

Abstract
The molecular chaperone heat shock protein 90 (Hsp90) has emerged as one of the most exciting targets for anticancer drug development and Hsp90 inhibitors are potentially useful chemotherapeutic agents in cancer. Within the current study, Hsp90 inhibitors that entered different phases of clinical trials were subjected to Zinc15 structure query to find similar compounds (≥ 78%). Obtained small molecules (1-29) with defined similarity cut-off were docked into ensemble of Hsp90-α NTDs. Docked complexes were ranked on the basis of binding modes and Gibbs free energies as Hsp90 binders (cut-off point; ΔGb ≤ −12 kcal/mol). Top-ranked compounds were subjected to energy decomposition analysis per residue of binding pocket via density functional theory (DFT) calculations in B3LYP level of theory. Subsequent MD simulations of the top-ranked complexes were performed for 100 ns to explore the stable binding modes during a reasonable period in explicit water. Results of molecular docking and intermolecular binding analysis indicated that H-bond, hydrophobic and salt bridge interactions were determinant forces in complex formation. Compounds 19 and 20 were well accommodated in binding pocket of Hsp90 via relatively varied conformations. It was revealed that Asn51 and Phe138 were key residues that interacted stably to 19 and 20. Although primary mechanism of action for proposed molecules are unknown and yet to be explored, results of the present study revealed key structural features for future structure-guided optimization toward potent inhibitors of Hsp90-α NTD.
Hsp90 inhibitors that entered different phases of clinical trials were subjected to Zinc15 based structure query to afford potential enzyme inhibitors 19 and 20.
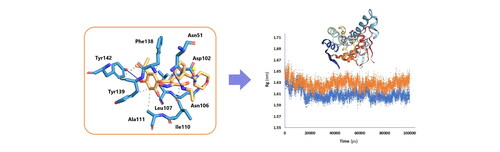
Quantum chemical calculations confirmed docking results and verified pivotal role of a conserved residues (Asn51, Leu103, Phe138 and Tyr139) in making effective hydrogen bonds.
MD simulations of top-ranked docked derivatives revealed the achievement of stable binding modes with less conformational variation of 20 than 19 in the active site of Hsp90-α NTD.
H-bond, hydrophobic contacts and salt bridge interactions were determinant forces in binding interactions of in silico hits.
Resorcinol and isoxazole were important structural motifs of in silico hits in binding to the active site of Hsp90-α NTD.
Highlights
Communicated by Ramaswamy H. Sarma
Disclosure statement
No potential conflict of interest was reported by the authors.