
Abstract
Hepatitis B virus (HBV) infection is an important problem threatening human health. After HBV virus invades human body, it may assemble a complete virus particle in the cytoplasm to trigger the immune reaction, especially the interaction between the HBV virus and the host that mediated by CD8+ T cell. We collected the sequences of HBV from the HBVdb database, then screened candidate mutation sites in Chinese, European and American populations based on conservation and physicochemical properties. After that we constructed the three-dimensional structure of Major histocompatibility complex class I (MHC I) -peptide complexes, performed molecular docking, run molecular dynamics to compare the binding free energy, stability, and affinity of MHC I-peptide complexes with the aim to estimate the effect of peptide mutation. The specific HBV virus subtypes of the Chinese, European and American population were studied and the candidate mutation sites were used to predict the mutant peptide antigen. Finally, based on physical and chemical properties and peptide antigen prediction scores, 21 HBV mutation sites were selected. Then combined with specific Human lymphocyte antigen (HLA) subtypes, 11 mutations were found to have a significant negative impact on affinity, stability and binding free energy. Overall, our work found important potential mutations, which provide an evaluation of HBV mutations and a clue of it in immunotherapy.
Communicated by Ramaswamy H. Sarma
Introduction
HBV infection is seriously threatening human health. According to the latest data released by the World Health Organization 2017, there are about 350 million people with chronic HBV infection in the world. There are about 1 million people die directly from HBV infection-related diseases every year. China is a high prevalence area of HBV with 93 million infected people, including 25-30 million chronic HBV patients(Francisco et al., Citation2017; Fu-Sheng et al., Citation2014). HBV virus infection and disease are mainly caused by the interaction between virus and specific HLA in different populations, which mediated by CD8+T cell (Levrero & Zucman-Rossi, Citation2016). Previous studies have shown that the genetic background of the host plays a crucial role in HBV infection and its prognosis (S. Li et al., Citation2012; Pan et al., Citation2013; Tan et al., Citation2008), different ethnic groups may be susceptible to different genotypes of HBV virus strains, and many tools used to study the interaction between HBV and HLA molecules in host immune system have been designed for the European and American populations currently (Rivino et al., Citation2013). However, the mechanisms of the interaction between susceptible HBV strains and specific MHC allotype in Chinese, European and American populations have not been systematically studied.
HBV is a DNA virus, whose genome is extremely small, so its protein is encoded by multiple overlapping reading frames, of which four open reading frames are S region, C region, P region, and X region (Cossart & Field, Citation1970). The S region encodes a surface antigen, an envelope protein of hepatitis B virus, which mediate the recognition between HBV virus and hepatocyte receptors (Gripon et al., Citation2005). The C region encodes the core antigen (HBcAg) and the hepatitis B e antigen (HBeAg) that buried inside HBcAg. HBcAg constitutes an icosahedral nucleocapsid, which encapsulates its mRNA to form core particles containing antigenic determinants, which is the key target for the immune response (Al-Qahtani et al., 2018; Tong & Revill, Citation2016; Wang et al., Citation1991).
After HBV infects into the host, the HBV virus genome has long been under the choice of immune pressure, and may generate new mutant strains through constant mutation to carry out immune escape, especially triggering the immune response associated with CD8+ T cells. HBV mutations are mainly found by sequencing, and those with a mutation frequency greater than 10% are defined as hotspot. In the current study, the correlation between hotspot and liver cancer is mainly assessed by meta-analysis and statistical methods. (Wei et al., Citation2017). The antigen produced by the intracellular HBV virus is processed into peptide antigen, which is then expressed on the cell surface by MHC I (Levrero & Zucman-Rossi, Citation2016; Webster et al., Citation2004), and is recognized by the T cell receptor (TCR) to form a pMHC-TCR ternary body molecule.
However, little is known about the mechanisms of immune-stress-related HBV mutations in T cell epitopes and their association with disease progression (Schuch et al., Citation2014). Previous studies have shown that many mutations reduce the antigenicity of the peptides they produce and affect the recognition of HBV virions by the immune system, this phenomenon may affect the formation of subsequent pMHC-TCR complex by reducing the binding of mutant antigenic peptides to MHC class I molecules, and induce the occurrence of immune escape (Bertoletti & Ferrari, Citation2016). In immunotherapy, one of the common methods is to promote the existing HBV-specific T cells, enhance the recognition of HBV-infected targets to achieve the goal of clearing the virus (Bertoletti & Le Bert, Citation2018). Antigen molecules from their initial state to the antigenic peptides that presented on the surface of MHCI molecules need to be degraded by proteasome, then further modified by heat shock proteins HSP70 and HSP90, and then selectively transported to the endoplasmic reticulum by the relevant transporter TAP. Then, the processed antigenic peptides will be submitted by gp69 to the newly synthesized class I molecules, and assembled under the action of chaperones such as tapasin, calanexin, calreticulin, etc. The formed complex is transported to the cell surface via the golgi apparatus for recognition by CD8 + T cells and stimulates immune escape (Riedl et al., Citation2006). Our focus is on the last but crucial final step-antigen presentation, to explore whether the mechanism of the binding between mutant antigenic peptides and MHC molecules can give a little inspiration to immunotherapy for liver cancer.
Through the investigation of the published literature, it is found that the existing HBV database is mostly functional, and the data are basically from Genbank (Hayer et al., Citation2013; Li et al., Citation2013 ). We choose HBVdb database, which source as European ENA (Nucleotide Archive), shares data with Genbank, DDBJ (DNA Data Bank of Japan), and keep updating to get data. We performed alignment and conservation analysis on 11,384 HBV virus sequences based on different HBV subtypes and coding regions, obtained 172 mutations that fell in the conserved regions. Finally, 21 key mutations were finally obtained through the physicochemical property analysis and antigenic peptides screening. Combined with the specific HLA subtypes of Chinese, European and American populations, the predicted antigenic peptides corresponding to the mutation was docked with HLA to construct a peptide-MHC complex and calculate the affinity and stability of the complex. Then, the molecular dynamics simulation was performed to calculate the binding free energy (MM/GBSA) and to predict the effect of the mutation on the complex.
Materials and methods
Data collection, conservation and physicochemical properties analysis
The amino acid sequences of the HBV S (SHBs) and C (PreC) regions of A, B and C subtypes in this study were downloaded from the HBVdb database (Hayer et al., Citation2013). There were 3349 sequences in the S region of subtype A, 1523 sequences in C region, 5594 sequences in S region of B subtype, 2862 sequences in C region, 9468 sequences in S region of C subtype, and 2801 sequences in C region. The highest frequency HLA-A subtype in Chinese and European or American populations comes from Han-MHC (Fusheng, Citation2016). The three-dimensional structure of HLA-A*1101 (PDB ID: 2HN7, 5GRG, 5GSD, 5GRD, 4UQ2) and HLA-A*0201(PDB ID: 1I1F, 5YXU, 4UQ3, 2HN7, 2V2W) were collected. Aligned amino acid sequences can be found in the DATASET in the HBVdb database. And PSIPRED 4.0 and TMHMM were used to predict the physicochemical properties of mutant amino acids, including helix and topology of transmembrane (Buchan & Jones, Citation2019; Krogh et al., Citation2001).
Prediction of antigenic peptide
Here, we utilized a new framework, NetMHCPan4.0, to predict antigenic peptides according to the specific HLA subtypes of Chinese and European or American populations. The length of predicted peptide antigen was across 8-14 amino acids and the optimal candidate was selected according to the score and %Rank provided by predict results. (Jurtz et al., Citation2017).
Protein preparation
The peptides in peptide-MHC complex were derived from the NetMHCPan4.0 predicted results, and the MHC molecules were obtained from the PDB. As for MHC protein, we used structures of HLA-A*11:01(PDB ID: 2HN7, 5GRG, 5GSD, 5GRD, 4UQ2) and HLA-A*02:01(PDB ID: 1I1F, 5YXU, 4UQ3, 2HN7, 2V2W).
The primary structures were performed with the following structural preparations. Firstly, the original structure was optimized using a hydrogen bond network. Secondly, the water molecules were manipulated by optimizing hydrogen bonding. Thirdly, exist strain was mitigated and the position of each set was fine tuned. Missing sides and loop were also filled in by Prime (Sastry et al., Citation2013). Finally, we obtained prepared proteins.
Protein interaction analysis and peptide docking
The interaction between original HLA and peptide was performed by the “Protein Interaction Analysis” workflow in Bioluminate. The docking was accomplished by Glide 5.7 program in Schrodinger Suite. Notably, the docking grid was generated using “Centroid of selected residues” with selected residue that is based on the results of Protein Interaction Analysis. In addition, GlideScore value was applied in the docking protocol, and the models of top scores were selected as the final docking poses. (Beard et al., Citation2013).
Molecular dynamics simulation, RMSD calculation and structural analysis
The docking poses were used as the initial structure in MD simulation, all MD simulations were carried out using Desmond module in Schrodinger Suite with OPLS3e force field. The simulation system was put in an orthorhombic periodic box of SPC water molecules, the distance between the complex and the box boundary was set to 10.0 Å along each dimension. Relaxing model system was carried out before simulations to optimize high energy, then the MD simulations were conducted at 1 atm and 300 K under the NPT (constant particles N, temperature T, and pressure P) ensemble for 100 ns (Piao et al., Citation2019). The trajectory snapshots were saved every 40 ps, and 1,250 frames were collected for further analyses (Ylilauri & Pentikainen, Citation2013).
RMSD was calculated by Desmond module, indicating the degree of molecular structure change. Residue Scanning was calculated by Bioluminate with default parameter to specify the mutations of interest.
MM/GBSA binding energy calculation
Molecular Mechanics/Generalized Born Surface Area (MM/GBSA) has been widely used to calculate binding free energy for protein-ligand systems. The calculations are done from the MD trajectories with Prime MM-GBSA, to predict the free energy of binding (ΔGbind) between protein-peptide or protein-protein complexes based on the molecular dynamics (Ylilauri & Pentikainen, Citation2013). The binding free energy of MM/GBSA was calculated as the following equation:
ΔGbind = Gcomplex – Gprotein– Gligand, where ΔGbind is the binding free energy, and Gcomplex, Gprotein, and Gligand are the free energies of complex, protein, and ligand. The equation ΔEMM + ΔGGB + ΔGnonpolar – TΔS is used to estimate the energy, ΔEMM is the gas-phase interaction energy between protein and the binding peptide, the polar and nonpolar components of the desolvation free energy represented by ΔGGB and ΔGnonpolar, and TΔS is the change of conformational entropy on ligand binding.
Results
Collection of HBV mutation sites in Chinese, European and American populations
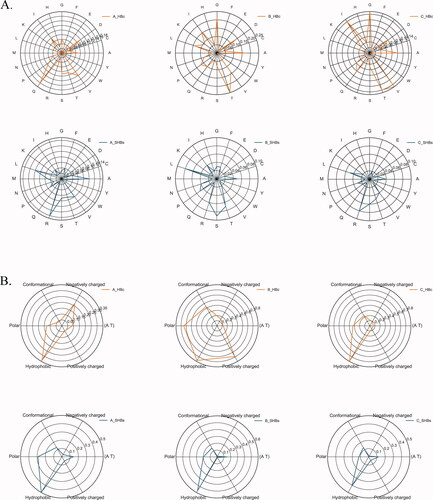
Due to the significant regional differences in HBV genotypes, the HBV genotypes in China are mainly B-type in the south, C-type in the north, and A-type as the main subtype in European and American populations (Sunbul, Citation2014), the amino acid sequences of HBV viruses of subtypes A, B, and C were collected in the HBVdb database. Then we focused on the S and C coding region, which encodes hepatitis B surface antigen and core antigen respectively. The sequences were aligned with the ClusterW module in Mega7.0 (Kumar et al., Citation2016) and the mutation at each locus was counted and displayed in the form of a radar chart (). The mutated amino acids were based on amino acid distribution and physicochemical properties: hydrophobicity (V, I, L, M, W, Y, C), polarity (N, Q, S), positive charge (R, K, H), negative charge (D, E), conformation (G, P), classification in the AT area (Ke, Citation2015; Shen & Vihinen, Citation2004). The statistical results showed that the mutations in the S and C regions of different ethnic groups was different. In the European and American populations, the C coding region of the HBV subtype was more likely to be mutated to Q, while the S coding region tended to be mutated to T. In contrast, the C coding region of the Chinese population susceptible to HBV subtypes was mutated to T, V, I, G, but the S coding region was L, S, R. We found the physicochemical properties indicate that the hydrophobic group has the highest mutation frequency in terms of functionality.
Figure 1. The Radar map of amino acid sequence alignment results of hepatitis B virus subtype A, B, and C.
A. The relative mutability of mutated residues in C and S regions of HBV virus based on 20-letter alphabet amino acids.
B. The relative mutability of mutated residues in the C and S regions of HBV viruses based on the definition of 6 groups, which can divide amino acids into following 6 groups: hydrophobicity (V, I, L, M, W, Y, C), polarity (N, Q, S), positive charge (R, K, H), negative charge (D, E), conformation (G, P), and AT area.
Identification of 21 potential candidate mutation sites
To find the mutation sites in conserved regions, the sequences were analyzed by ClusterW1.81 in the HBVdb database (). At the same time, considering the specificity of the viral genome subtype, ethnicity, and distribution area, we selected the most frequently occurring amino acids at each locus as a reference (). The hydrophobic interaction of amino acid residues is the main force for maintaining and stabilizing the conformation of the protein, so we used PSIPRED to screen for hydrophobic amino acid mutation sites, and finally found 37 mutation sites (, ). Peptide antigen prediction was performed on the screened mutation sites using NetMHCPan4.0. Conditions were set for HLA-A*0201 and HLA-A*1101. The peptide length was set to 8-14 amino acids. The threshold of the strong binder was set to 0.5, and the threshold of the weak binder was set to 2. At the same time, we searched for HLA-A*1101 and HLA-A*0201 subtypes in AFND (Allele Frequency Net Database) (González-Galarza et al., 2015). The results also showed that these two subtypes had the highest frequency in European and American populations and Southeast Asian populations.
Table 1. The mutation sites in the conserved region.
Table 2. 37 mutation sites selected based on hydrophobicity.
According to the simulation results of NetMHCPan4.0, HBV mutations with the highest predicted MHC binding affinity were preserved, and we obtained the epitope sequences of 21 mutation sites finally (). It was found that most of the predicted antigenic peptides were 9-10 amino acids in length, which was consistent with the distribution characteristics of T cell epitopes corresponding to specific HLA subtypes (Trolle et al., Citation2016).
Table 3. Mutation sites selected based on antigen peptide prediction score (Cutoff is rank%<2) The one that labeled red is an antigenic peptide that has appeared in the hepitopes database, the rank% threshold for strong binding peptides is 0.500, and the weak one is 2.000.
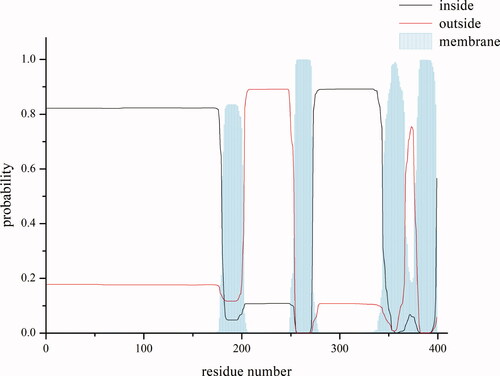
However, it has been reported that the PreS1 domain, buried in viral membrane, directly interacts with the sodium-taurocholate cotransporting polypeptide (NTCP). Whereas the α-determinant, which locates on the small S domain outside the membrane, interacts with heparin sulfate proteoglycan on hepatocytes, increasing the concentration of HBV virus particles on the surface of liver cells (Bertoletti & Ferrari, Citation2016). So, we want to explore whether the effects of mutations inside and outside the membrane are different, which play a key role in virus infection. Combined with the predicted result of transmembrane region using TMHMM server v.2.0 (), six conservative candidate mutation sites of transmembrane helical regions of subtype A, I199V, I202V/M, I256M, V351A, V354A and L389V, were found in the S region.
Figure 2. Prediction of transmembrane regions of S region.
This picture shows the predicted map of transmembrane region. The abscissa is the position of the amino acid residue, and the ordinate is the prediction possibility. The black line indicates that the position may be in the intracellular segment; the red line denotes the outside region; and the vertical line on the X axis represents the prediction of transmembrane region.
Moreover, 13 of the 21 antigenic peptides predicted from the mutation sites were included in the hepitopes database(Lumley et al., Citation2016), which collected all HBV epitopes corresponding to HLA class I genes based on published literature (). This indicates that our predictions have certain credibility. Seven antigenic peptides were not reported, and the corresponding mutation sites were I32F/V, L44F, L98F, L60I, V149A, I155V/M, L213F, and I256M. The Signal P server result reveals that (Almagro Armenteros et al., Citation2019) that our collected sites are not in the signal peptide region ().
Docking of the HLA and peptide
Structures used for docking, including HLA-A*0201 (PDB ID: 1I1F, 5YXU, 4UQ3, 2HN7, 2V2W) and HLA-A*1101 (PDB ID: 2HN7, 5GRG, 5GSD, 5GRD, 4UQ2), were obtained from PDB. These structures were conducted by protein interaction analysis, which analyzed the positions of the amino acid residues of MHC molecules in contact with peptides. We selected the repetitive sites as the sites in the binding pocket of MHC and incorporated these sites as constraints in the docking. In addition, we docked the peptides with each of the selected PDB structures. and selected the optimal structure according to the docking scores. then performed molecular dynamics simulations on the corresponding complexes, and finally selected the best receptor of each peptide based on the RMSD results ().
Figure 3. Binding models of peptide FLLTRILTI and MHC I molecular proteins.
MHC and peptide are separately colored in gray and magenta, key residues are shown in stick model. Hydrogen bonds are shown in yellow dash lines.
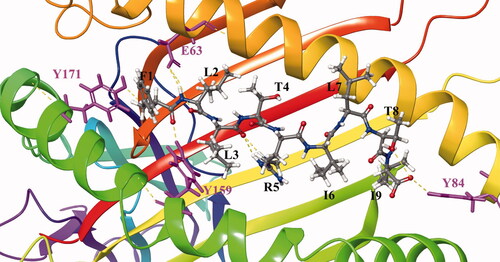
The scoring system of the peptide docking module is mainly evaluated by calculating the binding free energy in the implicit solvent. For example, the binding mode of the peptide antigen FLLTRILTI produced by I199 to HLA is shown in figure (). It can be seen that the peptide binds to the groove of the HLA. L3, I6, and L7 residues pointing to the outer space and do not contact HLA protein. F1, L2, and I9 which are inside the groove. Moreover, F1 contacts TYR171 and TYR159 on the groove to form hydrogen bonds, and L2 and I9 also form hydrogen bonds with GLU63 and TYR84, respectively. Clearly, HBV virus mutations mentioned above in these residues may affect the binding between peptide and HLA.
The impact of the 21 mutations on complex formation
Based on the docked MHC-peptide complex, we performed a residue scanning calculation on these 21 candidate mutations. The results showed that the eight mutations: L149V, I156L, L213F, I256M, V351A, V354A, M372V/I and L389V in subtype A and V149A, I155V/M in subtype B resulted in a change in affinity greater than +3 kcal/mol. It can be speculated that the mutations have a significant effect on the affinity between the peptide and MHC, which may result in weakening binding of the antigenic peptide to MHC and immunological escape. In addition, mutations of L213F, I256M, M372V/I and I155M also resulted in a decrease in the overall stability of the complex (). Among them, the mutation of M372V caused the most negative effects, and the changes of affinity and stability were as high as +10.96 kcal/mol and +8.22 kcal/mol, respectively.
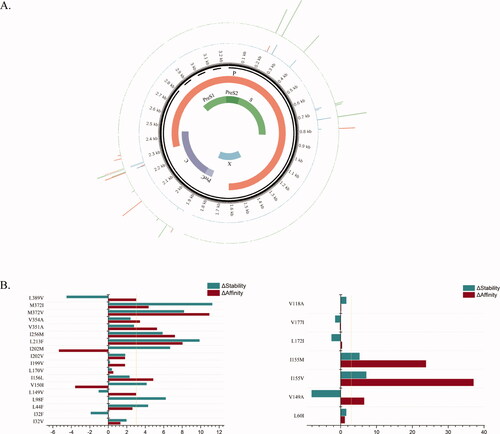
Figure 4. Residue Scanning Computation of 21 mutation sites.
A. Results of residue scanning are shown on the genome. The inner circle displays the HBV viral genome structure and its encoded protein. The first outer ring (green) indicates the change in affinity produced by the corresponding site mutation, and the outermost layer (blue) indicates the resulting change in stability. The degree of change is expressed in terms of the height of the column, where the negative result is marked in red.
B. Comparison of results. The left and right images represent mutations that occur in subtype A and B/C, respectively. The ordinate indicates the mutation site. The horizontal axis represents ΔΔGstability (kcal/mol) and ΔΔGaffinity (kcal/mol), marked with blue and red. The threshold is set at +3kcal/mol (yellow line). Positive values indicate that the mutation has a negative impact on affinity and stability.
In order to consider the relax process of the mutant protein and perform global analysis including interactions between residues or domains, molecular dynamics simulation was used for subsequent analysis. Mutation with affinity change greater than +3kcal/mol was selected, and the predicted peptide-MHC complex was simulated with 100 ns molecular dynamics. The RMSD of backbone evolution with time was calculated by using the initial structure as a reference structure, and the binding free energy of the period was calculated based on the trajectories with stable RMSD values. The effects of the mutation were explained by comparing the changes of binding free energy between wild type and mutant type. For example, for the wild type and mutation of L389V of HBV peptide, we conducted 100 ns MD simulations. Based on the RMSD analysis, we found that the system of wild type and L389V went to equilibrium after 20 and 30 ns, respectively (). The MM/GBSA binding free energy was calculated by using the snapshots extracted from the trajectory of 20-100 ns in the wild type system and 30-60 ns in the L389V system. It was found that the Leu mutated into Val cause the absolute values of the binding free energy to decrease from 69.48 kcal/mol to 61.73 kcal/mol. Consistent with the result of the residue scanning, the binding free energy analysis results confirmed that the mutation would weaken the affinity between the HLA molecule and the peptide antigen (, S3, and Table S4). We have also selected peptide antigen FLPSDFFPSV (hepatitis B core 18-27), which have confirmed the mutations at position P10 would reduce the binding of peptide to MHC I molecules by classical laboratory methods (Bertoletti et al., Citation1994). The mutation from Val to Ala resulted in the decrease of binding free energy from 81.99 kcal/mol to 72.79 kcal/mol, which was consistent with the experimental results, indicating that the bioinformatics analysis method we adopted had certain reliability.
Figure 5. RMSD of docking structure produced by mutant L389V.
A stable period (20ns∼100ns, 30 ∼ 60ns) demonstrates a stable structure. The arrow points the structure that we selected to run MM/GBSA calculation. The MM/GBSA values of the wild type (L389) and mutant type (L389V) were -69.48kcal/mol and -61.73kcal/mol, respectively.
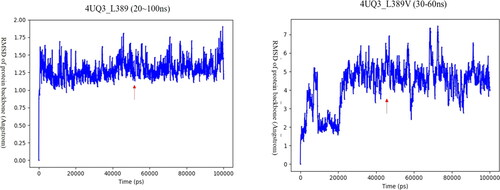
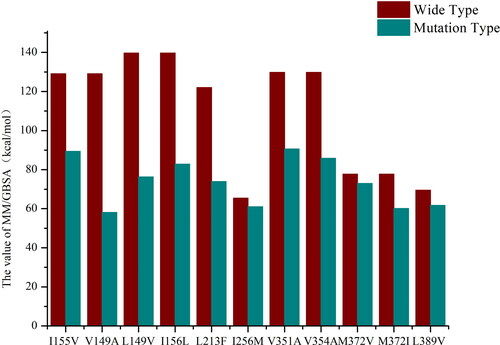
Figure 6. MM/GBSA calculation results of mutation sites.
The x-axis represents the mutation sites and the ordinate is the values (kcal/mol) of MM/GBSA. The red and blue columns represent the MM/GBSA values of the wild-type and the mutant-type, respectively.
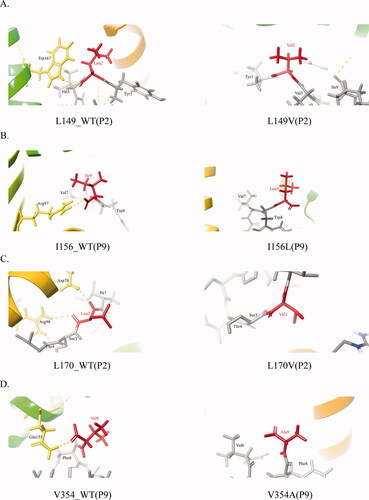
In general, the second (P2) and ninth (P9) residues of antigenic peptides bound to HLA-A *02:01 are usually hydrophobic residues, such as Leu, Val, or Ile, and their interactions are usually mediated by hydrogen bonds. Based on this, we chose mutations that occurred at positions 2 and 9 that were originally hydrophobic amino acids. Finally, four mutation sites L149, I156, L170 and V354 were selected. We analyzed the effect of anchor residues mutation on non-covalent bonds between peptides and MHC. It was found that the mutation at P2 of the mutant peptide antigen YL/VSFGVWI produced by L149V and the mutation at P9 of the mutant peptide YLVSFGVWI/L produced by I156L resulted in a reduction in the total number of hydrogen bonds generated. The L149V mutation caused the original hydrogen bond formed by P2 and TRP167 on HLA to disappear. Similarly, the I156L mutation at P9 of peptide led to the disappearance of the hydrogen bonds originally formed by Ile and ARG97 (). In the peptide antigen formed by L170V (IL/VSTLPETTV) mutation, Leu initially bound to ASP78 and ARG98 to form a hydrogen bond, which disappeared after mutation (). The same is true for the mutant peptide antigen WLSLLVPFV/A produced by V354A, the mutation caused the hydrogen bond generated by the original P9 and GLN155 to disappear, the non-covalent binding force between the anchor residue and HLA was reduced ().
Figure 7. Non-covalent pattern between peptide and HLA.
The picture shows the covalent bond between the antigenic peptide and HLA. MHC and peptide are separately colored in yellow and gray. The mutant amino acids are in red, the yellow dotted line is the hydrogen bond.
A-D. Non-covalent bond pattern between HLA and residue of P2or P9position of peptide antigen that corresponding to L149V, I156L, L170V, and V354A before and after mutation.
We also performed non-covalent bond analysis for other mutated sites, but their effect on the non-covalent binding force between peptide and HLA was not very significant. This may be due to the amino acid properties that did not change much before and after the mutation, and it is possibly because of the predicted antigenic peptides are not all standard nonapeptides. Previous studies have shown that when the length of the antigenic peptides is more than 9 amino acids, the anchor residue originally thought will migrate (Phillip, Citation2017).
Discussion
Previous work showed increased expression of MHC class I molecules in HBV-infected liver cancer cell lines and liver cancer tissues, but it did not promote CD8+ T cell-mediated immune clearance, which may be related to the immune escape of HBV virus (Lianhong, Citation2010; Shen et al., Citation2009). Moreover, different ethnic groups may be susceptible to different genotypes of HBV strains, so we still need to consider the different genotypes of HBV and the genetic background of the host.
To clarify the mechanism of immune escaping caused by HBV mutations in Chinese, European and American populations, the amino acid sequences of various regions of different HBV subtypes were collected from the HBVdb database. The B, C subtypes are dominant in Chinese and the A subtype is dominant in European and American population. After alignment, the mutations in the conserved regions of the HBV viral genome were identified. At the same time, we calculated the mutations of each subtype S region and C region as a whole, and found that the hydrophobic amino acids had the highest mutation rate. Furthermore, the hydrophobic amino acids in the conserved region were selected to predict antigenic peptides, and finally obtain 21 antigenic peptides. As far as the MHC structures are concerned, we used HLA-A*0201 and HLA-A*1101 subtypes which are the highest frequency HLA subtypes among Chinese and European and American populations (Gragert et al., Citation2013). Investigation results of these two subtypes in AFND (Allele Frequency Net Database) also confirmed this. Then the antigenic peptides were docked with the corresponding HLA structures, and the compounds were subjected to residue scanning to study the effect of the mutation on the affinity and stability of the MHC-peptide complex. The results showed that, for example, mutations of L213F and I256M both had significant effects on the affinity of the complex, with affinity changes as high as +8.06 kcal/mol and +7.23 kcal/mol respectively. Combined with transmembrane region analysis, we noticed that except for I256M, mutations at the other five sites had a negative effect on the binding of peptide antigen to MHC molecule, and the I256M, V351A, V354A and L389V mutations had a more significant effect on the decrease in affinity (). Since our focus is on the binding of antigenic peptides to MHC molecules, we chose mutations with affinity changes greater than +3kcal/mol, and finally collected 11 mutation sites, L149V, I156L, L213F, I256M, V351A, V354A, M372V/I, L389V, I155V and V149V. Among these mutation sites, antigenic peptides predicted by L213F, I256M, V149A and I155V were not present in the hepitopes database (Lumley et al., Citation2016). After that, molecular dynamics simulation was carried out on the corresponding complex, and the equilibrium trajectories were selected according to the RMSD analysis and the MM/GBSA calculation was performed to study the influence of the mutation.
The results showed that these mutations led to a decrease in binding free energy to varying degrees. For example, the binding free energy of I155V was reduced from 129.12 kcal/mol to 89.35 kcal/mol, which affected the binding of peptides to MHC molecules. At the same time, we combined the calculated results with transmembrane analysis and found that four of six mutations located in the transmembrane region were found to cause a decrease in the efficiency of presentation of antigenic peptides by MHC. Therefore, we supposed that mutations in the transmembrane region may play a more important and should be taken into account in the study of the immune escape mechanism of hepatitis B virus. Actually, it has been reported in the literature that by testing the mutations of the three variant antigenic peptides sequences, it is found that the natural single residue exchange of the HBsAg peptide antigen sequence IVSPFIPLL (L389V) bound to mouse or human HLA-A*0201 class molecules can change the immunogenicity of CD8+T cells (Riedl et al., Citation2006). The experimental results found that the mutation induced the efficient initiation of different but cross-reactive CD8+ T cell populations, and different T cell populations were specifically activated. Therefore, it is speculated that mutations may participate in cross-reaction in addition to cause instability of the peptide-MHC complex. It has also discovered that immune escape mutation may promote the spread of HBV and increase the adaptability of drug-resistant strains in vaccinated individuals (Luna, Citation2018).
The predicted scores of some peptides may differ from the experimental data, which may be due to the software is only based on affinity and mass spectral peptide data. While in the actual biological process, the stability of MHC complex, TAP transport, shear and other related processes may be affected by many factors. Although, the predicted affinity does not represent the actual situation of intermolecular interactions, it can still provide clues in understand the laws of immunology. As well as, the use of bioinformatic methods to study the affinity of peptides and HLA molecules also could provide a new insight to investigate the peptide vaccines, improves the efficiency of epitope peptides modification, which can greatly improve research efficiency. In summary, we identified important potential candidate mutations in large and comprehensive data collection that related to immune escape in Chinese, European and American populations through sequence and structural analysis, and mutations reduce the efficiency of MHC molecules to present antigenic peptides by altering the antigenic peptides produced by HBV viruses, which explains the mechanism that might leads to immune escape and provide a new insight for immunotherapy from the structural aspect. On the basis of the above work, we will continue to explore the binding mechanism between other MHC subtypes and different HBV genotypes in the future.
Authors’ contributions
JL and XL conceived this work, JL and WX supervised this work. SLG conducted data collection and data analysis. LL, XYL and XL assisted the data collection. SLG drafted the manuscript. JL, RK, JQZ, JCD and WX modified the manuscript. All authors read and approved the manuscript.
| Abbreviations | ||
| HBV | = | Hepatitis B virus |
| HLA | = | Human lymphocyte antigen |
| MHC | = | Major histocompatibility complex |
| MM/GBSA | = | Generalized Bonn surface area based on molecular mechanics |
| NTCP | = | sodium-taurocholate cotransporting polypeptide |
| RMSD | = | Root-Mean-Square Deviation |
| TCR | = | T cell receptor |
Sfigure.pdf
Download PDF (1.2 MB)Availability of data and materials
All sequences involved in this manuscript are available in HBVdb database (https://hbvdb.ibcp.fr/) and AFND (http://www.allelefrequencies.net/). Structure information could acquire from RCSB PDB (https://www.rcsb.org/).
Disclosure statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Additional information
Funding
References
- Almagro Armenteros, J. J., Tsirigos, K. D., Sønderby, C. K., Petersen, T. N., Winther, O., Brunak, S., von Heijne, G., & Nielsen, H. (2019). SignalP 5.0 improves signal peptide predictions using deep neural networks. Nature Biotechnology, 37(4), 420–423. https://doi.org/https://doi.org/10.1038/s41587-019-0036-z
- Al-Qahtani, A. A., Al-Anazi, M. R., Nazir, N., Abdo, A. A., Sanai, F. M., Al-Hamoudi, W. K., Alswat, K. A., Al-Ashgar, H. I., Khan, M. Q., Albenmousa, A., El-Shamy, A., Alanazi, S. K., Dela Cruz, D., Bohol, M. F. F., & Al-Ahdal, M. N. (2018). The correlation between hepatitis B virus precore/core mutations and the progression of severe liver disease. Frontiers in Cellular and Infection Microbiology, 8, 355. https://doi.org/https://doi.org/10.3389/fcimb.2018.00355
- Beard, H., Cholleti, A., Pearlman, D., Sherman, W., & Loving, K. A. (2013). Applying physics-based scoring to calculate free energies of binding for single amino acid mutations in protein-protein complexes. PLoS One, 8(12), e82849. https://doi.org/https://doi.org/10.1371/journal.pone.0082849
- Bertoletti, A., Costanzo, A., Chisari, F. V., Levrero, M., Artini, M., Sette, A., Penna, A., Giuberti, T., Fiaccadori, F., & Ferrari, C. (1994). Cytotoxic T lymphocyte response to a wild type hepatitis B virus epitope in patients chronically infected by variant viruses carrying substitutions within the epitope. The Journal of Experimental Medicine, 180(3), 933–943. https://doi.org/https://doi.org/10.1084/jem.180.3.933
- Bertoletti, A., & Ferrari, C. (2016). Adaptive immunity in HBV infection. Journal of Hepatology, 64(1 Suppl), S71–S83. https://doi.org/https://doi.org/10.1016/j.jhep.2016.01.026
- Bertoletti, A., & Le Bert, N. (2018). Immunotherapy for chronic hepatitis B virus infection. Gut and Liver, 12(5), 497–507. https://doi.org/https://doi.org/10.5009/gnl17233
- Buchan, D. W. A., & Jones, D. T. (2019). The PSIPRED protein analysis workbench: 20 years on. Nucleic Acids Research, 47(W1), W402–W407. https://doi.org/https://doi.org/10.1093/nar/gkz297
- Cossart, Y., & Field, A. (1970). Virus-like particles in serum of patients with Australia-antigen-associated hepatitis. The Lancet, 295(7651), 848.
- Francisco, J. C., Dai, Q., Luo, Z., Wang, Y., Chong, R. H.-H., Tan, Y. J., Xie, W., Lee, G.-H., & Lin, C. (2017). Transcriptional elongation control of hepatitis B virus covalently closed circular DNA transcription by super elongation complex and BRD4. Molecular and Cellular Biology, 37(19). https://doi.org/https://doi.org/10.1128/MCB.00040-17
- Fu-Sheng, W., Jian-Gao, F., Zheng, Z., Bin, G., & Hong-Yang, W. (2014). The global burden of liver disease: The major impact of China. Hepatology, 60(6), 2099–2108. https://doi.org/https://doi.org/10.1002/hep.27406
- Fusheng, Z. (2016). Deep sequencing of the MHC region in the Chinese population contributes to studies of complex disease. Nature Genetics, 7(48), 740–746.
- González-Galarza, F. F., Takeshita, L. Y., Santos, E. J., Kempson, F., Maia, M. H. T., Silva, A. L. S. D., Silva, A. L. T. E., Ghattaoraya, G. S., Alfirevic, A., Jones, A. R., & Middleton, D. (2015). Allele frequency net 2015 update: New features for HLA epitopes, KIR and disease and HLA adverse drug reaction associations. Nucleic Acids Research, 43, 784–788.
- Gragert, L., Madbouly, A., Freeman, J., & Maiers, M. (2013). Six-locus high resolution HLA haplotype frequencies derived from mixed-resolution DNA typing for the entire US donor registry. Human Immunology, 74(10), 1313–1320. https://doi.org/https://doi.org/10.1016/j.humimm.2013.06.025
- Gripon, P., Cannie, I., & Urban, S. (2005). Efficient inhibition of hepatitis B virus infection by acylated peptides derived from the large viral surface protein. Journal of Virology, 79(3), 1613–1622. https://doi.org/https://doi.org/10.1128/JVI.79.3.1613-1622.2005
- Hayer, J., Jadeau, F., Deleage, G., Kay, A., Zoulim, F., & Combet, C. (2013). HBVdb: A knowledge database for Hepatitis B virus. Nucleic Acids Research, 41(D1), D566–570. https://doi.org/https://doi.org/10.1093/nar/gks1022
- Jurtz, V., Paul, S., Andreatta, M., Marcatili, P., Peters, B., & Nielsen, M. (2017). NetMHCpan-4.0: Improved peptide-MHC class I interaction predictions integrating eluted ligand and peptide binding affinity data. Journal of Immunology (Baltimore, Md. : 1950)), 199(9), 3360–3368. https://doi.org/https://doi.org/10.4049/jimmunol.1700893
- Ke, S. (2015). Understanding amino acid mutations in hepatitis B virus proteins for rational design of vaccines and drugs. Advances in Protein Chemistry and Structural Biology, 99, 131–153.
- Krogh, A., Larsson, B., von Heijne, G., & Sonnhammer, E. L. (2001). Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. Journal of Molecular Biology, 305(3), 567–580. https://doi.org/https://doi.org/10.1006/jmbi.2000.4315
- Kumar, S., Stecher, G., & Tamura, K. (2016). MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Molecular Biology and Evolution, 33(7), 1870–1874. https://doi.org/https://doi.org/10.1093/molbev/msw054
- Levrero, M., & Zucman-Rossi, J. (2016). Mechanisms of HBV-induced hepatocellular carcinoma. Journal of Hepatology, 64(1 Suppl), S84–S101. https://doi.org/https://doi.org/10.1016/j.jhep.2016.02.021
- Lianhong, X. (2010). Up-regulate HLA class I expression following hepatitis B virus transfection in a hepatocellular carcinoma cell line BEL7405. Immunological Investigations, 6(39), 621–634.
- Li, X., Liu, Y., Xu, Z., Wan, Z., Bai, S., Mao, P., Mao, Y., Xin, S., & Xu, D. (2013). A complete genomic analysis of hepatitis B virus isolated from 516 Chinese patients with different clinical manifestations. Journal of Medical Virology, 85(10), 1698–1704. https://doi.org/https://doi.org/10.1002/jmv.23640
- Li, S., Qian, J., Yang, Y., Zhao, W., Dai, J., Bei, J.-X., Foo, J. N., McLaren, P. J., Li, Z., Yang, J., Shen, F., Liu, L., Yang, J., Li, S., Pan, S., Wang, Y., Li, W., Zhai, X., Zhou, B., … Zhou, W. (2012). GWAS identifies novel susceptibility loci on 6p21.32 and 21q21.3 for hepatocellular carcinoma in chronic hepatitis B virus carriers. PLoS Genetics, 8(7), e1002791. https://doi.org/https://doi.org/10.1371/journal.pgen.1002791
- Loras, C., Gisbert, J. P., Mínguez, M., Merino, O., Bujanda, L., Saro, C., Domenech, E., Barrio, J., Andreu, M., Ordás, I., & Vida, L. (2010). Liver dysfunction related to hepatitis B and C in patients with inflammatory bowel disease treated with immunosuppressive therapy. Gut, 59(10), 1340–1346.
- Lumley, S., Noble, H., Hadley, M. J., Callow, L., Malik, A., Chua, Y. Y., Duffey, O. J., Grolmusova, N., Kumar, A., Ravenscroft, S., Spencer, J. I., Neumann-Haefelin, C., Thimme, R., Andersson, M., Klenerman, P., Barnes, E., & Matthews, P. C. (2016). Hepitopes: A live interactive database of HLA class I epitopes in hepatitis B virus. Wellcome Open Research, 1, 9. https://doi.org/https://doi.org/10.12688/wellcomeopenres.9952.1
- Luna, C. (2018). Immune-escape mutations and stop-codons in HBsAg develop in a large proportion of patients with chronic HBV infection exposed to anti-HBV drugs in Europe. BMC Infectious Diseases, 1(18).
- Pan, N., Chen, K., Qiu, J., Sun, H., Xu, J., Miao, F., Shi, Q., Jiang, W., Jin, H., He, Y., Xie, W., & Zhang, J. (2013). Human leukocyte antigen class I alleles and haplotypes associated with primary hepatocellular carcinoma in persistent HBV-infected patients. Human Immunology, 74(6), 758–763. https://doi.org/https://doi.org/10.1016/j.humimm.2013.02.007
- Phillip, P. (2017). MHC-I peptides get out of the groove and enable a novel mechanism of HIV-1 escape. Nature Structural & Molecular Biology, 4(24), 387–394.
- Piao, L., Chen, Z., Li, Q., Liu, R., Song, W., & Kong, R. (2019). Molecular dynamics simulations of wild type and mutants of SAPAP in complexed with Shank3. International Journal of Molecular Sciences., 20(1). https://doi.org/https://doi.org/10.3390/ijms20010224
- Riedl, P., Bertoletti, A., Lopes, R., Lemonnier, F., Reimann, J., & Schirmbeck, R. (2006). Distinct, cross-reactive epitope specificities of CD8 T cell responses are induced by natural hepatitis B surface antigen variants of different hepatitis B virus genotypes. The Journal of Immunology, 176(7), 4003–4011.
- Rivino, L., Tan, A. T., Chia, A., Kumaran, E. A., Grotenbreg, G. M., MacAry, P. A., & Bertoletti, A. (2013). Defining CD8+ T cell determinants during human viral infection in populations of Asian ethnicity. Journal of Immunology (Baltimore, Md.: 1950), 191(8), 4010–4019.
- Sastry, G. M., Adzhigirey, M., Day, T., Annabhimoju, R., & Sherman, W. (2013). Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. Journal of Computer-Aided Molecular Design, 27(3), 221–234. https://doi.org/https://doi.org/10.1007/s10822-013-9644-8
- Schuch, A., Hoh, A., & Thimme, R. (2014). The role of natural killer cells and CD8(+) T cells in hepatitis B virus infection. Frontiers in Immunology, 5, 258. https://doi.org/https://doi.org/10.3389/fimmu.2014.00258
- Shen, B., & Vihinen, M. (2004). Conservation and covariance in PH domain sequences: Physicochemical profile and information theoretical analysis of XLA-causing mutations in the Btk PH domain. Protein Engineering Design and Selection, 17(3), 267–276. https://doi.org/https://doi.org/10.1093/protein/gzh030
- Shen, Y., Xia, M., Zhang, J., Xu, L., Yang, J., Chen, A., Miao, F., Ferrone, S., & Xie, W. (2009). IRF-1 and p65 mediate upregulation of constitutive HLA-A antigen expression by hepatocellular carcinoma cells. Molecular Immunology, 46(10), 2045–2053.
- Sunbul, M. (2014). Hepatitis B virus genotypes: Global distribution and clinical importance. World Journal of Gastroenterology, 20(18), 5427–5434.
- Tan, A. T., Loggi, E., Boni, C., Chia, A., Gehring, A. J., Sastry, K. S. R., Goh, V., Fisicaro, P., Andreone, P., Brander, C., Lim, S. G., Ferrari, C., Bihl, F., & Bertoletti, A. (2008). Host ethnicity and virus genotype shape the hepatitis B virus-specific T-cell repertoire. Journal of Virology, 82(22), 10986–10997. https://doi.org/https://doi.org/10.1128/JVI.01124-08
- Tong, S., & Revill, P. (2016). Overview of hepatitis B viral replication and genetic variability. Journal of Hepatology: The Journal of the European Association for the Study of the Liver, 64(1S), S4–S16.
- Trolle, T., McMurtrey, C. P., Sidney, J., Bardet, W., Osborn, S. C., Kaever, T., Sette, A., Hildebrand, W. H., Nielsen, M., & Peters, B. (2016). The length distribution of class I-restricted T cell epitopes is determined by both peptide supply and MHC allele-specific binding preference. Journal of Immunology (Baltimore, Md. : 1950)), 196(4), 1480–1487. https://doi.org/https://doi.org/10.4049/jimmunol.1501721
- Wang, J., Lee, A. S., & Ou, J. H. (1991). Proteolytic conversion of hepatitis B virus e antigen precursor to end product occurs in a postendoplasmic reticulum compartment. Journal of Virology, 65(9), 5080–5083. https://doi.org/https://doi.org/10.1128/JVI.65.9.5080-5083.1991
- Webster, G. J. M., Reignat, S., Brown, D., Ogg, G. S., Jones, L., Seneviratne, S. L., Williams, R., Dusheiko, G., & Bertoletti, A. (2004). Longitudinal analysis of CD8+ T cells specific for structural and nonstructural hepatitis B virus proteins in patients with chronic hepatitis B: Implications for immunotherapy. Journal of Virology, 78(11), 5707–5719. https://doi.org/https://doi.org/10.1128/JVI.78.11.5707-5719.2004
- Wei, F., Zheng, Q., Li, M., & Wu, M. (2017). The association between hepatitis B mutants and hepatocellular carcinoma: A meta-analysis. Medicine, 96(19), e6835 https://doi.org/https://doi.org/10.1097/MD.0000000000006835
- Ylilauri, M., & Pentikainen, O. T. (2013). MMGBSA as a tool to understand the binding affinities of filamin-peptide interactions. Journal of Chemical Information and Modeling, 53(10), 2626–2633. https://doi.org/https://doi.org/10.1021/ci4002475