citation1993). According to UV absorbance experiments, thermal unfolding of GAC RNA involves two distinct transitions each at different temperatures (Bukhman & Draper, Citation1997). Higher concentrations of magnesium and other divalent ions increase the temperature at which the first transition occurs, while not affecting the second transition (Bukhman & Draper, Citation1997). These experiments, when combined with crystallography at high magnesium concentrations, indicate specific divalent ion binding sites, and imply the first unfolding transition of GAC RNA is affected by divalent ion concentration. The available consensus crystal structures of the GAC RNA (PDBs: 1MMS and 1HC8) (Conn et al., Citation2002; Wimberly et al., Citation1999), as well as a hydroxyl radical foot printing experiment, suggest that a chelated magnesium is directly bound to the O4 of U1094 and OP2 of A1073 that belong to two loops (1092–1098 and 1070–1073, respectively) which are in close proximity in the tertiary structure; this magnesium is also 5.7 Å away from a buried monovalent ion (Figure (A)) (Leipply & Draper, Citation2011).
Figure 1. (A) Tertiary structure of the GAC RNA according to the 1HC8 crystal structure (Conn et al., Citation2002). A magnesium ion (green sphere) bridges between the two loops (from residues 1070–1073 and 1092–1098, red and blue ribbons, respectively) with direct interactions to O4 atom of U1094 and OP2 atom of A1073 (red spheres). The nearby bound potassium ion is shown as purple sphere. (B) The GAC structure from the last frame of the 40.0 Å distance umbrella of the simulation in absence of ions showing that the loops are completely disengaged.
Despite many experimental studies and theoretical calculations with non-linear Poisson–Boltzmann methods (Misra & Draper, Citation2001), a clear model has not been devised to physically or structurally describe the transitions observed in thermal unfolding of GAC RNA. The simultaneous roles of diffuse and bound cations in stabilizing this RNA tertiary structure, as well as the presence of both monovalent and divalent ions in experiments, makes it difficult to generate models that specifically distinguish the role of one ion type from the other, and to probe the related structural changes during folding and unfolding transitions. Advances in molecular dynamics (MD) simulations have provided the means for physical descriptions of conformational changes of RNA molecules as influenced by magnesium. For example, recent work in our lab demonstrated spontaneous conversion of the relatively dynamic magnesium free structure of the stem loop V (SLV) RNA to the magnesium-bound structure when Mg2+ was added, effectively stabilizing the structure (Bergonzo, Hall, & Cheatham, Citation2015). We also showed that various different and available additive point charge magnesium models can reproduce the magnesium-dependent conformational change, although some of the models appear to interact too favorably with the RNA leading to trapping of intermediates in long-lived non-native magnesium binding sites (Bergonzo, Hall, & Cheatham, Citation2016). Related examples use MD to describe the influence of the absence and presence of bound ligand on the breaking up of specific kissing loop interactions in the Add A-riboswitch (Allner, Nilsson, & Villa, Citation2013), and provide energetic insight into the migration of a specific catalytic magnesium from the C-site to the bridging site in both reactant and activated precursor states in the hammerhead ribozyme with all atom umbrella sampling simulations (Panteva, Giambaşu, & York, Citation2015).
The structure and dynamics of the GAC RNA is briefly studied with MD simulations as well (Beššeová, Réblová, Leontis, & Šponer, Citation2010; Li, Sengupta, Rath, & Frank, Citation2006; Razga, Koca, Mokdad, & Sponer, Citation2007). Li et al. used 16 ns MD to study the dynamics of the GAC complex structure and docked the representative structures from such simulations onto the cryo-EM maps (Li et al., Citation2006). They reported some detailed observations of the base configurational rearrangement in RNA during the simulations. Rázga et al. described the dynamics of three bigger regions of the GAC RNA relative to each other using 30.5 ns MD simulations (Razga et al., Citation2007). Beššeová et al. also extended that investigation to sub-μs simulations with the aim of understanding the functional of rRNA dynamics (Beššeová et al., Citation2010). However, our preliminary MD simulations of GAC RNA (data not shown) even in absence of the L11 protein showed that these simulations would potentially suffer from lack of convergence and also force field artifacts which would cause serious uncertainty about the accuracy of the results.
In this work, we use all atom MD simulations and umbrella sampling to probe the conformational changes related to the first unfolding transition of the GAC RNA, with regards to the role of a bound magnesium ion. Based on our initial observations in simulations in the absence of any ions, where loop interactions were rapidly destabilized, we hypothesized that the physical equivalent of the first transition event observed in the unfolding experiments (Bukhman & Draper, Citation1997) is separation of two loops, specifically the two loops from residues 1070–1073 and also 1092–1098. Given the presence of the chelated magnesium ion which bridges between these two loops in the crystal structures, we further hypothesized that this binding site plays the important role in the first GAC RNA unfolding transition as described by Bukhman and Draper (Citation1997). We show that the stability of the folded GAC RNA is increased by the presence of this chelated Mg2+ ion, and, despite increasing diffuse Mg2+ ion concentration, in the absence of the chelated ion the folded structure is less stable relative to the partially unfolded state.
Materials and methods
Ion-free simulations (IF-R and IF-UnR)
The GAC RNA in the crystal structure 1HC8 (Conn et al., Citation2002) was used as the initial structure for all simulations in this work. Hydrogen atoms were added to the RNA and all ions and crystallographic water residues were stripped using UCSF Chimera (Pettersen et al., Citation2004). The resulting structure was solvated using the tLEaP program in Amber 14 (Case et al., Citation2014) in TIP3P water (Jorgensen, Chandrasekhar, Madura, Impey, & Klein, Citation1983) in a truncated octahedral box with a minimum distance of 12 Å between its edges and the solute. The ff99 (Cheatham, Cieplak, & Kollman, Citation1999; Wang, Cieplak, & Kollman, Citation2000) force field with parmbsc0 α/γ (Pérez et al., Citation2007) and χOL3 (Zgarbová et al., Citation2011) modifications was used to build the topology. The structure was equilibrated in with a nine-step protocol as described in the following equilibration protocol section. Six production simulations were initiated from the same equilibrated structure, assigned different initial random velocities with random seeds to prevent synchronization (Sindhikara, Kim, Voter, & Roitberg, Citation2009) and extended to 10 ns in NPT conditions with pressure relaxation time of 5 ps (IF-UnR set). The time step for the production simulations was set to 2 fs and structures were saved to the trajectory file every 1 ps. The temperature was set to 298.15 K using Langevin algorithm (Loncharich, Brooks, & Pastor, Citation1992) with 5 ps−1 collision rate and SHAKE (Ryckaert, Ciccotti, & Berendsen, Citation1977) was used to constrain the bonds to hydrogen atoms. The cut off for direct space interactions was set to 8.0 Å and particle-mesh Ewald was used for calculating the long-range interactions (Essmann et al., Citation1995) with default parameters. PMEMD.MPI in Amber 14 was used to run the simulations. To prepare the IF-R set, six production simulations as described above were repeated with 0.5 kcal/mol-Å2 positional restraints on residues 1092–1098.
Equilibration protocol
The solvated initial structure was energy minimized in 1000 steps with steepest descent while RNA heavy atoms were restrained in position with 5 kcal/mol-Å2 force constant. 15 ps MD was performed with 5 kcal/mol-Å2 positional restraints on RNA heavy atoms in NVT conditions; 1000 steps of steepest descent energy minimization were performed with 2 kcal/mol-Å2 positional restraints on RNA heavy atoms. The same energy minimization as described in last step was repeated with decreased restraint weights of 0.1 kcal/mol-Å2 followed by the same minimization procedure without restraints. 5 ps MD was performed in NPT conditions with 1 kcal/mol-Å2 restraints on RNA heavy atoms. SHAKE algorithm (Ryckaert et al., Citation1977) was used to constrain bonds to hydrogen and was applied from this step on. 1 ns MD in NPT conditions was done with 0.5 kcal/mol-Å2 restraints on RNA heavy atoms. The same MD was performed with the same restraints on backbone RNA atoms only. Then, the same MD was done with no restraints. This last step was not performed for equilibrating the umbrella trajectories. The MD time steps were 1 fs for the steps prior to the last step and 2 fs in the last one. The weak-coupling algorithm was used to keep the temperature at 338.15 K in all MD steps (Berendsen, Postma, van Gunsteren, DiNola, & Haak, Citation1984) with temperature coupling constant of 1 ps, except the IF-R and IF-UnR which were done at 298.15 K with the same thermostat. All steps were done with particle-mesh Ewald (Essmann et al., Citation1995).
Preparing the initial structures for the umbrella sampling simulations
In all umbrella sampling simulations in this work, the distance from the center of mass of phosphorus atoms of A1070 and A1073 to the center of mass of phosphorus atoms of U1094 and U1097 was used as the reaction coordinate (Supporting Figure 4). Umbrella sampling simulations were performed in parallel with 1 ns simulations for 321 umbrellas with 0.1 Å spacing between the reaction coordinate distances of 8.0 and 40.0 Å. The initial structures for the umbrellas were prepared in three different ways for three ranges along the reaction coordinate.
For low-range distances (8.0–9.6 Å), which represent distances between the two loops closer than experimentally observed, simulations started from the initial crystal of the GAC RNA. The structure was prepared the same way as in ion-free simulations, except that the crystallographic bound K+ and the loop–loop bridging Mg2+ were retained, the system was neutralized by adding K+ and 1.6 M KCl (experimental KCl concentration in (Bukhman & Draper, Citation1997)) was also added. All ion positions (except the crystallographic K+ and Mg2+) were randomized using CPPTRAJ so that no ion was closer than 6 Å to any RNA atom and ions were not closer than 4 Å from each other. Joung–Cheatham parameters (Joung & Cheatham, Citation2008) were used for monovalent ions, and Allnér–Nilsson–Villa (Allnér et al., Citation2012) parameters were used for Mg2+. The system was equilibrated using the described equilibration protocol. Starting from the crystal reaction coordinate position (9.6 Å), the loops were pulled toward each other using 100 kcal/mol-Å2 distance restraint over 32 ps in NPT conditions. Other simulation parameters were set as in IF-R production simulations. The two closest frames with reaction coordinate values closest to each umbrella window value were extracted from the resulting trajectory using CPPTRAJ program (Roe & Cheatham, Citation2013) to be used as initial structures in two independent runs.
For windows in the medium range distance (9.7–12.5 Å), the loops in the equilibrated structure as described above were also pulled away from each other using 100 kcal/mol-Å2 distance restraint gradually in 60 ps in NPT conditions, while the charge of the chelated Mg2+ was temporarily set to zero using parmed.py program in Amber suite, and its distance to O4 atom of the U1094 was fixed at the crystallographic distance using 100 kcal/mol-Å2 distance restraint, so that the loops disengaged with the ion attached to one side while avoiding unwanted conformational disruptions. The initial structures for the umbrellas in this range were also extracted from the resulting trajectory using CPPTRAJ program as done for the low distance range.
The initial structures for umbrellas in the high range distance (12.6–40.0 Å) were extracted from one of the IF-R production simulations using CPPTRAJ program as done for the low and medium distance ranges. Where specified, the bound Mg2+ was added manually to the O4 atom of U1094 for all US-bound Mg runs.
The above methods were used to generate the initial structures for the umbrella sampling simulations sets indicated in Table .
Table 1. Description of the umbrella sampling simulations performed in this work.
Running umbrella sampling simulations
The initial structures in each umbrella for the US-IF, US-bound and the US-NeutK simulation sets without ion exclusion area were equilibrated using the described equilibration protocol. To initiate the umbrellas of US-unbound Mg simulations, the bound Mg2+ in the initial frame from the RNA backbone-restrained equilibration step of the corresponding US-bound Mg umbrella was randomized, and then, this step was repeated followed by the production simulation. Likewise, the US-NeutK simulations with ion exclusion region were initiated in the same way from the corresponding simulations without the ion exclusion region. The ions were restrained from entering the exclusion area in the last equilibration step and production steps.
Then, a 1 ns production simulation was performed for each umbrella, while the reaction coordinate distance was fixed with 100 kcal/mol-Å2 restraint. This restraint weight provided desirable overlap between umbrellas and kept the sampled distance close to the umbrella distance (Supporting Figure 5). The temperature in all equilibration and production runs was set to 338.15 K to match the highest temperature for the first GAC RNA unfolding transition in melting experiments done by Bukhman and Draper (Bukhman & Draper, Citation1997). In US-neutK simulations with ion-free zones, all K+ ions were kept out of the zone with 10 kcal/mol-Å2 distance restraints during the equilibration and production simulations. The simulations were run with PMEMD.CUDA (Salomon-Ferrer, Götz, Poole, Le Grand, & Walker, Citation2013) and PMEMD.MPI from Amber 14 (Case et al., Citation2014). Two independent copies of all umbrella sampling simulations were performed, each initiating from different initial structures which were extracted from the same trajectories.
PMF calculations and error analysis
The version of the WHAM method (Kumar, Rosenberg, Bouzida, Swendsen, & Kollman, Citation1992; Roux, Citation1995) implemented by Alan Grossfield (http://membrane.urmc.rochester.edu/sites/default/files/wham/doc.html) was utilized with a convergence tolerance of 10−9 to generate the potential of mean force (PMF) plots. Each umbrella sampling window was run for 1 ns, and only 100–300 ps portions of the trajectories were used by WHAM to generate PMFs. The first 100 ps was not used because it led to a very much higher energetic barrier in the PMF plots, which was significantly relaxed with removing this portion of data from WHAM calculations (Supporting Figure 6). Also the data beyond 300 ps were not used in WHAM calculations to enable comparison of the free energy profiles close to the same energetic surface, as the umbrella structures deviated from the initial structures significantly after 300 ps (Supporting Figure 1). All PMF plots reported here are averages obtained from the two independent runs.
Other analysis
The structure figures were generated using UCSF Chimera 1.9 (Pettersen et al., Citation2004). All analysis were done using CPPTRAJ (Roe & Cheatham, Citation2013) unless mentioned otherwise.
Results
The 1070–1073 and 1092–1098 loops dissociate in absence of ions
According to the melting experiments (Bukhman & Draper, Citation1997), the first conformational changes in GAC RNA unfolding is dependent on the ion environment. To probe those changes, we first performed MD simulations under nonphysical conditions where no ions were present in the simulation environment (specifically, the IF-UnR and IF-R simulations sets). The immediate and consistent major conformational change observed in the structure in six independent copies of the simulation was the separation of the two 1070–1073 and 1092–1098 loops, depicted in Figure (A) and (B), which happened spontaneously in the first few nanoseconds of each simulation (Figure (A)). Additionally, the U-turn conformation of the 1092–1098 loop was disrupted. Because of this, we further investigated whether the disengagement of loops was a consequence of the U-turn disruption by repeating the simulations with 0.5 kcal/mol-A2 positional restraints on the heavy atoms of residue 1092–1098. In these restrained MD simulations, the same loop–loop dissociation was observed as was seen in the unrestrained RNA simulations. Accordingly, we hypothesized that the distance between these loops can be used as a reaction coordinate describing the GAC RNA partial unfolding to its intermediate conformations, an event that seems to correlate with the first transition in previously described melting experiments because of its sensitivity to the presence of ions.
Figure 2. (A) The distance from the center of mass of phosphorus atoms of A1070 and A1073 to the center of mass of phosphorus atoms of U1094 and U1097, in GAC RNA simulations in absence of any ions, which is used as the reaction coordinate in all umbrella sampling simulations here. Each black line represents an independent copy of unrestrained simulation, while each red line represents an independent copy of simulations with positional restraints on 1092–1098 residues. (B) The PMF profile along the reaction coordinate described in (A) for partial unfolding of GAC RNA in the absence of any ions.
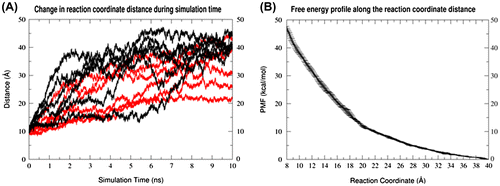
Umbrella sampling simulations were performed using the loop–loop distance as a reaction coordinate with no ions in the simulation environment to confirm the results of the short MD simulations (referred to as Umbrella Sampling-Ion Free simulations, US-IF). The reaction coordinate was chosen as the distance from the center of mass of phosphorus atoms of A1070 and A1073 to the center of mass of phosphorus atoms of U1094 and U1097. The resulting PMF along this reaction coordinate shows a significant drop in the calculated free energy as the two 1070–1073 and 1092–1098 loops are separated from each other, which is consistent with the very short time scale of the conformational change observed in unbiased MD simulations (Figure (B)).
The presence of ions is essential for the stability of the folded tertiary structure
To see the extent to which minimal amounts of ions can affect the stability of the tertiary structure, neutralizing potassium was added in random positions around the initial structure of each umbrella, and 1 ns of MD was performed after equilibration (US-neutK simulation set). Extending the umbrella simulations to 1 ns (in this run as well as other umbrella sampling runs) resulted in dramatic deviation of the structures from the initial structures (Supporting Figure 1). Therefore, the first 200 ps of data after discarding 100 ps for equilibration was used for PMF calculations. The black line in Figure shows that adding neutralizing potassium ions reversed the free energy profile in favor of the relative stability of the folded state, at a reaction coordinate value of 9.6 Å. The structures obtained from the umbrellas close to the folded state, corresponding to the global minimum value of the PMF plot, show accumulation of the K+ ions near the magnesium ion-binding site where the two loops closely interact (Supporting Figure 2).
Figure 3. PMF profiles along the loop–loop distance reaction coordinate with free neutralizing potassium ions (black) and increasing ion exclusion zones around the magnesium-binding site with ion exclusion radii of 5 (cyan), 10 (blue) and 15 Å (magenta). The zero point is adjusted to the PMF values at 40.0 Å. The plots represent the average PMF values between two independent runs and the error bars are calculated as the standard deviation between them.
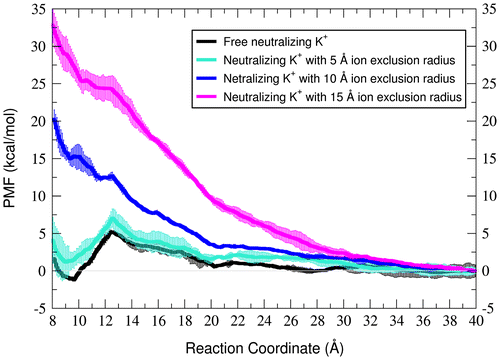
Due to the relative stability of the tertiary structure in the presence of neutralizing ions, and accumulation of ions near the magnesium-binding site, we hypothesized that ion interactions in this area are important to GAC RNA relative stability. To confirm this, we restrained, in separate simulations, the distance of all potassium ions to remain 15, 10 and 5 Å away, respectively, from the center of mass of the oxygen atoms which directly bind the bridging magnesium in the crystal structure (OP2 of A1073 and O4 of U1094). Such restraints create ion-exclusion zones around the loop interaction site. The resulting free energy profiles in Figure indicate that the relative stability of the folded state with reference to the partially unfolded states is decreased as the ion exclusion zone expands. This trend implies that some ion density surrounding crystallographic magnesium binding site is required to stabilize the folded RNA structure.
Chelation of a single Mg2+ ion increases the stability of the folded GAC RNA at different Mg2+ concentrations
Based on the fact that binding of a Mg2+ ion is involved in the first transition during thermal unfolding of the GAC RNA at different Mg2+ concentrations (Bukhman & Draper, Citation1997; Leipply & Draper, Citation2011), we expect to see the folded structure (at 9.6 Å) is stabilized relative to the intermediate structure (at >20 Å) in the presence of a chelated Mg2+ which bridges the two 1070–1073 and 1092–1098 loops. Therefore, we set up umbrella sampling simulations in neutralizing potassium with additional concentrations of diffuse MgCl2 (0, 10, 20, and 50 mM) while retaining the chelated magnesium at its crystallographic position (US-bound Mg simulation sets, Figure , left). The initial equilibrium steps of each umbrella were performed with restraints on the bound magnesium so that it is not pulled into the closely located monovalent ion pocket (Conn et al., Citation2002). To analyze the effect of the bound Mg2+, that ion was randomized to a position at minimum 6 Å away from any RNA atom prior to the last equilibration step in each umbrella, and the last equilibration step as well as the production step were repeated in the absence of the bound Mg2+ for a third set of independent simulations (US-unbound Mg simulation sets, Figure , right). As indicated in the Supporting Figure 7, few Mg2+ binding events were observed in some umbrellas of the simulations. These occasional bindings happened at random positions along the RNA (data not shown) in different umbrella windows, but not at the bridging Mg2+ binding site when it is supposed to be free. However, the umbrella simulations were short enough that the diffuse ions remained mostly dissociated from the RNA atoms and no consistent ion binding happened to any RNA atom (Supporting Figure 8). Figure represents the PMF plots for the change along the reaction coordinate in presence (left) and absence (right) of the chelated Mg2+.
Figure 4. PMF profiles along the loop–loop separation distance reaction coordinate with neutralizing potassium and increasing amount of excess magnesium chloride. (A) One Mg2+ ion is chelated in the crystallographic position as depicted in Figure (A) close to the global minimum and bound to only the O4 of U1094 in longer distances. (B) Continuation of the simulations in (A) after the chelated Mg2+ ion is displaced to random minimum of 6 Å distances from the RNA. All the PMFs are zeroed at the closest local minimum to the crystallographic reaction coordinate distance (9.6 Å). The error bars are calculated as the standard deviation between two independent runs.
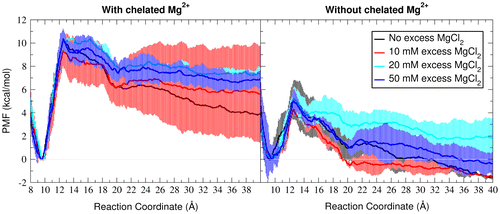
In all Mg2+ concentrations, the free energy of unfolding decrease significantly with removing the chelated Mg2+ from its crystallographic position. Also the PMFs have sharper slopes from 9.7 to 12 Å in the presence of the bound Mg2+. This sharper slope in low umbrellas correlate with dissociation of the loops as the bridging magnesium is pulled from A1073.
Description of the first unfolding transition
The simulations in this work provide insight into the possible conformational changes that are observed as the folded GAC RNA partially unfolds to an intermediate state. The unfolding transition, which is described first by the unrestrained and restrained MD simulation and then further by the PMFs, initially involves disengagement of the 1070–1073 and 1092–1098 loops. According to the GAC crystal structures, these loops are tightly linked together by a chelated Mg2+ ion which bridges the O4 atom of the U1094 and OP2 atom of the A1073. This is in agreement with the PMF plots in Figure , where the energy barrier for unfolding is significantly higher in presence of chelated Mg2+. However, there are other stabilizing interactions between RNA atoms that are unraveled during unfolding as well.
A major structural change that significantly contributes to RNA unfolding is the loss of contact between A1088 and U1060 (Figure (B) and Supporting Figure 3). In the crystal structure, A1088 from one strand intercalates into the adjacent helix and forms a Hoogsteen base pair with U1060 while stacking on top of C1079 (Conn et al., Citation1999). According to the thermal unfolding and L11 binding experiments performed on the GAC RNA mutants, a GAC RNA carrying A1088U mutation is described as lethal and is both unable to fold to the tertiary structure and to bind to the L11 protein (Maeder, Conn, & Draper, Citation2006) The described contacts of A1088 are broken immediately in the ion-free simulations (IF-R) and therefore, they are missing in all umbrella initial structures which are extracted from those trajectories (See Material and Methods and Supporting Figure 3). This suggests that the base pairing of A1088 and U1060 and stacking between A1088 and C1079 are among the first key interactions that break during the unfolding pathway. This highlights the importance of these interactions and is consistent with the A1088U mutation experiments.
Figure 5. (A) The global structure of GAC RNA according to the 1HC8 crystal structure (Conn et al., Citation2002) with important residues which undergo major structural changes during unfolding transition highlighted. (B) Hoogsteen base pair between A1088 and U1060 (hydrogen bonds represented as solid black lines) and stacking interaction between A1088 and C1079 according to the 1HC8 crystal structure (Conn et al., Citation2002). (C) Stacking interaction between G1071 and A1089 according to the 1HC8 crystal structure (Conn et al., Citation2002).
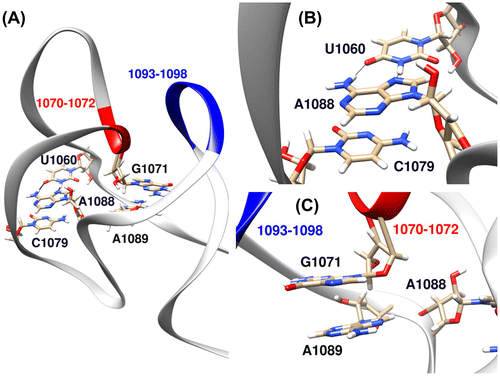
Another change in the RNA upon partial unfolding is the loss of a stacking interaction between G1071 and A1089 (Figure (C) and Supporting Figure 3). The backbone of G1071 contributes to the formation of the buried monovalent ion pocket. Because of the proximity of A1089 and A1088 in sequence, loss of base stacking between A1089 and G1071 might potentially allow a concerted conformational change in the neighboring A1088 as described above. This stacking interaction is also lost in the IF-R simulations in absence of ions and early in the unfolding pathway (Supporting Figure 3). This is in agreement with a recent 2-aminopurine fluorescence experiment which reports that stacking of 2-aminopurine derivative of A1089 in GAC structure is dependent on the presence of Mg2+ (Rau, Welty, Tom Stump, & Hall, Citation2015).
Structural changes after 12.5 Å on the unfolding pathway include breaking of many interactions between these two parts of the RNA finishing with total disruption of the U-turn 1082–1086.
Discussion
GAC RNA is an extensively studied RNA model system in terms of how the stability of its tertiary structure is affected by different ion interactions. However, there is little structural information about how these ions play their roles in the pathway during which this RNA unfolds. Here, we employed umbrella sampling MD simulations to qualitatively describe an important step in the unfolding pathway of the GAC RNA, with an emphasis on the role of a bound magnesium ion. The results of unrestrained simulations in the absence of ions show that unfolding step is characterized by a loss of loop-loop interactions in the GAC RNA; and a chelated Mg2+ ion that bridges between the 1070–1073 and 1092–1098 loops can relatively stabilize the folded state. The PMF plots resulted from the simulations with different Mg2+ concentrations show qualitatively similar stability of the folded state relative to the PMF plot with no Mg2+ (the right panel of the Figure and the black PMF plot in Figure ) unless one Mg2+ is chelated between the loops. This is consistent with the first step of the multistep pathway for unfolding of GAC RNA which has been experimentally shown to be ion-sensitive and involve binding of a divalent cation (Bukhman & Draper, Citation1997; Leipply & Draper, Citation2011). We expect that dissociation of these loops is the first event in the GAC RNA unfolding pathway. Also we showed that separation of these loops in the unfolding pathway is accompanied by breaking of the A1088-U1060 Hoogsteen base paring and A1088-C1079 stacking interactions and also the simultaneous loss of stacking between A1089 and G1071.
Although results of this work are qualitatively consistent with the available experimental data, one should avoid over-interpretation of the umbrella sampling outcomes of folding/unfolding pathways. One reason is that the folding/unfolding pathways involve complex rearrangements of intramolecular interactions which might not be fully represented by any chosen one-dimensional reaction coordinate. The second reason is that converging the umbrella simulation, especially in the unfolded states is a nontrivial task and requires incredible amount of sampling. Since full sampling of the conformational space along the reaction coordinate is almost impossible for a macromolecule of this size with the current technology, using short simulations that sample close to the initial conformations is preferred (Supporting Figure 1). Therefore, the results of umbrella sampling simulations that involve breaking and making of so many interactions tend to be dependent on the initial conditions (Di Palma, Bottaro, & Bussi, Citation2015). Even starting from the same initial conditions, two independent runs might have relatively different destinies causing an imperfect match between the PMF plots (Allner et al., Citation2013). The third reason for avoiding over-interpretation of the PMF plots in a quantitative way is the difficulty of equilibrating the ion environment. The occasional ion-binding events, although are not very close to the loop–loop interaction region, could potentially affect the PMF plots and contribute to increasing the error bars.
Increasing the stability of the folded tertiary structure relative to the intermediate structure could occur through either stabilizing the tertiary structure or destabilizing the intermediate structures in higher magnesium concentrations. We have shown stabilization of the GAC RNA is only possible in the presence of cations that bridge between these two loops. A single Mg2+ ion with direct interactions with the OP2 atom of A1073 and O4 atom of U1094 can keep the loops in close proximity, stabilizing the folded state relative to the unfolded state. This magnesium ion is present in all available GAC crystal structures and its presence has been reconfirmed by hydroxyl radical foot printing experiments (Conn et al., Citation2002; Leipply & Draper, Citation2011; Wimberly et al., Citation1999). Regarding the observation that the GAC RNA folds in the presence of other divalent ions as well, using modified GAC RNA that selectively binds to some divalent ions can validate the proposed role for the chelated magnesium in the folding pathway in future experiments. However, one should also note that the RNA might fold and unfold through different pathways (Onoa & Tinoco, Citation2004), and therefore, we should not necessarily expect that simulations initiated from the intermediate structures in which the loops are dissociated lead us to the correct folded state through capturing of the bridging Mg2+ in physiological conditions.
Supplemental data
The supplementary material for this article is available online at http://dx.doi.org/10.1080/07391102.2016.1274272
Funding
This work was supported by the National Institute of General Medical Sciences [GM-098102].
JBSD-revised-SI.docx
Download MS Word (2.2 MB)Acknowledgements
This research has become possible by using the computational resources of National Science Foundation Extreme Science and Engineering Discovery Environment (XSEDE) allocation MCA01S027P and computational resources from the Center for High Performance Computing at the University of Utah. We also acknowledge funding by the NIH GM-098102.
Disclosure statement
No potential conflict of interest was reported by the authors.
Related Research Data
References
- Allner, O., Nilsson, L., & Villa, A. (2013). Loop-loop interaction in an adenine-sensing riboswitch: A molecular dynamics study. RNA, 19, 916–926. doi:10.1261/rna.037549.112
- Allnér, O., Nilsson, L., & Villa, A. (2012). Magnesium ion-water coordination and exchange in biomolecular simulations. Journal of Chemical Theory and Computation, 8, 1493–1502. doi:10.1021/ct3000734
- Berendsen, H. J. C., Postma, J. P. M., van Gunsteren, W. F., DiNola, A., & Haak, J. R. (1984). Molecular dynamics with coupling to an external bath. The Journal of Chemical Physics, 81, 3684–3690. doi:10.1063/1.448118
- Bergonzo, C., Hall, K. B., & Cheatham 3rd, T. E. (2015). Stem-loop V of Varkud satellite RNA exhibits characteristics of the Mg2+ bound structure in the presence of monovalent ions. The Journal of Physical Chemistry B, 119, 12355–12364. doi:10.1021/acs.jpcb.5b05190
- Bergonzo, C., Hall, K. B., & Cheatham 3rd, T. E. (2016). Divalent ion dependent conformational changes in an RNA stem-loop observed by molecular dynamics. Journal of Chemical Theory and Computation, 12, 3382–3389. doi:10.1021/acs.jctc.6b00173
- Beššeová, I., Réblová, K., Leontis, N. B., & Šponer, J. (2010). Molecular dynamics simulations suggest that RNA three-way junctions can act as flexible RNA structural elements in the ribosome. Nucleic Acids Res, 38, 6247–6264. doi:10.1093/nar/gkq414
- Bukhman, Y. V.. & Draper, D. E. (1997). Affinities and selectivities of divalent cation binding sites within an RNA tertiary structure. Journal of Molecular Biology, 273, 1020–1031. doi:10.1006/jmbi.1997.1383
- Case, D., Babin, V., Berryman, J., Betz, R., Cai, Q., Cerutti, D., … Kollman, P. A. (2014). Amber 14.
- Cheatham 3rd, T. E., Cieplak, P., & Kollman, P. A. (1999). A modified version of the Cornell et al. force field with improved sugar pucker phases and helical repeat. Journal of Biomolecular Structure and Dynamics, 16, 845–862. doi: 10.1080/07391102.1999.10508297
- Conn, G. L., Draper, D. E., Lattman, E. E., & Gittis, A. G. (1999). Crystal structure of a conserved ribosomal protein-RNA complex. Science, 284, 1171–1174.10.1126/science.284.5417.1171
- Conn, G. L., Gittis, A. G., Lattman, E. E., Misra, V. K., & Draper, D. E. (2002). A compact RNA tertiary structure contains a buried backbone–K+ complex. Journal of Molecular Biology, 318, 963–973. doi:10.1016/S0022-2836(02)00147-X
- Di Palma, F., Bottaro, S., & Bussi, G. (2015). Kissing loop interaction in adenine riboswitch: Insights from umbrella sampling simulations. BMC Bioinformatics, 16, S6. doi:10.1186/1471-2105-16-s9-s6
- Draper, D. E. (2004). A guide to ions and RNA structure. RNA, 10, 335–343.10.1261/rna.5205404
- Essmann, U., Perera, L., Berkowitz, M. L., Darden, T., Lee, H., & Pedersen, L. G. (1995). A smooth particle mesh Ewald method. The Journal of Chemical Physics, 103, 8577–8593. doi:10.1063/1.470117
- Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W., & Klein, M. L. (1983). Comparison of simple potential functions for simulating liquid water. The Journal of Chemical Physics, 79, 926–935. doi:10.1063/1.445869
- Joung, I. S., & Cheatham 3rd, T. E. (2008). Determination of alkali and halide monovalent ion parameters for use in explicitly solvated biomolecular simulations. The Journal of Physical Chemistry B, 112, 9020–9041. doi:10.1021/jp8001614
- Kumar, S., Rosenberg, J. M., Bouzida, D., Swendsen, R. H., & Kollman, P. A. (1992). THE weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. Journal of Computational Chemistry, 13, 1011–1021. doi:10.1002/jcc.540130812
- Leipply, D., & Draper, D. E. (2011). Evidence for a thermodynamically distinct Mg2+ ion associated with formation of an RNA tertiary structure. Journal of the American Chemical Society, 133, 13397–13405. doi:10.1021/ja2020923
- Li, W., Sengupta, J., Rath, B. K., & Frank, J. (2006). Functional conformations of the L11–ribosomal RNA complex revealed by correlative analysis of cryo-EM and molecular dynamics simulations. RNA, 12, 1240–1253. doi:10.1261/rna.2294806
- Loncharich, R. J., Brooks, B. R., & Pastor, R. W. (1992). Langevin dynamics of peptides: The frictional dependence of isomerization rates of N-acetylalanyl-N′-methylamide. Biopolymers, 32, 523–535. doi:10.1002/bip.360320508
- Maeder, C., Conn, G. L., & Draper, D. E. (2006). Optimization of a ribosomal structural domain by natural selection. Biochemistry, 45, 6635–6643. doi:10.1021/bi052544p
- Misra, V. K., & Draper, D. E. (2001). A thermodynamic framework for Mg2+ binding to RNA. Proceedings of the National Academy of Sciences, 98, 12456–12461. doi:10.1073/pnas.221234598
- Onoa, B., & Tinoco Jr, I. (2004). RNA folding and unfolding. Current Opinion in Structural Biology, 14, 374–379. doi:10.1016/j.sbi.2004.04.001
- Panteva, M. T., Giambaşu, G. M., & York, D. M. (2015). Force field for Mg2+, Mn2+, Zn2+, and Cd2+ ions that have balanced interactions with nucleic acids. The Journal of Physical Chemistry B, 119, 15460–15470. doi:10.1021/acs.jpcb.5b10423
- Pérez, A., Marchán, I., Svozil, D., Sponer, J., Cheatham, T. E., 3rd, Laughton, C. A., & Orozco, M. (2007). Refinement of the AMBER force field for nucleic acids: Improving the description of α/γ conformers. Biophysical Journal, 92, 3817–3829. doi:10.1529/biophysj.106.097782
- Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C., & Ferrin, T. E. (2004). UCSF chimera?A visualization system for exploratory research and analysis. Journal of Computational Chemistry, 25, 1605–1612. doi:10.1002/jcc.20084
- Rau, M. J., Welty, R., Tom Stump, W., & Hall, K. B. (2015). Formation of tertiary interactions during rRNA GTPase center folding. Journal of Molecular Biology, 427, 2799–2815. doi:10.1016/j.jmb.2015.07.013
- Razga, F., Koca, J., Mokdad, A., & Sponer, J. (2007). Elastic properties of ribosomal RNA building blocks: Molecular dynamics of the GTPase-associated center rRNA. Nucleic Acids Research, 35, 4007–4017. doi:10.1093/nar/gkm245
- Roe, D. R., & Cheatham, T. E., 3rd (2013). PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. Journal of Chemical Theory and Computation, 9, 3084–3095. doi:10.1021/ct400341p
- Roux, B. (1995). The calculation of the potential of mean force using computer simulations. Computer Physics Communications, 91, 275–282. doi:10.1016/0010-4655(95)00053-I
- Ryckaert, J.-P., Ciccotti, G., & Berendsen, H. J. C. (1977). Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. Journal of Computational Physics, 23, 327–341. doi:10.1016/0021-9991(77)90098-5
- Salomon-Ferrer, R., Götz, A. W., Poole, D., Le Grand, S., & Walker, R. C. (2013). Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. Explicit solvent particle Mesh Ewald. Journal of Chemical Theory and Computation, 9, 3878–3888. doi:10.1021/ct400314y
- Sergiev, P. V., Bogdanov, A. A., & Dontsova, O. A. (2005). How can elongation factors EF-G and EF-Tu discriminate the functional state of the ribosome using the same binding site? FEBS Letters, 579, 5439–5442. doi:10.1016/j.febslet.2005.09.010
- Shiman, R., & Draper, D. E. (2000). Stabilization of RNA tertiary structure by monovalent cations. Journal of Molecular Biology, 302, 79–91. doi:10.1006/jmbi.2000.4031
- Sindhikara, D. J., Kim, S., Voter, A. F., & Roitberg, A. E. (2009). Bad seeds sprout perilous dynamics: Stochastic thermostat induced trajectory synchronization in biomolecules. Journal of Chemical Theory and Computation, 5, 1624–1631. doi:10.1021/ct800573m.
- Wang, Y. X., Lu, M., & Draper, D. E. (1993). Specific ammonium ion requirement for functional ribosomal RNA tertiary structure. Biochemistry, 32, 12279–12282.10.1021/bi00097a002
- Wang, J., Cieplak, P., & Kollman, P. A. (2000). How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? Journal of Computational Chemistry, 21, 1049–1074. doi:10.1002/1096-987x(200009)21:12<1049::aid-jcc3>3.0.co;2-f
- Wimberly, B. T., Guymon, R., McCutcheon, J. P., White, S. W., & Ramakrishnan, V. (1999). A detailed view of a ribosomal active site: The structure of the L11-RNA complex. Cell, 97, 491–502.10.1016/S0092-8674(00)80759-X
- Woodson, S. A. (2010). Compact intermediates in RNA folding. Annual Review of Biophysics, 39, 61–77. doi:10.1146/annurev.biophys.093008.131334
- Zgarbová, M., Otyepka, M., Šponer, J., Mládek, A., Banáš, P., Cheatham 3rd, T. E., & Jurečka, P. (2011). Refinement of the Cornell et al. nucleic acids force field based on reference quantum chemical calculations of glycosidic torsion profiles. Journal of Chemical Theory and Computation, 7, 2886–2902. doi:10.1021/ct200162x