
Abstract
Metabotropic glutamate receptor 5 (mGluR5) is a class C G protein-coupled receptor (GPCR) with both an extracellular ligand binding site and an allosteric intrahelical chamber located similarly to the orthosteric ligand binding site of Class A GPCRs. Ligands binding to this ancestral site of mGluR5 can act as positive (PAM), negative (NAM) or silent (SAM) allosteric modulators, and their medicinal chemistry optimization is notoriously difficult, as subtle structural changes may cause significant variation in activity and switch in the functional response. Here we present all atom molecular dynamics simulations of NAM, SAM and PAM complexes formed by closely related ligands and analyse the structural differences of the complexes. Several residues involved in the activation are identified and the formation of a continuous water channel in the active complex but not in the inactive ones is recognized. Our results suggest that the mechanism of mGluR5 activation is similar to that of class A GPCRs.
Communicated by Ramaswamy H. Sarma
Introduction
G protein-coupled receptors (GPCR) are a diverse set of seven transmembrane (TM) proteins that mediate various extracellular stimuli to the interior of the cell. They are involved in numerous disorders and their structure and activation mechanism are subject to intensive research. Recent advances in GPCR structural biology together with other experimental and computational tools improved our understanding of the complex mechanism of GPCR activation (Hilger, Masureel, & Kobilka, Citation2018; Wang, Qiao, & Li, Citation2018; Weis & Kobilka, Citation2018). Despite the large variations in GPCR functions, there are common features in their overall structure and activation. Agonist binding induces significant change in the protein conformation, including the positions of the helices, their interactions with each other, with the membrane and with water, and results in the binding to G-protein, and promotes downstream signalling via changes affecting G-protein structure and interactions. In addition, ligand binding to GPCRs may activate β-arrestin-mediated signalling pathways. Metabotropic glutamate (mGlu) receptors (Niswender & Conn, Citation2010) belong to the class C family of GPCRs. They contain an extracellular domain of glutamate binding site and are able to bind allosteric ligands within the transmembrane domain corresponding to the orthosteric site of class A GPCRs. The mGlu family include eight receptors divided into three groups that act through different intracellular pathways. mGluR5 (Sengmany & Gregory, Citation2016) belongs to group I whose members are coupled to Gq proteins and activate phospholipase C. The optimization of allosteric mGluR ligands is highly challenging owing to frequently observed steep structure–activity relationships (SAR) and the variation of functional response upon subtle structural changes of the allosteric ligands (molecular switch) (Lindsley et al., Citation2016). It has been demonstrated recently that steep SAR can be attributed to the flexibility of the allosteric pocket leading to induced binding and to the interaction of the allosteric ligand with the water network in the intrahelical chamber (Christopher et al., Citation2019).
Computational studies are increasingly applied to explore the structural and dynamical properties of biological macromolecules including GPCRs (Bartuzi, Kaczor, & Matosiuk, Citation2018; Latorraca, Venkatakrishnan, & Dror, Citation2017) and among them mGluR receptors (Llinas del Torrent, Pérez-Benito, & Tresadern, Citation2019). Molecular dynamics (MD) simulations are particularly valuable in investigating ligand binding (Bernetti, Masetti, Rocchia, & Cavalli, Citation2019; De Vivo, Masetti, Bottegoni, & Cavalli, Citation2016; Guo & IJzerman, Citation2018) and activation mechanism of GPCRs (Miao & McCammon, Citation2018; Yuan, Chan, et al., Citation2016; Yuan, Peng, Palczewski, Vogel, & Filipek, Citation2016). Here we used all-atom MD simulations to examine the effect of closely related allosteric ligands on the structure and activation of the mGluR5 receptor.
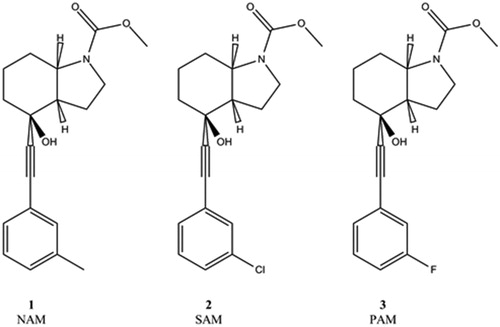
Mavoglurant is the prototype of mGluR5 negative allosteric modulators (NAMs), and the replacement of a single Me substituent to Cl and F yields a silent allosteric modulator (SAM) and a positive allosteric modulator (PAM), respectively (). Our all atom MD simulations of the transmembrane domains of active and inactive mGluR5 complexes provide information complement to recent experimental structures (Koehl et al., Citation2019) of the mGluR5 receptor dimer containing extracellular domains. In contrast to the cryo-electron microscopy provided big picture of signal propagation from ligand binding to domain reorganization, our MD simulations focus on changes in the intradomain atomic interactions upon activation and explicitly include the G-protein bound to the active form of the receptor. Previous computational studies on the activation of mGluR receptors include MD simulations for mGluR2 that confirmed the role of the trigger switch amino acids (F6433.40c, N7355.47c and W7736.50c) in allosteric modulation (Doornbos et al., Citation2016; Pérez-Benito et al., Citation2017). MD simulations with mGluR5 NAM and PAM complexes revealed the critical role of the transmission switch residues (S6583.343c, Y6593.44c and T7816.46c) in allosteric activation (Llinas del Torrent, Casajuana-Martin, Pardo, Tresadern, & Pérez-Benito, Citation2019). In a recent MD study, dual binding mode of the investigated PAM was found and although elements of the activation mechanism in comparison with class A GPCRS were analysed, limited differences between the hydration of the allosteric sites of active and inactive receptors were observed (Cong, Chéron, Golebiowski, Antonczak, & Fiorucci, Citation2019). Our MD simulations for the inactive receptor are based on the mavoglurant (NAM) complex for which X-ray structure is available, while the active receptor complex is modelled together with the Gq protein and with a PAM having close structural similarity to mavoglurant. In analysing the results, we pay particular attention to analogy with class A GPCR activation and to the role of water within the allosteric pocket, as water was found to affect both activation in class A GPCRs (Angel, Chance, & Palczewski, Citation2009; Yuan, Filipek, Palczewski, & Vogel, Citation2014) and ligand binding in mGluR5 (Christopher et al., Citation2019).
Figure 1. mGlur5 ligands used in the MD simulations.
Simulation of the mavoglurant (NAM)–mGluR5 complex started from the X-ray structure (PDB: 4OO9) (Doré et al., Citation2014). The complex of the SAM was constructed by replacing the Me group by Cl. The PAM complex was built using the same protein structure with a procedure established for activated class C GPCRs (Doornbos et al., Citation2016). The TM6 helix was replaced by a model based on the active state μ-opioid receptor in complex with Gi protein (PDB: 6DDE). The Gq protein was modelled using the Gi coordinates of the same complex. The initial position of the PAM ligand was obtained by replacing the Me group by F. 3 × 500 ns MD simulations were performed for each complex. An additional simulation with mavoglurant (NAM) placed into the active mGluR5 model not containing the Gi protein was also performed to investigate if simulations restore the inactive state.
Methods
Model building
NAM (ID:4OO9) and SAM (ID:4OO9-Cl) complex structures: The 3 D structure of the inactive metabotropic glutamate receptor 5 (mGlu5, PDB code: 4OO9) (Doré et al., Citation2014) was acquired from Protein Data Bank (Berman et al., Citation2000) database. The T4-lysozyme which was used to facilitate the crystallization was removed from the structure. The proteins were prepared using default settings of Protein Preparation Wizard (Maestro version: 11.5.011) (Madhavi Sastry, Adzhigirey, Day, Annabhimoju, & Sherman, Citation2013), the missing loops (ICL 2 (721-728 AA) and ECL 2 (679-688 AA)) were built using Prime (“Schrödinger Release 2018-4, Schrödinger, LLC, New York, NY, 2018,” n.d.).
PAM (ID:4OO9-F) structure: The 3 D structure of the inactive metabotropic glutamate receptor 5 (mGlu5, PDB code: 4OO9) (Doré et al., Citation2014) and the μ-opioid receptor–Gi protein complex (PDB code: 6DDE) (Koehl et al., Citation2018) were acquired from the Orientations of Proteins in Membranes (OPM) (Lomize, Pogozheva, Joo, Mosberg, & Lomize, Citation2012) and Protein Data Bank (Berman et al., Citation2000) databases, respectively. The mGlu5 template contains the Lysozyme coordinates, which were deleted from the file manually (deleted residues: 1–567 and 833–1212).
The sequence of Gq-protein was obtained from the Universal Protein Resource (UniProt ID: P50148) (The UniProt Consortium, Citation2007).
When combining the mGlu5 and μ-opioid structures, a local structure-based sequence alignment was made for TM6 using GPCRdb generic numbers (Isberg et al., Citation2015; Pándy-Szekeres et al., Citation2018). Based on this, three pairs of residues were selected for superimposing the μ-opioid TM6 region (TRP785-TRP293, ILE783-VAL291, THR780-VAL288; 4OO9-6DDE respectively) together with the Gi protein from the μ-opioid–Gi complex structure.
The overall sequence alignment was done by Advanced Homology Modelling package of Schrödinger Suites. The knowledge-based procedure was applied to build 100 multi-template models. The 4OO9 coordinates served as template for the whole receptor except the starting sequence of TM6 (FNEALYIAFTMYTTCI) where the corresponding sequence of 6DDE TM6 (RRITRMVLVVVAVFIV) was the template. The Gq α subunit structure was generated using Gi α subunit coordinates of the 6DDE complex. The Mavoglurant – co-crystallized ligand of 4OO9 – was also included in the model building. The best model was chosen on the basis of normalized Discrete Optimized Protein Energy score (Shen & Sali, Citation2006). The chosen model was refined by Protein Preparation Wizard Wizard (Maestro version: 11.5.011) (Madhavi Sastry et al., Citation2013).
The ligand in the X-ray structure was used for 4OO9, and the 2-methyl group of the phenyl ring was changed to chloride for creating the SAM, and fluorine for creating the PAM ligands.
Water molecules were generated for each protein cavities using iterative water hot spot identification with molecular interaction field analysis (GRID) (Goodford, Citation1985; Sciabola et al., Citation2010) and short molecular dynamics simulations (WaterFLAP version 2.2.1). The energies of the water molecules were calculated using OH2 and CRY (combining C1 and DRY) probes (Mason et al., Citation2013) and an entropy analysis of the degrees of freedom and the shape of the energy wells of the water molecules.
MD simulation
Molecular dynamics simulations were conducted for three different protein–ligand complexes of the metabotropic glutamate receptor 5. The first one (4OO9) was the crystal structure of the receptor complexed with the negative allosteric modulator, mavoglurant(Doré et al., Citation2014).The second one was the same structure with a modified ligand (resulted in silent allosteric modulator) where the methyl group was replaced by Cl (4OO9-Cl). The third one was the active state receptor structure with positive allosteric modulator, where the methyl group was replaced by F (ID: 4OO9-F).
The previously prepared structures were placed into fully hydrated POPC membrane by means of the Desmond/System Setup module using the orientation parameters obtained from “Orientations of Proteins in Membranes” website (Lomize et al., Citation2012). During the preparation the minimum distance between the wall of the periodic box and any protein atoms was set at least 16 Å in the xy plane and an additional waterslab was also added in the z dimension (15 Å). Appropriate number of ions (Na + and Cl−) were added, as well, to ensure a 0.15 mol/dm3 salt concentration and electroneutrality of the whole system. The number of atoms and the composition of the systems are presented in .
Table 1. Characteristic parameters of the complex structures investigated by molecular dynamics simulations.
After preparation, the membrane protein equilibration protocol was used with GPU code of the DESMOND program package (Bowers et al., Citation2006), OPLSAA2005 (Banks et al., Citation2005; Jensen & Jorgensen, Citation2006; Kaminski, Friesner, Tirado-Rives, & Jorgensen, Citation2001) parameter set and TIP3P potential for water molecules (Jorgensen et al., Citation1983). For water molecules present in the initial structure, the Gaussian Barrier Potential was not applied to keep them inside the receptor.
After the equilibration the structure was converted to AMBER format, and the following force field parameters were assigned: protein–ff14SB (Maier et al., Citation2015), lipid–lipid14 (Dickson et al., Citation2014), ligand–gaff1.4 (Wang, Wolf, Caldwell, Kollman, & Case, Citation2004), waters–TIP3P potential, ion–parameters developed by Joung and Cheatham (Citation2008, Citation2009).
Assignment of gaff1.4 parameters for the ligands was performed with the antechamber (Wang, Wang, Kollman, & Case, Citation2006) module of AmberTools18 (Case et al., Citation2018). RESP charges (Dupradeau et al., Citation2010) were calculated for ligands according to the resp protocol using R.E.D. version III.52 for geometry optimizations and Gaussian 09, Revision A.01 (Frisch et al., Citation2016) for molecular electrostatic map calculations.
3 × 625-ns-long molecular dynamics simulations were performed for each structure. In the first step, each structure was minimized and two consecutive NVT simulations were conducted at 10 and at 310 K, each step taking 20 ps long. After the NVT calculation, we turned to NPT calculations. The temperature was kept constant using Langevin dynamics (γln = 5.0 ps-1 in NVT and γln = 1.0 ps-1 in NPT) (Bussi & Parrinello, Citation2007; Melchionna, Citation2007) and in the NPT step, Berendsen barostat was used (T = 310.0 K, semiisotropic pressure scaling, p = 1.0 bar, τp = 1.0 ps-1, γten = 0.0 dyne/cm, number of interfaces = 2) (Berendsen, Postma, van Gunsteren, DiNola, & Haak, Citation1984), the cut-off value was set to 10 Å. We applied the hydrogen mass repartition scheme (Hopkins, Le Grand, Walker, & Roitberg, Citation2015) as well, allowing the use of 4 fs time step.
The GPU version (Götz et al., Citation2012; Le Grand, Götz, & Walker, Citation2013; Salomon-Ferrer, Götz, Poole, Le Grand, & Walker, Citation2013) of pmemd module of AMBER16 (Case et al., Citation2016) was used in the simulations, achieving 80–90 ns/day and 37 ns/day performance for 4OO9, 4OO9-Cl and 4OO9-F, respectively, with a GeForce GTX 1080 graphics card.
Results and discussion
Complex structures () and binding modes were found to be stable during the simulations (Supplementary Material, Figure S1). The NAM and SAM complexes are very close to the antagonist bound experimental structure (Doré et al., Citation2014) with a 0.90 Å TM region RMSD. The 2.12 Å TM region RMSD between the PAM complex and the cryo-electron microscopy structure of the active receptor (Koehl et al., Citation2019) represents a reasonable agreement taking into account that the latter has a 4 Å resolutions and does not include the G-protein (Supplementary Material, Table S1).
Figure 2. Global view of the TM domain of the PAM complex. Residues found to be involved in activation and discussed in the text are shown. Switch residues Y6593.44c, I7515.51c T7816.46c and S6543.39c are shown with grey surfaces. Ionic/polar lock of K6653.50c, E7706.35c, S613ICL1 and K8217.51c is shown with red surfaces and are partially obscured by water. W7856.50c is shown with violet surface. Water is in blue. The water channel under the ligand is characteristic to the PAM complex.
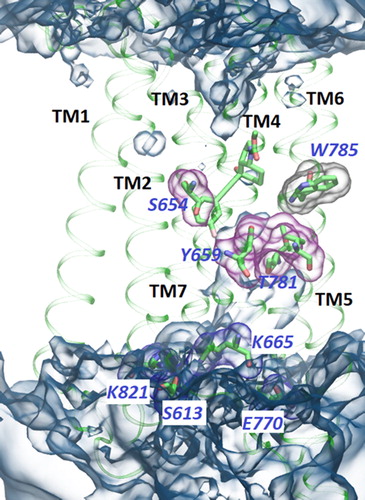
A comparative analysis of the MD trajectories of the NAM, SAM and PAM complexes reveals several intriguing differences in the positions, conformations and interactions of the residues (see for an overall view of the TM domain of the PAM complex). Considering the binding site, its solvent-accessible surface area (SASA) in the presence of PAM is larger, and the ∼40% increase can be attributed primarily to polar uncharged residues and to a lesser extent to nonpolar residues (Supplementary Material, Figure S2).
W7856.50c was observed to turn transiently towards the ligand in the PAM complex. This is not possible in the complexes of 1 (NAM) and 2 (SAM), as the octahydro-1H-indole group of mavoglurant and its Cl derivative occupies a space would be needed for the indole ring of W7856.50c in the inward position. In the complex of 3 (PAM), however, the outward movement of the TM6 intracellular end makes this rotamer state available, although in our simulation of the active state, it is less populated then the other rotamer pointing away from the allosteric ligand. This highly conserved Trp residue was found to be part of the activation in several class A GPCRs (Trzaskowski et al., Citation2012), including rhodopsin (Ahuja & Smith, Citation2009). Nevertheless, its conformational change is not a universal toggle of GPCR activation (Rasmussen et al., Citation2011), and the conformation is ligand dependent in NAM complexes (Christopher et al., Citation2015, Citation2019; Doré et al., Citation2014) and both conformations are accessible in activated mGluR5 in complex with 3 (PAM).
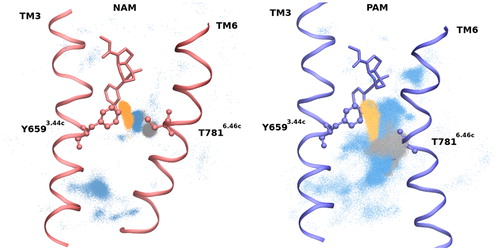
Y6593.44c is close to the varying aromatic substituent (Me/Cl/F; Scheme 1) of the complexes studied and its analogous 3.40 residue in class A GPCRs is part of the transmission switch (Trzaskowski et al., Citation2012). Y6593.44c forms either a direct or a water mediated H-bond with T7816.46c in the NAM and SAM complexes. The side chain of Y6593.44c donates an H-bond to the side chain of T7816.46c with an occupancy of 13% (10%) in NAM (SAM) and a water-mediated H-bond is observed with an occupancy of 33% (43%) (). The structural and functional importance of these interactions is underlined by several findings. The water molecule mediating the H-bond between Y6593.44c and T7816.46c is identified in all available X-ray structures of mGluR5-NAM complexes (Christopher et al., Citation2015, Citation2019; Doré et al., Citation2014) and the H-bond was also observed in MD simulations of the mGluR2 (Pérez-Benito et al., Citation2017) and mGluR5 receptors in complex with NAMs (Llinas del Torrent, Casajuana-Martin, et al., Citation2019). Moreover, it was shown that mutations of Y6593.44c and T7816.46c in mGluR5 significantly impact the activity of allosteric ligands by either reducing (Gregory et al., Citation2014; Malherbe et al., Citation2006) or inverting (Turlington et al., Citation2013) their functional effect. Our simulations show that this H-bond stabilizes the structure by interconnecting TM3 and TM6. These residues are near to the bottom of the ligand in the intracellular side. Although T7816.46c is located above the kink of TM6, below which TM6 points outward from the helical bundle in the active structure, nevertheless, a ∼2Å shift in the backbone atoms of the active structure prevents the formation of a direct H-bond between residues Y6593.44c and T7816.46c. A water-mediated H-bond between the side chains is observed only with 7% occupancy, and Y6593.44c donates an H-bond to the backbone of T7816.46c with an occupancy of 13% in the simulations of the PAM complex (). Therefore, while the direct and water mediated side chain interactions between these residues tend to fix TM3 and TM6 in the inactive state, the propensity of these structure stabilizing interactions is significantly lower in the active complex resulting in a larger separation of the helices. The increased intrahelical space allows the formation of a continuous water channel from the ligand and Y6593.44c toward the intracellular side of the TM helices (see later). The above mentioned shift of TM6 in the PAM complex leads to the loss of contacts between the ligand and T7816.46c as it is shown by ligand-protein contact probability analysis (Deriu et al., Citation2014, Citation2016) (Supplemental Material, Figure S3). Another ligand–protein contact probability significantly differing in NAM and PAM complexes is the one with I6212.46c. The contact probability with the methyl group of NAM substantially reduces for the PAM where the methyl group is replaced with an F-atom having smaller van der Waals radius (Supplemental Material, Figure S3).
Figure 3. Receptor sidechain positions in NAM and PAM complexes. Distribution of OG1 of T781 (orange), O of Y659 (grey) and water (blue). Direct and water mediated H-bond is characteristic in NAM complex and rare in PAM complex.
S6543.39c encloses the intracellular end of the ligand from the opposite side than does Y6593.44c and adopts different conformations in PAM than in NAM and SAM complexes. This residue is adjacent in sequence to P6553.40c, the latter is identified as part of the transmission switch in several class A receptors, including histamine H1 and cannabinoid CB1 receptors (Jongejan et al., Citation2005; McAllister et al., Citation2004), and found to play a role in mGluR2 activation (Pérez-Benito et al., Citation2017). In mGluR5, 3.40c is a proline with limited conformational flexibility. However, the adjacent S6543.39c, a residue with ∼4 Å separation from the intracellular end of the bound ligand is found to adopt dominantly g+ conformation in NAM and SAM complexes, while the t position that has an OH group closer to the ligand and is stabilized by an H-bond to G6242.45c backbone carbonyl is important only in the PAM complex (Supplemental Material, Figure S4).
Ile7515.51c is in contact with Y6593.44c and its analogous 5.51a residue in class A GPCRs is part of the transmission switch. This residue shows distinct conformational preference in PAM versus NAM and SAM complexes (Supplemental Material, Figure S5). While the side chain of Ile7515.51c takes a well-defined conformation with a narrow range of dihedral angles in the NAM complex, it has a larger flexibility in the more opened conformation of the PAM complex. Similar difference for the analogous Ile7395.51c residue was observed for the NAM versus PAM complexes in mGluR2 (Pérez-Benito et al., Citation2017).
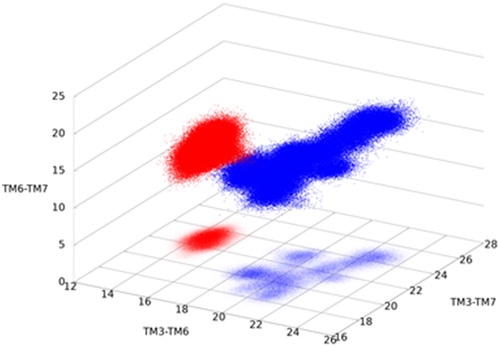
Ionic and polar interactions were observed among residues K6653.50c, E7706.35c, S613ICL1 and K8217.51c in the NAM and SAM complex structures, while they are not representative in the PAM complex (Supplemental Material, Figure S6). These residues correspond to three different helices, TM3, TM6 and TM7, while S613ICL1 is the first residue of ICL1 connecting to TM2. The interactions assure close separations for the residues and for the corresponding helices. Analogous locking interactions were observed in the mGluR1 X-ray structure (Wu et al., Citation2014) and found to be persistent in MD simulations of an inactive mGluR4 model (Dalton, Pin, & Giraldo, Citation2017). They appear also in GABAB, the other subgroup of class C receptors, and in class A receptors (Binet et al., Citation2007). However, the distances between the relevant atoms in the PAM complex are significantly higher for each TM pairs, and span a wide range for TM3-TM7, both indicating the loosening of interactions and the critical role of these residues in the activation of both class A and class C GPCRs. Helices TM3, TM6 and TM7 directly interact with Gq, and their interhelical distances at the intracellular side are able to distinguish active from inactive states as it was proposed also for the P2Y1 (class A) receptor(Yuan, Chan, et al., Citation2016) ().
Figure 4. Interhelix separations distinguish active (blue) from inactive (red) structure. TM3, TM6 and TM7 are in direct contact with the G-protein and their increased separation and surface, characteristic to the active structure, facilitates G-protein binding.
A continuous water channel is observed under the ligand in the PAM complex (). No similar water molecules are present in the NAM and SAM complexes (Supplemental Material, Figure S7) owing to the more compact helical bundle in these structures. A comparison of the abundance of water molecules near to the residues in the PAM versus the NAM complexes identifies residues delimiting the water channel in the PAM complex (Supplemental Material, Table S2). L6623.47c, A7716.36c, I7746.39c, T7776.42c, M7786.43c, T7816.46c and C8167.46c were found to have increased number of neighbouring water molecules and are situated under the ligand toward the cytosolic region. An analogous water channel in class A GPCRs was recognized as a characteristic component of active structures(Angel et al., Citation2009; Yuan et al., Citation2014). To check if the formation of the water channel is a distinctive feature of activated mGluR5, an additional MD study was performed in which compound 3 (PAM) in the activated receptor complex was replaced by 1 (NAM) and the G-protein was removed. The water channel disappeared in the simulation and other features of the NAM-complex were restored. In particular, direct and water mediated H-bonds between Y6593.44c and T7816.46c, and the ionic and polar interactions among residues K6653.50c, E7706.35c, S613ICL1 and K8217.51c assure close contacts for helices TM3, TM6 and TM7. The SASA of the binding site was reduced to the size previously obtained for the inactive receptor complexed with NAM.
Figure 5. Continuous water channel in the PAM–receptor complex under the ligand at the intracellular side.

Conclusion
MD simulations expose structural differences between NAM and PAM complexes that are associated with the activation of the mGluR5 receptor. This class C GPCR has a chamber in the TM region where class A receptors have their orthosteric binding site, and this chamber can function as an allosteric site. No endogenous ligand is known to bind to this site, rather it is occupied by water molecules. Our simulations show that the activation of the receptor assisted by a PAM exhibits structural transformations similar to those in class A activation. The backbone movements of TM helices are accompanied by the breaking of interactions like the ionic lock of residues K6653.50c, E7706.35c, S613ICL1 and K8217.51c and the hydrogen bond between Y6593.44c and T7816.46c. In addition, residues enclosing the allosteric site, namely, S6543.39c and Y6593.44c, both close to the aromatic substituent causing functional switch of the ligands, change their conformation that leads to an increased and more polar chamber. Interestingly, the role of intrahelical waters has not been investigated in the recent studies (Llinas del Torrent, Casajuana-Martin, Pardo, Tresadern, & Pérez-Benito, Citation2019). We found that the TM bundle becomes less tightly packed and a continuous water channel is formed among the TM helices in the intracellular side below the PAM, analogously to the formation of a water channel upon activation of class A GPCRs (Angel et al., Citation2009; Yuan et al., Citation2014). Moreover, the extended surface created by TM helices, in particular TM3, TM6 and TM7, facilitates G-protein binding leading to signal transduction. These intramolecular changes revealed by the MD simulations contribute to the activation that is also characterized by intermolecular structural rearrangements as observed for mGluR1 (Hlavackova et al., Citation2012) and mGluR5 (Koehl et al., Citation2019) receptors.
Residues showing characteristic structural differences in the NAM and PAM complex structures are recognized as part of the activation mechanism. These residues are sequentially and spatially close to or identical with the analogous switches identified in class A receptors as key residues in the activation process (Binet et al., Citation2007; Nygaard, Frimurer, Holst, Rosenkilde, & Schwartz, Citation2009; Trzaskowski et al., Citation2012). Our comparative MD simulations of mGluR5 complexes with NAM, SAM and PAM ligands strongly suggest that mGluR5 activation proceeds via similar changes in residue interactions and the activation mechanism of class A receptors is preserved at a large extent in mGluR5.
| Abbreviations | ||
| MD | = | molecular dynamics |
| mGluR5 | = | metabotropic glutamate receptor 5 |
| GPCR | = | G-protein-coupled receptor |
| NAM | = | negative allosteric modulator |
| PAM | = | positive allosteric modulator |
| RMSD | = | root mean square deviation |
| SAM | = | silent allosteric modulator |
| SAR | = | structure–activity relationship |
| SASA | = | solvent-accessible surface area |
| TM | = | transmembrane |
Supplemental Material
Download PDF (1 MB)Acknowledgements
The support of the National Brain Research Program (KTIA_NAP_13-1-2013-0001 and 2017-1.2.1-NKP-2017-00002) for O. Z., G.G.F. and G.M.K. is acknowledged.
Disclosure statement
No potential conflict of interest was reported by the authors.
Related Research Data
References
- Ahuja, S., & Smith, S. O. (2009). Multiple switches in G protein-coupled receptor activation. Trends in Pharmacological Sciences, 30(9), 494–502. doi: 10.1016/j.tips.2009.06.003
- Angel, T. E., Chance, M. R., & Palczewski, K. (2009). Conserved waters mediate structural and functional activation of family A (rhodopsin-like) G protein-coupled receptors. Proceedings of the National Academy of Sciences, 106(21), 8555–8560. doi: 10.1073/pnas.0903545106
- Banks, J. L., Beard, H. S., Cao, Y., Cho, A. E., Damm, W., Farid, R., … Levy, R. M. (2005). Integrated modeling program, applied chemical theory (IMPACT). Journal of Computational Chemistry, 26(16), 1752–1780. doi: 10.1002/jcc.20292
- Bartuzi, D., Kaczor, A. A., & Matosiuk, D. (2018). Opportunities and challenges in the discovery of allosteric modulators of GPCRs. Methods in Molecular Biology (Clifton, N.J.), 1705, 297–319. doi:10.1007/978-1-4939-7465-8_13
- Berendsen, H. J. C., Postma, J. P. M., van Gunsteren, W. F., DiNola, A., & Haak, J. R. (1984). Molecular dynamics with coupling to an external bath. The Journal of Chemical Physics, 81(8), 3684–3690. doi: 10.1063/1.448118
- Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., … Bourne, P. E. (2000). The Protein Data Bank. Nucleic Acids Research, 28(1), 235–242. doi: 10.1093/nar/28.1.235
- Bernetti, M., Masetti, M., Rocchia, W., & Cavalli, A. (2019). Kinetics of drug binding and residence time. Annual Review of Physical Chemistry, 70(1), 143–171. doi: 10.1146/annurev-physchem-042018-052340
- Binet, V., Duthey, B., Lecaillon, J., Vol, C., Quoyer, J., Labesse, G., … Prézeau, L. (2007). Common structural requirements for heptahelical domain function in class A and class C G protein-coupled receptors. Journal of Biological Chemistry, 282(16), 12154–12163. doi: 10.1074/jbc.M611071200
- Bowers, K. J., Chow, E. C., Xu, H., Dror, R. O., Eastwood, M. P., Gregersen, B. A., … Sacerdoti, F. D. (2006). Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the ACM/IEEE Conference on Supercomputing (SC06).
- Bussi, G., & Parrinello, M. (2007). Accurate sampling using Langevin dynamics. Physical Review E, 75(5), 056707. doi: 10.1103/PhysRevE.75.056707
- Case, D. A., Ben-Shalom, I. Y., Brozell, S. R., Cerutti, D. S., T.E. Cheatham, I., Cruzeiro, V. W. D., … Kollman, P. A. (2018). AMBER 2018. San Francisco, CA: University of California.
- Case, D., M. Betz, R., Cerutti, D. S., Cheatham, T., Darden, T., Duke, R., … A. Kollman, P. (2016). AMBER 2016. San Francisco, CA: University of California.
- Christopher, J. A., Aves, S. J., Bennett, K. A., Doré, A. S., Errey, J. C., Jazayeri, A., … Congreve, M. (2015). Fragment and structure-based drug discovery for a class C GPCR: Discovery of the mGlu5 negative allosteric modulator HTL14242 (3-chloro-5-[6-(5-fluoropyridin-2-yl)pyrimidin-4-yl]benzonitrile). Journal of Medicinal Chemistry, 58(16), 6653–6664. doi: 10.1021/acs.jmedchem.5b00892
- Christopher, J. A., Orgován, Z., Congreve, M., Doré, A. S., Errey, J. C., Marshall, F. H., … Keserű, G. M. (2019). Structure-based optimization strategies for G protein-coupled receptor (GPCR) allosteric modulators: A case study from analyses of new metabotropic glutamate receptor 5 (mGlu 5) X‑ray structures. Journal of Medicinal Chemistry, 62(1), 207–222. doi: 10.1021/acs.jmedchem.7b01722
- Cong, X., Chéron, J.-B., Golebiowski, J., Antonczak, S., & Fiorucci, S. (2019). Allosteric modulation mechanism of the mGluR5 transmembrane domain. Journal of Chemical Information and Modeling, 59(6), 2871. doi: 10.1021/acs.jcim.9b00045
- Dalton, J. A. R., Pin, J. P., & Giraldo, J. (2017). Analysis of positive and negative allosteric modulation in metabotropic glutamate receptors 4 and 5 with a dual ligand. Scientific Reports, 7(1), 1–14. doi: 10.1038/s41598-017-05095-5
- De Vivo, M., Masetti, M., Bottegoni, G., & Cavalli, A. (2016). Role of molecular dynamics and related methods in drug discovery. Journal of Medicinal Chemistry, 59(9), 4035–4061. doi: 10.1021/acs.jmedchem.5b01684
- Deriu, M. A., Grasso, G., Licandro, G., Danani, A., Gallo, D., Tuszynski, J. A., & Morbiducci, U. (2014). Investigation of the Josephin domain protein-protein interaction by molecular dynamics. PLoS ONE, 9(9), e108677. doi: 10.1371/journal.pone.0108677
- Deriu, M. A., Grasso, G., Tuszynski, J. A., Massai, D., Gallo, D., Morbiducci, U., & Danani, A. (2016). Characterization of the AXH domain of Ataxin-1 using enhanced sampling and functional mode analysis. Proteins: Structure, Function, and Bioinformatics, 84(5), 666–673. doi: 10.1002/prot.25017
- Dickson, C. J., Madej, B. D., Skjevik, Å. A., Betz, R. M., Teigen, K., Gould, I. R., & Walker, R. C. (2014). Lipid14: The Amber lipid force field. Journal of Chemical Theory and Computation, 10(2), 865–879. doi: 10.1021/ct4010307
- Doornbos, M. L. J., Pérez-Benito, L., Tresadern, G., Mulder-Krieger, T., Biesmans, I., Trabanco, A. A., … Heitman, L. H. (2016). Molecular mechanism of positive allosteric modulation of the metabotropic glutamate receptor 2 by JNJ-46281222. British Journal of Pharmacology, 173(3), 588–600. doi: 10.1111/bph.13390
- Doré, A. S., Okrasa, K., Patel, J. C., Serrano-Vega, M., Bennett, K., Cooke, R. M., … Marshall, F. H. (2014). Structure of class C GPCR metabotropic glutamate receptor 5 transmembrane domain. Nature, 511(7511), 557–562. doi: 10.1038/nature13396
- Dupradeau, F.-Y., Pigache, A., Zaffran, T., Savineau, C., Lelong, R., Grivel, N., … Cieplak, P. (2010). The R.E.D. tools: Advances in RESP and ESP charge derivation and force field library building. Physical Chemistry Chemical Physics, 12(28), 7821. doi: 10.1039/c0cp00111b
- Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., … Fox, D. J. (2016). Gaussian09 Revision A. Wallingford, CT: Gaussian, Inc.
- Goodford, P. J. (1985). A computational procedure for determining energetically favorable binding sites on biologically important macromolecules. Journal of Medicinal Chemistry, 28(7), 849–857. doi: 10.1021/jm00145a002
- Götz, A. W., Williamson, M. J., Xu, D., Poole, D., Le Grand, S., & Walker, R. C. (2012). Routine microsecond molecular dynamics simulations with AMBER on GPUs. 1. Generalized born. Journal of Chemical Theory and Computation, 8(5), 1542–1555. doi: 10.1021/ct200909j
- Gregory, K. J., Nguyen, E. D., Malosh, C., Mendenhall, J. L., Zic, J. Z., Bates, B. S., … Conn, P. J. (2014). Identification of specific ligand–receptor interactions that govern binding and cooperativity of diverse modulators to a common metabotropic glutamate receptor 5 allosteric site. ACS Chemical Neuroscience, 5(4), 282–295. doi: 10.1021/cn400225x
- Guo, D., & IJzerman, A. P. (2018). Molecular basis of ligand dissociation from G protein-coupled receptors and predicting residence time. Methods in Molecular Biology (Clifton, N.J.), 1705, 197–206. doi:10.1007/978-1-4939-7465-8_9
- Hilger, D., Masureel, M., & Kobilka, B. K. (2018). Structure and dynamics of GPCR signaling complexes. Nature Structural & Molecular Biology, 25(1), 4–12. doi: 10.1038/s41594-017-0011-7
- Hlavackova, V., Zabel, U., Frankova, D., Batz, J., Hoffmann, C., Prezeau, L., … Lohse, M. J. (2012). Sequential inter- and intrasubunit rearrangements during activation of dimeric metabotropic glutamate receptor 1. Science Signaling, 5(237), ra59. doi: 10.1126/scisignal.2002720
- Hopkins, C. W., Le Grand, S., Walker, R. C., & Roitberg, A. E. (2015). Long-time-step molecular dynamics through hydrogen mass repartitioning. Journal of Chemical Theory and Computation, 11(4), 1864–1874. doi: 10.1021/ct5010406
- Isberg, V., de Graaf, C., Bortolato, A., Cherezov, V., Katritch, V., Marshall, F. H., … Gloriam, D. E. (2015). Generic GPCR residue numbers – Aligning topology maps while minding the gaps. Trends in Pharmacological Sciences, 36(1), 22–31. doi: 10.1016/j.tips.2014.11.001
- Jensen, K. P., & Jorgensen, W. L. (2006). Halide, ammonium, and alkali metal ion parameters for modeling aqueous solutions. Journal of Chemical Theory and Computation, 2(6), 1499–1509. doi: 10.1021/ct600252r
- Jongejan, A., Bruysters, M., Ballesteros, J. A., Haaksma, E., Bakker, R. A., Pardo, L., & Leurs, R. (2005). Linking agonist binding to histamine H1 receptor activation. Nature Chemical Biology, 1(2), 98–103. doi: 10.1038/nchembio714
- Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W., Klein, M. L., Chandrasekhar, J., Madura, J. D., … Lein, M. L. (1983). Comparison of simple potential functions for simulating liquid water. The Journal of Chemical Physics, 79(2), 926–935. doi: 10.1063/1.445869
- Joung, I. S., & Cheatham, T. E. (2008). Determination of alkali and halide monovalent ion parameters for use in explicitly solvated biomolecular simulations. The Journal of Physical Chemistry B, 112(30), 9020–9041. doi: 10.1021/jp8001614
- Joung, I. S., & Cheatham, T. E. (2009). Molecular dynamics simulations of the dynamic and energetic properties of alkali and halide ions using water-model-specific ion parameters. The Journal of Physical Chemistry B, 113(40), 13279–13290. doi: 10.1021/jp902584c
- Kaminski, G. A., Friesner, R. A., Tirado-Rives, J., & Jorgensen, W. L. (2001). Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. The Journal of Physical Chemistry B, 105(28), 6474–6487. doi: 10.1021/jp003919d
- Koehl, A., Hu, H., Feng, D., Sun, B., Zhang, Y., Robertson, M. J., … Kobilka, B. K. (2019). Structural insights into the activation of metabotropic glutamate receptors. Nature, 566(7742), 79–84. doi: 10.1038/s41586-019-0881-4
- Koehl, A., Hu, H., Maeda, S., Zhang, Y., Qu, Q., Paggi, J. M., … Kobilka, B. K. (2018). Structure of the µ-opioid receptor–Gi protein complex. Nature, 558(7711), 547–552. doi: 10.1038/s41586-018-0219-7
- Latorraca, N. R., Venkatakrishnan, A. J., & Dror, R. O. (2017). GPCR dynamics: Structures in motion. Chemical Reviews, 117(1), 139–155. doi: 10.1021/acs.chemrev.6b00177
- Le Grand, S., Götz, A. W., & Walker, R. C. (2013). SPFP: Speed without compromise—A mixed precision model for GPU accelerated molecular dynamics simulations. Computer Physics Communications, 184(2), 374–380. doi: 10.1016/j.cpc.2012.09.022
- Lindsley, C. W., Emmitte, K. A., Hopkins, C. R., Bridges, T. M., Gregory, K. J., Niswender, C. M., & Conn, P. J. (2016). Practical strategies and concepts in GPCR allosteric modulator discovery: recent advances with metabotropic glutamate receptors. Chemical Reviews, 116(11), 6707–6741. doi: 10.1021/acs.chemrev.5b00656
- Llinas del Torrent, C., Casajuana-Martin, N., Pardo, L., Tresadern, G., & Pérez-Benito, L. (2019). Mechanisms underlying allosteric molecular switches of metabotropic glutamate receptor 5. Journal of Chemical Information and Modeling, 59(5), 2456–2466. doi: 10.1021/acs.jcim.8b00924
- Llinas del Torrent, C., Pérez-Benito, L., & Tresadern, G. (2019). Computational drug design applied to the study of metabotropic glutamate receptors. Molecules (Basel, Switzerland), 24(6), 1098. doi: 10.3390/molecules24061098
- Lomize, M. A., Pogozheva, I. D., Joo, H., Mosberg, H. I., & Lomize, A. L. (2012). OPM database and PPM web server: Resources for positioning of proteins in membranes. Nucleic Acids Research, 40(D1), D370–D376. doi: 10.1093/nar/gkr703
- Madhavi Sastry, G., Adzhigirey, M., Day, T., Annabhimoju, R., & Sherman, W. (2013). Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. Journal of Computer-Aided Molecular Design, 27(3), 221–234. doi: 10.1007/s10822-013-9644-8
- Maier, J. A., Martinez, C., Kasavajhala, K., Wickstrom, L., Hauser, K. E., & Simmerling, C. (2015). ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. Journal of Chemical Theory and Computation, 11(8), 3696–3713. doi: 10.1021/acs.jctc.5b00255
- Malherbe, P., Kratochwil, N., Muhlemann, A., Zenner, M.-T., Fischer, C., Stahl, M., … Porter, R. H. P. (2006). Comparison of the binding pockets of two chemically unrelated allosteric antagonists of the mGlu5 receptor and identification of crucial residues involved in the inverse agonism of MPEP. Journal of Neurochemistry, 98(2), 601–615. doi: 10.1111/j.1471-4159.2006.03886.x
- Mason, J. S., Bortolato, A., Weiss, D. R., Deflorian, F., Tehan, B., & Marshall, F. H. (2013). High end GPCR design: Crafted ligand design and druggability analysis using protein structure, lipophilic hotspots and explicit water networks. In Silico Pharmacology, 1(1), 23. doi: 10.1186/2193-9616-1-23
- McAllister, S. D., Hurst, D. P., Barnett-Norris, J., Lynch, D., Reggio, P. H., & Abood, M. E. (2004). Structural mimicry in class A G protein-coupled receptor rotamer toggle switches: The importance of the F3.36(201)/W6.48(357) interaction in cannabinoid CB1 receptor activation. Journal of Biological Chemistry, 279(46), 48024–48037. doi: 10.1074/jbc.M406648200
- Melchionna, S. (2007). Design of quasisymplectic propagators for Langevin dynamics. The Journal of Chemical Physics, 127(4), 044108. doi: 10.1063/1.2753496
- Miao, Y., & McCammon, J. A. (2018). Mechanism of the G-protein mimetic nanobody binding to a muscarinic G-protein-coupled receptor. Proceedings of the National Academy of Sciences, 115(12), 3036–3041. doi: 10.1073/pnas.1800756115
- Niswender, C. M., & Conn, P. J. (2010). Metabotropic glutamate receptors: Physiology, pharmacology, and disease. Annual Review of Pharmacology and Toxicology, 50(1), 295–322. doi: 10.1146/annurev.pharmtox.011008.145533
- Nygaard, R., Frimurer, T. M., Holst, B., Rosenkilde, M. M., & Schwartz, T. W. (2009). Ligand binding and micro-switches in 7TM receptor structures. Trends in Pharmacological Sciences, 30(5), 249–259. doi: 10.1016/j.tips.2009.02.006
- Pándy-Szekeres, G., Munk, C., Tsonkov, T. M., Mordalski, S., Harpsøe, K., Hauser, A. S., … Gloriam, D. E. (2018). GPCRdb in 2018: Adding GPCR structure models and ligands. Nucleic Acids Research, 46(D1), D440–D446. doi: 10.1093/nar/gkx1109
- Pérez-Benito, L., Doornbos, M. L. J., Cordomí, A., Peeters, L., Lavreysen, H., Pardo, L., & Tresadern, G. (2017). Molecular switches of allosteric modulation of the metabotropic glutamate 2 receptor. Structure, 25(7), 1153–1162.e4. doi: 10.1016/j.str.2017.05.021
- Rasmussen, S. G. F., Choi, H.-J., Fung, J. J., Pardon, E., Casarosa, P., Chae, P. S., … Kobilka, B. K. (2011). Structure of a nanobody-stabilized active state of the β2 adrenoceptor. Nature, 469(7329), 175–180. doi: 10.1038/nature09648
- Salomon-Ferrer, R., Götz, A. W., Poole, D., Le Grand, S., & Walker, R. C. (2013). Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. Explicit solvent particle mesh Ewald. Journal of Chemical Theory and Computation, 9(9), 3878–3888. doi: 10.1021/ct400314y
- Sciabola, S., Stanton, R. V., Mills, J. E., Flocco, M. M., Baroni, M., Cruciani, G., … Mason, J. S. (2010). High-throughput virtual screening of proteins using GRID molecular interaction fields. Journal of Chemical Information and Modeling, 50(1), 155–169. doi: 10.1021/ci9003317
- Sengmany, K., & Gregory, K. J. (2016). Metabotropic glutamate receptor subtype 5: Molecular pharmacology, allosteric modulation and stimulus bias. British Journal of Pharmacology, 173(20), 3001–3017. doi: 10.1111/bph.13281
- Shen, M.-Y., & Sali, A. (2006). Statistical potential for assessment and prediction of protein structures. Protein Science, 15(11), 2507–2524. doi: 10.1110/ps.062416606
- The UniProt Consortium. (2007). The Universal Protein Resource (UniProt). Nucleic Acids Research, 36(Database), D190–D195. doi:10.1093/nar/gkm895
- Trzaskowski, B., Latek, D., Yuan, S., Ghoshdastider, U., Debinski, A., & Filipek, S. (2012). Action of molecular switches in GPCRs–theoretical and experimental studies. Current Medicinal Chemistry, 19(8), 1090–1109. doi: 10.2174/092986712799320556
- Turlington, M., Noetzel, M. J., Chun, A., Zhou, Y., Gogliotti, R. D., Nguyen, E. D., … Stauffer, S. R. (2013). Exploration of allosteric agonism structure–activity relationships within an acetylene series of metabotropic glutamate receptor 5 (mGlu5) positive allosteric modulators (PAMs): Discovery of 5-((3-fluorophenyl)ethynyl)-N-(3-methyloxetan-3-yl)picolinamide (ML254). Journal of Medicinal Chemistry, 56(20), 7976–7996. doi: 10.1021/jm401028t
- Wang, J., Wang, W., Kollman, P. A., & Case, D. A. (2006). Automatic atom type and bond type perception in molecular mechanical calculations. Journal of Molecular Graphics and Modelling, 25(2), 247–260. doi: 10.1016/j.jmgm.2005.12.005
- Wang, J., Wolf, R. M., Caldwell, J. W., Kollman, P. A., & Case, D. A. (2004). Development and testing of a general Amber force field. Journal of Computational Chemistry, 25(9), 1157–1174. doi: 10.1002/jcc.20035
- Wang, W., Qiao, Y., & Li, Z. (2018). New insights into modes of GPCR activation. Trends in Pharmacological Sciences, 39(4), 367–386. doi: 10.1016/j.tips.2018.01.001
- Weis, W. I., & Kobilka, B. K. (2018). The molecular basis of G protein–coupled receptor activation. Annual Review of Biochemistry, 87(1), 897–919. doi: 10.1146/annurev-biochem-060614-033910
- Wu, H., Wang, C., Gregory, K. J., Han, G. W., Cho, H. P., Xia, Y., … Stevens, R. C. (2014). Structure of a class C GPCR metabotropic glutamate receptor 1 bound to an allosteric modulator. Science, 344(6179), 58–64. doi: 10.1126/science.1249489
- Yuan, S., Chan, H. C. S., Vogel, H., Filipek, S., Stevens, R. C., & Palczewski, K. (2016). The molecular mechanism of P2Y1 receptor activation. Angewandte Chemie International Edition, 55(35), 10331–10335. doi: 10.1002/anie.201605147
- Yuan, S., Filipek, S., Palczewski, K., & Vogel, H. (2014). Activation of G-protein-coupled receptors correlates with the formation of a continuous internal water pathway. Nature Communications, 5(1), 1–10. doi: 10.1038/ncomms5733
- Yuan, S., Peng, Q., Palczewski, K., Vogel, H., & Filipek, S. (2016). Mechanistic studies on the stereoselectivity of the serotonin 5-HT1A receptor. Angewandte Chemie International Edition, 55(30), 8661–8665. doi: 10.1002/anie.201603766