
Abstract
Lipases are enzymes of industrial importance responsible for the hydrolysis of ester bonds of triglycerides. A lipolytic fungus was isolated and subsequently identified based on the ITS sequence analysis as putative Aspergillus flavus with accession number LC424503. The gene coding for extracellular triacylglycerol lipase was isolated from Aspergillus flavus species, sequenced, and characterised using bioinformatics tools. An open reading frame of 420 amino acid sequence was obtained and designated as Aspergillus flavus lipase (AFL) sequence. Alignment of the amino acid sequence with other lipases revealed the presence GHSLG sequence which is the lipase consensus sequence Gly-X1-Ser-X2-Gly indicating that it a classical lipase. A catalytic active site lid domain composed of TYITDTIIDLS amino acids sequence was also revealed. This lid protects the active site, control the catalytic activity and substrate selectivity in lipases. The 3-Dimensional structural model shared 34.08% sequence identity with a lipase from Yarrowia lipolytica covering 272 amino acid residues of the template model. A search of the lipase engineering database using AFL sequence revealed that it belongs to the class GX-lipase, superfamily abH23 and homologous family abH23.02, molecular weight and isoelectric point values of 46.95 KDa and 5.7, respectively. N-glycosylation sites were predicted at residues 164, 236 and 333, with potentials of 0.7250, 0.7037 and 0.7048, respectively. O-glycosylation sites were predicted at residues 355, 358, 360 and 366. A signal sequence of 37 amino acids was revealed at the N-terminal of the polypeptide. This is a short peptide sequence that marks a protein for transport across the cell membrane and indicates that AFL is an extracellular lipase. The findings on the structural and molecular properties of Aspergillus flavus lipase in this work will be crucial in future studies aiming at engineering the enzyme for biotechnology applications.
Communicated by Ramaswamy H. Sarma
1. Introduction
Lipases (triacylglycerol acyl hydrolases EC 3.1.1.3) are enzymes produced by a variety of living organisms, ranging from plants to animals, and micro-organism and are widely used as biocatalysts capable of contributing towards improving the underexploited fat and oil biotechnology industry (Sharma & Kanwar, Citation2014). This family of enzymes have also been used as markers for the diagnosis of diseases such as cystic fibrosis and atherosclerosis (Etschmaier et al., 2011; Loli et al., Citation2015) thus underpinning their relevance in the biomedical industry. On the other hand, lipases are useful in multiple industrial processes, such as biodiesel production, in the textile industry for improving absorbency during dying, in the leather industry for dehairing and degreasing in leather making, and in the food production such as baking and in food flavouring (Mehta et al., Citation2017; Raveendran et al., Citation2018). Lipases catalyse the hydrolysis of long-chain triacylglycerols to generate fatty acids and glycerol (Schmid, Citation1998). In addition to hydrolysis, lipases catalyse other reactions which includes esterification, transesterification, and interesterification under reduced water environments (Geoffry & Achur, Citation2018; Sharma et al., Citation2016). Their catalytic versatility is a driver for the increased demand for lipases in chemical, detergent, pharmaceutical, food and oil industries (Elemuo et al., Citation2019; Jaiswal et al., Citation2017).
In the last decades, lipases of microbial origin have attracted much attention within the fast-growing enzyme technology in comparison to plant and animal lipases due to their varied catalytic activities, high stability, ease of genetic manipulation, regular supply due to absence of seasonal fluctuation and the rapid growth of microbes (Singh et al., Citation2016). Fungi are widely recognized as reliable sources of lipases and in particular, filamentous fungi are the preferred choice (Joshi & Kuila, Citation2018; Kumar, Citation2015) largely due to the high catalytic activity and ease of extraction from fermentation broth (Mehta et al., Citation2017).
Numerous reports described the production and biochemical characterisation of lipases from the filamentous fungi Aspergillus flavus (Colla et al., Citation2015; Kareem et al., Citation2017; Toscano et al., Citation2013). However, there is limited information available on the molecular and structural characteristics of Aspergillus flavus lipases.
To adequately understand the biochemical properties of lipase from Aspergillus flavus, the knowledge of (i) the molecular structure and (ii) the interaction between amino acid sequence, structure and function; are required to pave the way for the improvement of their catalytic efficiency and industrial applicability which can be achieved through protein engineering (Bassegoda et al., Citation2012; Mala & Takeuchi, Citation2008).
The evolutionary history, structure and functions of proteins can be predicted using bioinformatic techniques and information available in databases. For example, new sequences can be compared to proteins with known function to establish evolutionary hierarchies and to predict their structures (Dalal & Atri, Citation2014; Edwards & Cottage, Citation2003; Menichelli et al., Citation2018; Zhang et al., Citation2018). A wide range of online bioinformatic tools and software are available for use in protein studies such as clustalX (Larkin et al., Citation2007); BLAST-P and BLAST-N for protein and nucleotide sequence and Swiss model (Waterhouse et al., Citation2018); PyMOL, signalP (Almagro Armenteros et al., Citation2019; Schrödinger, Citation2010) and InterPro scan for structural studies and; Pfamscan for functional studies of proteins (Jones et al., Citation2014; Mitchell et al., 2019). Additionally, bioinformatics tools can be used to analyse and manipulate native proteins in order to produce more stable variants through the selection of important residues to be altered to produce functionally diverse proteins, providing predictions for experimental evaluation with reduced analysis time (Cristoni & Mazzuc, Citation2011; Suplatov et al., Citation2015; Zou et al., Citation2012).
Several studies report the use of bioinformatic tools for molecular and structural characterization of microbial lipases (Batumalaie et al., Citation2018; Chakravorty et al., Citation2011; Martínez-Corona et al., Citation2020; Panda et al., Citation2016; Sharma et al., Citation2018). However, the structure of numerous known and new microbial lipases has yet to be resolved, including the AFL, the subject of this study.
In this work, we have extracted and analysed the DNA sequence of the gene that codes an extracellular triacylglycerol lipase (AFL) from a putative Aspergillus flavus isolated from decaying oil seeds of Cucumeropsis mannii (white melon). Isolation of lipase-producing strains from the decaying seed of C. mannii (a neglected and underutilized seed) will allow isolation of lipases with high specificity to oil from the seed. This will in turn offer the possibility of improving its industrial applications as biodiesel feedstock. Aspergillus flavus can produce lipase at a relatively high yield (Colla et al., Citation2015; Kareem et al., Citation2017), hence there is a need to investigate the structural and molecular characteristics of the enzyme. Importantly, lipases produced by Aspergillus flavus possess a great potential for industrial and biotechnological applications due to its stability in a broad range of pH and high temperature compared to other fungal lipases (Kareem et al., Citation2017; Rajeswari et al., Citation2010). In this work, we have determined the molecular and structural characteristics using bioinformatics. To this end, different tools have been used for the identification of the conserved motifs, to predict the molecular weight and the predicted 3-D structure model. This work will provide the knowledge base for the resolution of the structure of a newly isolated putative Aspergillus flavus lipase and subsequent structural studies for the engineering of the enzyme for specific industrial application.
2. Materials and methods
2.1. Strain isolation and identification
The putative fungal species used for this study was isolated from decaying seeds of Cucumeropsis mannii (white seed melon), in Enugu State South East, Nigeria. Isolation of fungi from the decaying seeds was carried out by serial dilution agar plate method as described elsewhere ( Shreya et al., Citation2018). Ten grams of decaying seeds were placed in 90 ml of sterile distilled water. The tube was vortexed for 5 min to make a suspension of the decaying material. A serial dilution of the suspension (10−1 to 10−6 dilutions) was prepared.
Five mL of each dilution was aseptically and uniformly spread on sterile Petri plates containing potato dextrose agar medium (PDA, Merck, Darmstadt, Germany) supplemented with streptomycin (Mercks Darmstadt, Germany, 25 mg/100 mL). The inoculated PDA plates were maintained at room temperature for 7 days. Subsequently, fungi colonies that were observed on PDA plates were further purified by point inoculation on a freshly prepared PDA plate. The process of point inoculation was carried out until pure cultures of each fungus isolate were obtained. The isolates were stored on 4% (w/v) PDA slants throughout.
Subsequently, the isolates were screened for lipase activity in a phenol red (Merck, Darmstadt, Germany) agar plate (Singh et al., Citation2006). Briefly, phenol red agar plates were prepared by incorporating phenol red 0.001% (w/v) along with olive oil 2% (w/v), 0.1 g CaCl2 and agar 2% (w/v). The pH was adjusted to 7.0 using 0.1 N NaOH. Plates were inoculated at the centre by streak plate method with a loop full of the fungi isolated from the pure culture and subsequently incubated at 28 ºC for 72 h. Colour changes occur due to the change in the pH of the media (from neutral to acidic) because of the release of fatty acids produced during the degradation of the triacylglycerols (olive oil) to fatty acids and glycerol. The isolate which showed high lipase activity as indicated by the size of yellow halos formed around the colony was used for DNA isolation.
2.2. Molecular biology procedures
2.2.1. DNA Isolation
Genomic DNA was obtained from 200 mg of the fungi spores treated with cetyltrimethylammonium bromide (CTAB, MP Biomedicals, Illkirch, France) buffer composed of 0.1 M Tris-HCl pH 8.0, 10 mM EDTA pH 8.8, 2.5 M NaCl, 3.5% (w/v) CTAB, 30 µg/ml proteinase K. The mixture vortexed and incubated for 1 h at 37 °C (Umesha et al., Citation2016). Genomic DNA was isolated using Monarch gDNA extraction kit (New England Biolabs, Hitchin, Hertfordshire United Kingdom) following the manufacturer's instruction. Molecular identification of the isolate was carried out by amplifying the gene sequence coding for the internal transcribed spacer (ITS) region and the lipase coding region respectively using appropriate primers.
2.2.2. Polymerase chain reaction for identification of the lipase producer organism
The isolated DNA was subjected to a polymerase chain reaction (PCR) in a thermocycler (Multigene Greadient, Labnet, Edison USA). A pair of primers, ITS1_Fwd (TCC GTA GGT GAA CCT GCG) and 1TS2_Rev (TCCTCC GCT TAT TGA TAT GC) (Eurofins genomic, Esbersberg, Germany) were used to target the ITS region of the fungus in the genomic DNA extract. The PCR reaction mixture consisted of 25 μL of Q5 High fidelity 2x Master Mix Polymerase (New England Biolabs, Hitchin, United Kingdom), 2.5 μL each primer (10 μM), 10 μL of template DNA. The volume was made up to 50 μL using MiliQ H20. PCR conditions were as follows: Initiation at 98 °C for 30 s, denaturation at 98 °C for 10 s, annealing temperature of 63 °C extension at 72 °C for 30 s, for 35 cycles. The final extension was carried out at 72 °C for 2 min. The holding temperature was 4 0C. The PCR product was mixed with 1 μL of DNA loading buffer (New England Biolabs, Hitchin, United Kingdom) and subsequently subjected to 1% (w/v) agarose gel electrophoresis (incorporating 10 μL of ethidium bromide per 100 mL of gel to aid visualization) at 130 V for 50 min. DNA bands were visualized using UV dual intensity transilluminator (Model TM-02, UVP, Cambridge, United Kingdom), excised and purified using NEB gel purification kit (New England Biolabs Hitchin, United Kingdom) following the manufacturer's instruction. The purified DNA was sequenced by the Sanger sequencing method (Eurofins Genomics, Ebersberg, Germany). The ITS sequence of the fungus was deposited in the DNA Data Bank of Japan (DDBJ).
2.2.3. Polymerase chain reaction for lipase gene isolation
To isolate the lipase coding gene from the genomic DNA extract, the isolated DNA was subjected to a polymerase chain reaction (PCR) in a thermocycler (Multigene Greadient, Labnet, Edison USA). A pair of primers, AFL_Fwd (ATGATAATGGCATTTGATGAAGTTGT) and AFL_Rev (CTAGGATTGATAATTCTTTTCCTTG) (Eurofins genomic, Esbersberg, Germany) were used to amplify the lipase coding gene in a 50 µL PCR mixture following the steps as described in section 2.3 above except that the melting temperature was maintained at 60 °C. At the end of PCR the product was subjected to 1% (w/v) agarose gel electrophoresis following the steps already described in section 2.3 above. The DNA band was excised and purified using NEB gel purification kit (New England Biolabs Hitchin, United Kingdom) following the manufacturer's instruction. The purified DNA was sequenced (Eurofins Genomics, Ebersberg, Germany).
2.2.4. Lipase identification
To determine the sequence of the extracellular triacylglycerol lipase of the isolate, a lipase coding nucleotide sequence from Aspergillus flavus strain CA14 (Accession Number QQZZ01000114.1) available from NCBI (National Centre for Biotechnology Information) was adopted as the template for designing primers for PCR. Primers were designed and synthesized (Eurofins genomics, Esbersberg Germany) to amplify the lipase coding region. The melting temperature (Tm) and annealing temperatures (Ta) of the primers were estimated using the NEB Tm calculator version 1.12.0. (https://tmcalculator.neb.com/#!/main). In silico PCR design and validation was performed using Serial Cloner version 2.6.1. (http://serialbasics.free.fr/Serial_Cloner.ht). The PCR reaction mixture consisted of 25 μL of Q5 High fidelity 2x Master Mix Polymerase (New England Biolabs, Hitchin, United Kingdom), 2.5 μL each primer (10 μM), 10 μL of template DNA. The volume was made up to 50 μL using MiliQ H20. PCR conditions were: Initiation at 98 °C for 30 s, denaturation at 98 °C for 10 s, annealing temperature (Ta) for the primers targeting different gene segments are shown in . The extension at 72 °C for 30 s for 35 cycles. The final extension was performed at 72 °C for 2 min. The holding temperature was 4 °C. The PCR product was subjected to 1% (w/v) agarose gel electrophoresis at 130 V for 50 min. The gel was visualized using a UV dual intensity transilluminator (Model TM-02, UVP, Cambridge, United Kingdom). For lipase gene extraction and sequencing, bands (1327 bp for the lipase gene and 1727 bp for the lipase gene including 200 bp upstream and 200 bp downstream overhangs of the expected lipase gene sequence) were carefully cut out using a scalpel and subsequently purified using the Monarch gel extraction kit (New England Biolabs, Hitchin, Hertfordshire United Kingdom) according to the manufacturer's instruction. Purified gene fragments were sequenced (Eurofins Genomic, Anzinger, Ebersberg, Germany). The obtained nucleotide sequence was aligned with the template sequence from Aspergillus flavus strain CA14 (Accession Number QQZZ01000114.1) using Serial Cloner version 2.6.1 to identify the introns and exons and any possible variation from the template. The gene was designated as Aspergillus flavus lipase gene (AFL). The lipase nucleotide sequence can be found in .
Figure 1. Isolation of fungi from the decaying seeds of C. mannii and qualitative lipase activity assay. (A) impure culture and (B) pure culture of putative Aspergillus flavus. Qualitative Screening of Aspergillus flavus for lipolytic activity on phenol red agar plate incorporated with olive oil (C) After 24 hr (D) After 72 hr of growth. The yellow halo around the colony indicates lipase activity.
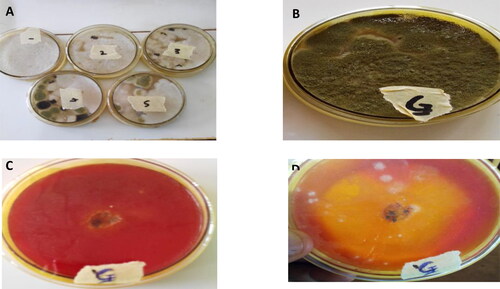
Table 1. Predicted O-glycosylation positions.
2.3. DNA and protein sequence analysis
2.3.1. Phylogenetic analysis
To establish the evolutionary path for the lipase isolated from the putative Aspergillus flavus, the AFL nucleotide sequence, as well as the protein sequence (), were used to generate phylogenetic trees based on the minimum percentage identity 95% (maximum sequence difference of 0.5) using the NCBI blast tools BlastN Nucleotide-Blast) and BlastP (Protein-protein blast) set at the default parameters. The fast maximum evolution trees were generated using the pairwise alignment.
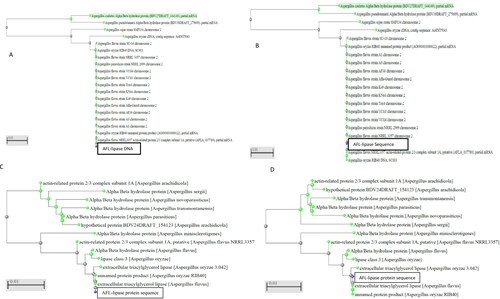
Figure 2. Phylogenetic trees of AFL nucleotide (A) is the Neighbour Joining tree for AFL-Lipase nucleotide. (B) is the Fast-minimum evolution tree for AFL-Lipase nucleotide obtained from NCBI. (C) is the neighbour-joining tree for the AFL-lipase amino acid sequence. (D) is the fast-minimum evolution tree for AFL-lipase amino acid sequence. The query sequence for the putative Aspergillus flavus sequence isolated in this work clustered with other Aspergillus flavus and are highlighted in yellow. The type material (ancestor) is highlighted in grey. Scale 0.01 represents the maximum sequence difference.
2.3.2. Multiple sequence alignment of the predicted AFL protein sequences
To identify the conserved amino acid region in the AFL protein sequence. The amino acid sequence for AFL was aligned with the amino acid sequence of other lipases listed below. Sequence alignment was carried out using Clustal X software (Larkin et al., Citation2007) Lipases from Aspergillus oryzae RIB40, accession number XP_001819264.1; Aspergillus flavus, accession number RAQ_47979.1; Aspergillus oryzae 3.042, accession number EIT_72839.1), Talaromyces marneffei ATCC 18224, accession number XP_002151838.1, Byssochlamys spectabilis, accession number GAD_93521.1, Rasamsonia emersonii CBS.393.64 (XP-013327259.1; Penicillium roqueforti FM164 (CDM34977.1). These selected lipase protein sequence has varying levels of similarities with Aspergillus flavus lipase (AFL). This will enable the identification of conserved regions of catalytic importance across lipases.
2.3.3. Characterization of the amino acid sequence of AFL-lipase
The amino acid sequence of AFL was translated from the DNA using ExPASy online tool https://web.expasy.org/translate/. The deduced protein sequence was aligned with other lipases as described in section 2.3.3. The molecular weight and isoelectric point of AFL were predicted using ExPASy compute PI/Mw tool (Gasteiger et al., Citation2003) using the default settings. Glycosylation sites were predicted using two tools; NetNGlyc Version 1.0 server for N-glycosylation (Asn-Xaa-Ser/Thr) (Gupta et al., Citation2004) and NetOglyc version 4.0 server (Steentoft et al., 2013). The presence of a secretion signal peptide was verified using signal P version 5.0 and InterPro bioinformatics tools (Almagro Armenteros et al., Citation2019; Mitchell et al., 2019) with the parameters set as default. To predict the active site lid, which has the molecular function of enabling the binding of the substrate onto the active site of the lipase enzymes superfamily, a comparative search on the Conserved Domain Database (CDD) (Lu et al., Citation2020) was performed using the default optional parameters.
2.3.4. Three-dimensional modelling of AFL (Aspergillus flavus lipase)
The modelling of AFL (Aspergillus flavus lipase) structure was performed using the SwissModel tool (Waterhouse et al., Citation2018). The template identification was performed by searching the Swiss-model interactive workplace using AFL amino acid sequence while the tool was set at the default parameters. The template with the highest percentage identity was used to generate three models. The generated models were evaluated using quality parameters such as the global model quality estimate GMQE and the quality model energy analysis QMEAN values for the model. GMQE and QMEAN are parameters commonly used for estimating model quality: The GMQE score is generated using information from the template alignment and structure to predict the accuracy of the model and it is expressed as a number ranging from 0 to 1, whereby values close to 1 indicate high reliability of the model (Biasini et al., Citation2014); The QMEAN is a scoring function that describes an important geometrical aspect of protein structure. It provides information on both the global (entire structure) and local (per residue) quality estimate (Santhoshkumar & Yusuf, Citation2020). The QMEAN score values typically range from 0 to −4.0, where scores closer to 0 indicates a good model. The best performing 3-D structure was adopted based on GMQE and QMEAN values. The stereochemical quality check of the Model was performed using PROCHECK (Laskowski et al., Citation1993) using the default parameters. The domains present in the AFL model structure were annotated and the model was superposed with the template (Yarrowia lipolytica) lipase using a trial version of PyMOL molecular graphics system version 2.0 (Schrödinger, Citation2010).
2.3.2. Codon optimization of AFL-Lip DNA sequence using different optimization tools
Three different tools for codon optimisation were used to codon optimize and adapt the AFL-lipase DNA sequence for heterologous expression in yeast (Pichia pastoris). These tools namely, Gensmart codon optimization (version Beta 1.0) https://www.genscript.com/tools/gensmart-codon-optimization,
GENEius (http://www.geneius.de/GENEius/Orf_start.action) and GENEWIZ (https://clims4.genewiz.com/Toolbox/CodonOptimization) were employed and their outputs were compared.
3. Results and discussion
3.1. Strain isolation and identification
The putative Aspergillus flavus strain was isolated from the decaying oilseed of Cucumeropsis mannii. Oxidation and release of fatty acids increases as the seed deteriorates, providing a good condition for organism capable of metabolising lipid substrates to colonize. As a result, lipolytic fungi strains were isolated from the decaying seed (). The isolates were tested for lipolytic activity in a phenol agar plate containing olive oil. After 72 h incubation at room temperature, yellow halos were seen around the isolates. Lipolytic activity of the isolates produced free fatty acids, which caused a drop in pH leading to a change in colour of the phenol red from red to yellow. The isolate that showed maximum lipolytic activity as indicated by the size of the yellow halos around the colonies was selected for this study (). The isolate showing the highest lipolytic activity under the tested conditions was identified to be Aspergillus flavus according to the sequencing results of the ITS region. The ITS sequence for the putative Aspergillus flavus selected for this work was then deposited in the DNA Data Bank of Japan (DDBJ) and the (Accession Numbers LC424503) was assigned. In agreement with the result obtained here, there have been reports on the isolation of lipolytic fungi from decaying oil seeds elsewhere. Sandi et al., Citation2020 isolated lipolytic fungi from decaying tropical oilseed. Lipase producing strains have been isolated from the decaying oilseed, Ginkgo biloba (Yuan et al., Citation2010). It has been demonstrated that microbial lipase exhibit specificity to fatty acids (Baillargeon et al., Citation1989; .). Isolation of lipase producing strains from the decaying seed of C.manni will make it possible to obtain lipases with high specificity to the C. mannii seed oil, which in turn offers the possibility to be used to improve its industrial applications.
3.2. Analysis and characterisation of AFL nucleotide sequence
After treating the fungi spores with CTAB and vortexing to break the cell wall as detailed in section 2.1, the gDNA was extracted using the Monarch gDNA isolation kit. The lipase coding gene and ITS sequence were amplified using the appropriately designed primers in separate PCR reactions.
Gel electrophoresis of the PCR products showed bright nucleotide bands for the targeted regions of the putative Aspergillus flavus lipase coding gene (1327 bp) and the ITS region (560 bp) as shown in .
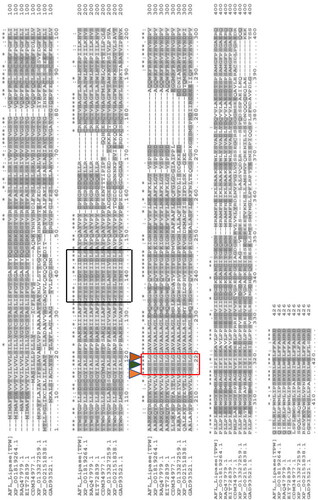
Figure 3. Multiple alignments of AFL-Lipase protein sequence with other fungi triacylglycerol lipases. Highly conserved regions are stared (*) Conserved amino acids at the catalytic site are indicated by a red box and the catalytic triad (G-X-S-X-G) is indicated with small triangles. The predicted active site lid is indicated by a black box. The dashed line represents gaps in the sequence. In the above alignment, AFL-lipase (TPW): Amino acid sequence from AFL-lipase being studied in this work; XP_001819264, Amino acid for extracellular Lipase from Aspergillus oryzae; RAQ_47979.1, Amino acid for Extracellular lipase from Aspergillus flavus; EIT-72839.1, Amino acid for lipase from Aspergillus oryzae; CDM-34977.1, Amino acid sequence coding for lipase from Penicillium roqueforti FM164; XP_013327259.1, Amino acid sequence for lipase from Rasamsonia emersonii CBS.393.64; XP_002151838.1, Amino acid sequence for lipase from Talaromyces marneffei ATC18224; GAD_93521.1, Amino acid sequence for lipase from Byssochlamys spectabilis.
A BLAST [ref] of the nucleotide sequence obtained from the ITS region in the NCBI showed 100% similarity with Aspergillus flavus thus, suggesting that the isolate which was selected for this study is Aspergillus flavus. A consensus nucleotide sequence of 1263 bp was obtained from the sequencing result after the introns and overhangs were identified by aligning the consensus sequence with the template lipase gene (Aspergillus flavus strain CA14 Accession Number QQZZ01000114.1) from the NCBI data bank. The consensus sequence shows 100% similarity with the template DNA. A neighbour-joining and Fast minimum evolution trees for the lipase DNA sequence showed the gene forming a clade with the Aspergillus flavus and Aspergillus oryzae indicating a common gene ancestry with the consensus gene ().
The nucleotide sequence was translated into a protein of 420 amino acid sequences using the ExpaSy online tool. The 420 amino acid sequence formed a single open reading frame (ORF). A blast of the amino acid sequence of AFL showed that it shares a high percentage similarity with extracellular triacylglycerol lipases from other Aspergillus species (Aspergillus oryzae − 99.76%, Aspergillus fumigatus − 63.27% Aspergillus niger − 61.25%, Aspergillus sclerotiicarbonarius − 60.59%) and extracellular triacylglycerol lipases from non -Aspergillus fungi (Talaromyces stipitatus ATCC 10500 − 55.38%, Rasamsonia emersonii CBS393.64 − 58.07%, Byssochlamyc spectabilis − 55.99%) and a host of other Aspergillus and non-Aspergillus fungi α/β hydrolases. The phylogenetic tree showed the AFL formed a clade with lipases from Aspergillus flavus and Aspergillus oryzae, which indicates that they could be originate from the same ancestor ()
3.3. Analysis and characterisation of AFL protein sequence
The AFL (Aspergillus flavus lipase) amino acid sequence was aligned with sequence from other lipase selected from a BlastP similarity search outcome in the NCBI. The aligned protein sequence was selected in such a way that a wide range of lipases was covered, including highly identical lipases, non-Aspergillus lipases and lipase with lower percentage identity with AFL-lipase. This selection was aimed at finding the conserved regions of catalytic and structural importance in the AFL-lipase. The alignment results revealed a conserved pentapeptide GHSLG (, red box) which is believed to be the conserved amino acid residues of the nucleophilic elbow. Another conserved region was also observed comprising of amino acids TYITDTIIDLS (, black box) which was in the active site lid. Gupta et al. (Citation2015) reported that lipases are usually conserved mainly in the signature sequence or motif such as the nucleophilic elbow, and oxyanion holes. The enzyme has been shown to possess catalytic triad formed by Serine-Histidine-Aspartate residues with the Serine residue appearing in the conserved pentapeptide (GXSXG where X can be any amino acid) (Contesini et al., Citation2017). The start codon (MIM sequence) in AFL-lipase was observed in some of the known lipase genes it was aligned with.
To further investigate the conserved features of the AFL-lipase, a domain search was carried out using the CDD. The CDD-search revealed the pentapeptide GHSLG in the nucleophilic elbow to be located between residue 202-206 and the active site lid at position 129-140 comprising of amino acids TYITDTIIDLS (). The sequence of amino acids obtained in this study showed a non-conserved lid region with the aligned sequence (, black box). This is in agreement with the report by (Albayati et al., Citation2020; Khan et al., Citation2017), which states that the active site lid domain of lipases varies in their amino acid sequence length and structures. Hence suggesting that the lid sequences are not strictly conserved in lipases. Lipases usually have their lids situated over their active site which contributes to both, the catalytic properties and stability of the lipase such as substrate recognition and binding (Khan et al., Citation2017; Tan & Miller, Citation1992). The movement of the lid associated with catalysis helps to expose the hydrophobic residues of the active site. When the lid is closed, it prevents the hydrophobic patches to be accessible to the solvents (Fojan et al., Citation2000). Substrate specificity by lipases has been shown to be controlled by the molecular properties of the protein and the structure of substrates (Jensen et al., Citation1983). This can be achieved by rational design (site directed mutagenesis) on the active site lid to fit a particular substrate In previous studies, modification in the lid region of lipases has caused a change in substrate specificity and stability (Yu et al., Citation2012, Citation2014) and lead to inability of interfacial activation by the enzyme (Tang et al., Citation2015) thus, emphasizing the importance of the active site lid in lipases. A search of the lipase engineering database (LED) with the AFL sequence showed that this lipase belongs to lipase Class GX, superfamily abH23 (filamentous fungal lipase) and homologous family abH23.02 (saccharomyces like lipases). These results are in accordance with the observation made by (Pleiss et al., Citation2000) that fungi lipases have oxyanion hole GX. The GX-class comprises a lipase superfamily, where X is the oxyanion forming amino acid preceded by a conserved glycine residue (Fischer et al., Citation2006).
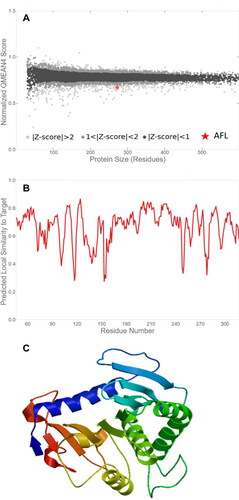
Figure 4. Prediction of putative Aspergillus flavus-like lipase (AFL) structure of the best fit model with a global model quality estimate (GMQE) value of 0.48 and a Qualitative Model Energy Analysis (QMEAN) value of -2.47. (A) Comparison with the non-redundant set of Proteins Database (PDB) structures, (B) Local quality estimate and (C) Three-dimensional structure representation.
3.4. Three-dimensional (3D) modelling of AFL-lipase
The 3-Dimensional model of AFL-lipase was generated using the SWISS-MODEL tool. The modelling was aimed at getting more insights into the structure of the putative Aspergillus flavus lipase. A 3-Dimensional model generated using the Swiss-Model tool revealed a structure comparable to the triacylglycerol lipase from Yarrowia lipolytic. Three top models were selected from the search outcome and the best fit model was chosen based on the sequence similarity, sequence coverage, global model quality estimate (GMQE) and qualitative Model Energy Analysis (QMEAN) values () Results of the analysis of the other two models with lower reliability can be found in and and were not adopted for further analysis. The structure for the best fit model () was retrieved and visualized using Pymol version 2.0. Raw data generated from the Swiss model tool revealed a percentage identity of (34.08%), query cover 272 residues of the model template. GMQE value of 0.48 and QMEAN score −2.47 were obtained. Both GMQE and QMEAN are used for estimating model quality. GMQE score is generated using information aligning the template and its structure to predict the accuracy of the model and it is expressed as a number ranging from 0 to 1, values closer to 1 indicate high reliability of the model (Biasini et al., Citation2014). QMEAN score, on the other hand, is a scoring function that describes an important geometrical aspect of protein structure. It provides information on both the global (entire structure) and local (per residue) quality estimate (Santhoshkumar & Yusuf, Citation2020). The QMEAN score values typically range from 0 to −4.0, where scores closer to 0 indicates a good model while values of −4 and bellow are highly unreliable and indicate a model of low-quality (Waterhouse et al., Citation2018). Though the 3-Dimensional model generated using the Swiss-Model tool revealed a structure comparable to the triacylglycerol lipase from Yarrowia lipolytic, the GMQE and QMEAN score predicted for the AFL model falls within the acceptable range, though the model is not of the best quality. The Ramachandran plot of the AFL model indicated that 88.9% (209) residues are in the most favourable region, 21(8.9%) of the residue in the additional allowed region, 5(2.1%) residues in the generously allowed region and 0.0% in the disallowed region (). This represents the statistical distribution of the torsional angle of the amino acid backbone for the structure. Amino acids with the torsional values in the permitted region give insight into the structure of the protein. The number of residues in the most favourable region of 88.9% which is slightly below the acceptable value of 90% further indicated that the model is not of the best quality. This finding is not surprising given the low identity of the model and this can be attributed to the relatively non-availability of published crystal structural information on Aspergillus flavus lipases as revealed by the PDB search. Contesini et al. (Citation2017) observed that information is still not adequate on the crystal structures of lipases from Aspergillus species. In addition, a superposed structure of our model for Aspergillus flavus lipase (AFL) with that of the template (lipase from Y. lipolytica) is presented in within the supplementary materials to show the similarities and differences at the 3-d level.
Figure 5. (A) Three-dimensional (3-D) structure of AFL-Lipase generated with PyMol software v2.0 along with the identified amino acid at the active site. The nucleophilic elbow is represented in sticks; Glycine 202 and 206 (red), Serine 204 (blue), Histidine (green), Lysine (cyan). Active site lid (residues 129-140, purple). (B) Zoomed catalytic site (Glycine 202 and 206, red); Serine 204 (blue) and the active site lid (purple).
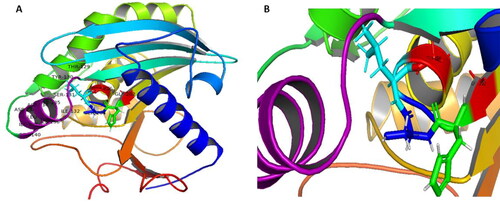
Figure 6. Ramachandran plot for the Aspergillus flavus lipase (AFL) model structure.
Elucidating the X-ray crystallographic structure of AFL is one of the objectives in this project in the future to enable the engineering of AFL possibly through site-directed mutagenesis, as it could contribute towards improving its catalytic efficiency, stability and substrate specificity which will, in turn, enhance the industrial and biotechnological applications.
The major structural characteristics determined through the modelling of AFL are the identification of the conserved motifs, the pentapeptide sequence GHSLG which is peculiar to the abH23.02 family (lipases of Saccharomyces type) and the catalytic important active site lid
PyMOL software was used to identify the amino acid residues in the nucleophilic elbow, which included the catalytic serine, in the predicted mode (). However, the information from the CDD blast search using the AFL amino acid sequence showed that the conserved lipase active site/nucleophilic elbow is located between amino acid sequence 202-206 (GHSLG) as observed in . The active site lid at the sequence position 129-140 consists of amino acid sequence TYITDTIIDLS (). To aid in the graphical identification of the active site and the catalytic site lid, a zoom-in is provided in .
depicts the Ramachandran plot for the Aspergillus flavus lipase (AFL) model structure in order to check the stereo quality of the model. This plot shows the torsional angles - phi (φ) and psi (ψ) - of the residues (amino acids) contained in the peptide sequence and it is therefore useful to obtain insights into the peptide structure. As can be observed, the plot statistics showed that out of 272 residues, 209 (88.9%) of the residues are in the most favourable region; 21 (8.9%) residues are in additional allowed region, 5 (2.1%) residues in the generously allowed region and 0 (0.0%) residues in the disallowed region. Despite the number of residues within the most favourable region in slightly below the acceptable value (90%), this observation supports our earlier observation (see section 3.4) that the model is not fully reliable. Nonetheless, this can be attributed to the non-availability of crystal structural information on Aspergillus flavus lipase in the protein data bank. This is in agreement with the report by Contesini et al. (Citation2017) that ‘there is no enough information on the crystal structures of lipases from Aspergillus species’ in general. A superposed 3d-level structure of our model for Aspergillus flavus lipase (AFL) with that of the template (lipase from Y. lipolytica) is presented in within the supplementary materials showing the similarities and differences at the 3-d level; where the AFL and the template models are coloured in blue and red, respectively.
3.5. Glycosylation analysis of the AFL sequence
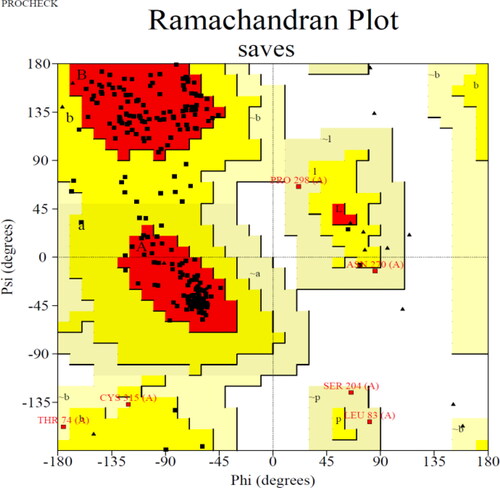
Glycosylation analysis of the AFL sequence using the NetNglyc bioinformatics tool revealed three strong asparagine-linked (++) N-glycosylation sites at amino acid positions 164, 236 and 333 with the strongest glycosylation potential values of 0.7250, 0.7037 and 0.7048, respectively; being values well above the threshold value of 0.5 as shown in and Table S1. Moreover, four O-glycosylation sites were predicted at amino acids positions 355, 358, 360 and 366 as shown in Table 2. Both N-glycosylation and O-glycosylation have been shown to play crucial roles in the stability and modulation of secreted proteins including lipases (Goto, Citation2007; Huang et al., Citation2014; Rubio et al., Citation2019). Multiple N-glycosylation sites were observed by Yang et al. (Citation2015) to be vital in the excretion and expression of lipase from Rhizopus chinensis expressed in Pichia pastoris. The presence of N-glycosylation and O-glycosylation sites in the protein sequence of AFL confirms that it is an extracellular lipase and this is in agreement with previous works that report that the glycosylation of protein (both N-glycosylation and O-glycosylation) occurs in eukaryotes (fungi and yeast) as part of posttranslational modification process (Dell et al., Citation2010). Glycosylation in Aspergillus and yeasts (i.e. P. pastoris) are similar, in their pattern of N-linked glycosylation which is characterized by polymannosylation and are attached to the N- amide of the asparagine at an Asn-Xaa-Ser/Thr sequence where X may be any other amino acid (Aebi, 2013; Weerapana & Imperiali, Citation2006). The O-linked glycosylation in Aspergillus are composed of GalNAc sugars attached to a beta-hydroxyl group of Serine or Threonine (Tran & Ten Hagen, Citation2013). The consensus sequences for O-linked glycosylation in Pichia pastoris are not fully established (Zhan & An, Citation2010). This suggests that the AFL-lipase is suitable for extracellular heterologous expression in yeast Pichia pastoris considering the similarity in the N-glycosylation pattern.
Figure 7. Predicted N-glycosylation using NetNglyc version 1.0. High glycosylation potentials were predicted at positions 164, 236 and 333 with a potential of 0.7250, 0.7037 and 0.7048, respectively. Minimum threshold = 0.5.
3.6. Prediction of the AFL signal peptide and isoelectric point
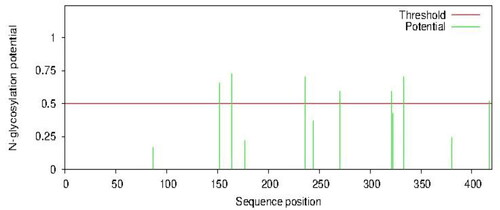
SignalP analysis of the AFL-lipase predicted that the enzyme has a signal sequence of 37 amino acid residues, from residue 1 to 37 at the N-terminal of the protein (). Signal sequences are short peptides that are located on the N-terminus of some proteins which marks these proteins for transport across cell membranes out of the cell (Clark & Pazdernik, Citation2013; Kunze & Berger, Citation2015). They are required for recombinant protein secretion (Freudl, Citation2018). Fungal lipases are extracellular and are expected to have signal peptides for their efficient secretion. The signal sequence of different amino acid lengths has been reported in the protein sequence of lipases from different microbial sources. Therefore, our results indicating the presence of a signal sequence is in line with previous works. Martínez-Corona et al., Citation2020, reported 33 amino signal sequence in yeast, Aspergillus oryzae triacylglycerol lipase, 30 amino acids signal sequence (Toida et al., Citation2000), 26 amino acid signal peptide was reported for Pseudomonas aeruginosa lipase (Wohlfarth et al., Citation1992). The signal sequence length of 37 amino from in AFL is in the same range as that from other lipases ().
Figure 8. Predicted signal peptide position in AFL-Lipase protein sequence using signal version 5.0 tool.
The Aspergillus flavus lipase in this work was classified as α/β hydrolase, lipase class GX, superfamily abH23 and homologous family abH23.02 according to the lipase engineering database (LED) search. While the molecular weight of the AFL protein was predicted to be 46950.47DA (46.9KDa) and an isoelectric point value of 5.73 using the ExPASy compute tool.
3.7. Codon optimisation of the AFL-Lipase for recombinant expression in the yeast Pichia pastoris
Aspergillus flavus lipase could be produced from the native producer Aspergillus flavus species at a high yield, however, considering the advantages offered by heterologous recombinant protein production such as higher yield and ease of purification, (Rosano & Ceccarelli, Citation2014; Sanchez & Demain, Citation2012), the AFL nucleotide sequence was optimized for expression in Pichia pastoris. Several microbial species exhibit a preference for codon usage. Hence, codon optimization is important in ensuring the proper elongation and accuracy of the translation and, can be achieved by using a range of bioinformatics tools. Here, three different codon optimization tools namely; Genesmart, GENeius and Genewiz optimization were used for optimizing the AFL gene sequence as detailed in Section 2. The optimized sequences obtained using the tools were aligned and results were compared (). Differences in the optimized sequences were observed (as indicated by the non-stared nucleotides). Such differences may be largely due to the different codons which naturally code for the same amino acid as well as different algorithms used by the different tools.
Figure 9. Multiple alignment codon-optimized AFL (Aspergillus flavus lipase) nucleotide sequence for expression in Pichia pastoris using Genesmart, GENEWIZ and GENEius optimization tools. Star symbol (*) indicates that the four sequences share the same nucleotides at a given position. A (adenine) coloured in orange; C (cytosine), coloured in cyan; G (guanine) and T (thymine), not coloured. The black box represents the active site lid sequence.

For example, Genesmart codon optimization tool algorithm optimizes parameters that are critical to (i) protein translation such as GC content, CpG dinucleotide content, cryptic splicing sites, and terminal sequence; (ii) translation efficacy, codon usage preference, structure of the mRNA, premature polyA tails, unstable RNA motifs, stable free energy mRNA, internal chi sites and ribosomal binding sites and; (iii) protein refolding parameters such as the interaction of codon and anticodon, codon context and RNA secondary structure.
In contrast, the GENEius optimization tool uses an algorithm that randomly assembles the DNA sequence and analyses it in relation to the codon usage of the expression host (Pichia pastoris in this study) by comparing it to the codon usage table taken from the Kazusa codon usage database (https://www.kazusa.or.jp/codon/) (GENEius also harmonizes the codon usage by checking for bad motifs and distributing the GC content to avoid GC rich sub-sequences (https://www.eurofinsgenomics.eu/en/gene-synthesis-molecular-biology/geneius/).
Finally, the GENEWIZ codon optimization tool optimizes parameters that stabilizes the DNA sequence, such as codon usage preference, GC content, mRNA secondary structure, repeat sequence, restriction enzyme recognition sites and ribosomal binding sites to improve gene expression.
When AFL nucleotide sequence obtained using these optimization tools was compared with the original sequence (non-optimized), our results showed that Genesmart, GENEius and GENEWIZ optimized sequences have 74.52, 74.52 and 72.52% similarities, respectively, in comparison to the non-optimized sequence. When these optimised sequences were compared with each other, the Genesmart optimized sequence showed 73.81 and 73.49% similarity with the GENEius and GENEWIZ optimized sequences respectively, while the GENEius and GENEWIZ optimized sequence shared 75.0% similarity. On multiple alignments of the three sequences optimized using the different tools, the same nucleotides in the same position are represented with stared, while different nucleotide in the same sequence position is not stared as shown in .
Further analysis of the active site lid sequence for optimized and non-optimized DNA revealed that the active site lid for AFL-lipase is located between sequence number 385 to 420. Percentage similarities of 77.78%, 72.22% and 69.44% with the non-optimised active site lid sequence were obtained for Genesmart, GENEius and GENEWIZ, respectively.
In principle, the Genesmart tool appears to be more robust in the optimization and adaptation of DNA sequence considering the parameters that it takes into consideration as outlined above. Therefore, to investigate the best optimization tool for the adaptation of the AFL sequence for expression in Pichia pastoris, it will be required that these optimized sequences be tested experimentally.
4. Conclusions
A 420 amino acid residues (ORF) lipase with extracellular lipase activity was identified and sequenced from a putative new Aspergillus flavus strain isolated from white melon seeds. We have employed bioinformatics tools for the characterization of the lipase. Data obtained from the lipase model showed that it belongs to class GX, superfamily abH23 (filamentous fungi lipase) and homologous family abH23.02 (saccharomyces-like lipase). The protein contained two predicted N-glycosylation sites and four O-glycosylation sites which are believed to play important roles in the protein modification as extracellular protein. A 37 amino acid residue peptide sequence was identified at the N-terminus of the protein which is vital for the export of the AFL-lipase across the cell membrane as an extracellular enzyme.
Though the important molecular and structural characteristics of the lipase was revealed, the 3-dimensional structural model produced low percentage similarity with lipase structures available in the PDB. This can be potentially resolved in future studies by X-ray crystallographic technique to establish the crystal structure of Aspergillus flavus lipase, which will pave the way for a more structural understanding of the protein. Whilst structural studies reveal that this new lipase is similar to previously described lipases from other fungi, only by understanding the structure of the enzyme, we will be able to engineer the protein for industrial application. To the best of our knowledge, just a few lipases from Aspergillus flavus have been characterised in terms of their biochemical characteristics but this is the first work where the structural elucidation of an Aspergillus flavus lipases is described.
Supplementary_Material_TBSD-2021-2104.pdf
Download PDF (4.6 MB)Disclosure statement
The authors declare that there is no conflict of interest.
Additional information
Funding
References
- Aebi, M. (2013,). N-linked protein glycosylation in the ER. Biochimica et Biophysica Acta – Molecular Cell Research, 1833(11), 2430-2437. https://doi.org/10.1016/j.bbamcr.2013.04.001
- Albayati, S. H., Masomian, M., Ishak, S. N. H., Mohamad Ali, M. S. b., Thean, A. L., Mohd Shariff, F. b., Muhd Noor, N. D. B., & Raja Abd Rahman, R. N. Z. (2020). Main structural targets for engineering lipase substrate specificity. Catalysts, 10(7), 747. https://doi.org/10.3390/catal10070747
- Almagro Armenteros, J. J., Tsirigos, K. D., Sønderby, C. K., Petersen, T. N., Winther, O., Brunak, S., von Heijne, G., & Nielsen, H. (2019). SignalP 5.0 improves signal peptide predictions using deep neural networks. Nature Biotechnology, 37(4), 420–423. https://doi.org/10.1038/s41587-019-0036-z
- Baillargeon, M. W., Bistline, R. G., & Sonnet, P. E. (1989). Evaluation of strains of Geotrichum candidum for lipase production and fatty acid specificity. Applied Microbiology and Biotechnology, 30(1), 92–96. https://doi.org/10.1007/BF00256003
- Bassegoda, A., Cesarini, S., & Diaz, P. (2012). Lipase improvement: Goals and strategies. Computational and Structural Biotechnology Journal, 2(3), e201209005. https://doi.org/10.5936/csbj.201209005
- Batumalaie, K., Edbeib, M. F., Mahat, N. A., Huyop, F., & Wahab, R. A. (2018). In silico and empirical approaches toward understanding the structural adaptation of the alkaline-stable lipase KV1 from acinetobacter haemolyticus. Journal of Biomolecular Structure & Dynamics, 36(12), 3077–3093. https://doi.org/10.1080/07391102.2017.1377635
- Biasini, M., Bienert, S., Waterhouse, A., Arnold, K., Studer, G., Schmidt, T., Kiefer, F., Cassarino, T. G., Bertoni, M., Bordoli, L., & Schwede, T. (2014). SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Research, 42(W1), W252–W258. https://doi.org/10.1093/nar/gku340
- Chakravorty, D., Parameswaran, S., Dubey, V. K., & Patra, S. (2011). In silico characterization of thermostable lipases. Extremophiles, 15(1), 89–103. https://doi.org/10.1007/s00792-010-0337-0
- Clark, D. P., & Pazdernik, N. J. (2013). Protein synthesis. Molecular Biology. https://doi.org/10.1016/b978-0-12-378594-7.00047-0.
- Colla, L. M., Ficanha, A. M. M., Rizzardi, J., Bertolin, T. E., Reinehr, C. O., & Costa, J. A. V. (2015). Production and characterization of lipases by two new isolates of Aspergillus through solid-state and submerged fermentation. BioMed Research International, 2015, 1–9. https://doi.org/10.1155/2015/725959
- Contesini, F. J., Calzado, F., Valdo, J., Jr., M., Rubio, M. V., & Zubieta, M. P. (2017). Fungal Metabolites. In J.-M., Merillo and K.G. Ramawat (Eds.), Reference series in phytochemistry (pp. 639–666). https://doi.org/10.1007/978-3-319-25001-4
- Cristoni, S., & Mazzuc, S. (2011). Bioinformatics Applied to Proteomics. In N.-S., Yang (Ed.), Systems and Computational Biology (pp. 25-50). https://doi.org/10.5772/23680.
- Dalal, A., & Atri, A. (2014). An Introduction to Sequence and Series. International Journal of Research, 1(10), 1286–1292. https://doi.org/10.1002/0471250953.bi0301s42.An
- Dell, A., Galadari, A., Sastre, F., & Hitchen, P. (2010). Similarities and differences in the glycosylation mechanisms in prokaryotes and eukaryotes. International Journal of Microbiology, 2010, 1–14. https://doi.org/10.1155/2010/148178
- Edwards, Y. J. K., & Cottage, A. (2003). Bioinformatics methods to predict protein structure and function: A practical approach. Molecular Biotechnology, 23(2), 139–166. https://doi.org/10.1385/MB:23:2:139
- Elemuo, N. G., Ikiensikimama, S. S., Ubani, C. E., Egwim, E. C., & Osuoha, J. O. (2019). Production and Characterization of Lipase Enzyme Expressed by Crude Oil Contaminated Soil Isolates. Universal Journal of Microbiology Research, 7(1), 1–6. https://doi.org/10.13189/ujmr.2019.070101
- Etschmaier, K., Becker, T., Eichmann, T. O., Schweinzer, C., Scholler, M., Tam-Amersdorfer, C., Poeckl, M., Schuligoi, R., Kober, A., Chirackal Manavalan, A. P., Rechberger, G. N., Streith, I. E., Zechner, R., Zimmermann, R., & Panzenboeck, U. (2011). Adipose triglyceride lipase affects triacylglycerol metabolism at brain barriers. Journal of Neurochemistry, 119(5), 1016–1028. https://doi.org/10.1111/j.1471-4159.2011.07498.x
- Fischer, M., Thai, Q. K., Grieb, M., & Pleiss, J. (2006). DWARF – A data warehouse system for analyzing protein families. BMC Bioinformatics, 7(1), 10. https://doi.org/10.1186/1471-2105-7-495
- Fojan, P., Jonson, P. H., Petersen, M. T. N., & Petersen, S. B. (2000). What distinguishes an esterase from a lipase: A novel structural approach. Biochimie, 82(11), 1033–1041. https://doi.org/10.1016/S0300-9084(00)01188-3
- Freudl, R. (2018). Signal peptides for recombinant protein secretion in bacterial expression systems. Microbial Cell Factories, 17(1), 1–10. https://doi.org/10.1186/s12934-018-0901-3
- Gasteiger, E., Gattiker, A., Hoogland, C., Ivanyi, I., Appel, R. D., & Bairoch, A. (2003). ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Research, 31(13), 3784–3788. https://doi.org/10.1093/nar/gkg563
- Geoffry, K., & Achur, R. N. (2018). Screening and production of lipase from fungal organisms. Biocatalysis and Agricultural Biotechnology, 14, 241–253. https://doi.org/10.1016/j.bcab.2018.03.009
- Goto, M. (2007). Protein O-glycosylation in fungi: Diverse structures and multiple functions. Bioscience, Biotechnology and Biochemistry, 71(6), 1415–1427. https://doi.org/10.1271/bbb.70080
- Gupta, R., Jung, E., & Brunak, S. (2004). NetNGlyc: Prediction of N-glycosylation sites in human proteins. in preparation.
- Gupta, R., Kumari, A., Syal, P., & Singh, Y. (2015). Molecular and functional diversity of yeast and fungal lipases: Their role in biotechnology and cellular physiology. Progress in Lipid Research, 57(January), 40–54. https://doi.org/10.1016/j.plipres.2014.12.001
- Huang, J., Xia, J., Yang, Z., Guan, F., Cui, D., Guan, G., Jiang, W., & Li, Y. (2014). Improved production of a recombinant Rhizomucor miehei lipase expressed in Pichia pastoris and its application for conversion of microalgae oil to biodiesel. Biotechnology for Biofuels, 7(1), 1–11. https://doi.org/10.1186/1754-6834-7-111
- Jaiswal, A., Preet, M., & Tripti, B. (2017). Production and optimization of lipase enzyme from mesophiles and thermophiles. Journal of Microbial & Biochemical Technology, 09(03), 126–131. https://doi.org/10.4172/1948-5948.1000355
- Jensen, R. G., DeJong, F. A., & Clark, R. M. (1983). Determination of lipase specificity. Lipids, 18(3), 239–252. https://doi.org/10.1007/BF02534556
- Jones, P., Binns, D., Chang, H.-Y., Fraser, M., Li, W., McAnulla, C., McWilliam, H., Maslen, J., Mitchell, A., Nuka, G., Pesseat, S., Quinn, A. F., Sangrador-Vegas, A., Scheremetjew, M., Yong, S.-Y., Lopez, R., & Hunter, S. (2014). InterProScan 5: Genome-scale protein function classification. Bioinformatics (Bioinformatics), 30(9), 1236–1240. https://doi.org/10.1093/bioinformatics/btu031
- Joshi, R., & Kuila, A. (2018). Lipase and their different industrial applications: A review. Brazilian Journal of Biological Sciences, 5(10), 237–247. https://doi.org/10.21472/bjbs.051004
- Kareem, S. O., Adebayo, O. S., Balogun, S. A., Adeogun, A. I., & Akinde, S. (2017). Purification and Characterization of Lipase from Aspergillus flavus PW2961 using Magnetic Nanoparticles. Nigerian Journal of Biotechnology, 32(1), 77. https://doi.org/10.4314/njb.v32i1.11
- Khan, F. I., Lan, D., Durrani, R., Huan, W., Zhao, Z., & Wang, Y. (2017). The lid domain in lipases: Structural and functional determinant of enzymatic properties. Frontiers in Bioengineering and Biotechnology, 5(MAR), 1–13. https://doi.org/10.3389/fbioe.2017.00016
- Kunze, M., & Berger, J. (2015). The similarity between N-terminal targeting signals for protein import into different organelles and its evolutionary relevance. Frontiers in Physiology, 6(SEP), 1–27. https://doi.org/10.3389/fphys.2015.00259
- Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., Valentin, F., Wallace, I. M., Wilm, A., Lopez, R., Thompson, J. D., Gibson, T. J., & Higgins, D. G. (2007). Clustal W and Clustal X version 2.0. Bioinformatics (Bioinformatics), 23(21), 2947–2948. https://doi.org/10.1093/bioinformatics/btm404
- Laskowski, R. A., MacArthur, M. W., Moss, D. S., & Thornton, J. M. (1993). PROCHECK: A program to check the stereochemical quality of protein structures. Journal of Applied Crystallography, 26(2), 283–291. https://doi.org/10.1107/S0021889892009944
- Loli, H., Narwal, S., Saun, N., & Gupta, R. (2015). Lipases in Medicine: An Overview. Mini-Reviews in Medicinal Chemistry, 15(14), 1209–1216. https://doi.org/10.2174/1389557515666150709122722
- Lu, S., Wang, J., Chitsaz, F., Derbyshire, M. K., Geer, R. C., Gonzales, N. R., Gwadz, M., Hurwitz, D. I., Marchler, G. H., Song, J. S., Thanki, N., Yamashita, R. A., Yang, M., Zhang, D., Zheng, C., Lanczycki, C. J., & Marchler-Bauer, A. (2020). CDD/SPARCLE: The conserved domain database in 2020. Nucleic Acids Research, 48(D1), D265–D268. https://doi.org/10.1093/nar/gkz991
- Mala, J. G. S., & Takeuchi, S. (2008). Understanding structural features of microbial lipases - An overview. Analytical Chemistry Insights, 3(3), ACI.S551–19. https://doi.org/10.4137/ACI.S551
- Martínez-Corona, R., Vázquez Marrufo, G., Cortés Penagos, C., Madrigal-Pérez, L. A., & González-Hernández, J. C. (2020). Bioinformatic characterization of the extracellular lipases from Kluyveromyces marxianus. Yeast, 37(1), 149–162. https://doi.org/10.1002/yea.3449
- Mehta, A., Bodh, U., & Gupta, R. (2017). Fungal lipases: A review. Journal of Biotech Research, 8(1), 58–77.
- Menichelli, C., Gascuel, O., & Bréhélin, L. (2018). Improving pairwise comparison of protein sequences with domain co-occurrence. PLOS Computational Biology, 14(1), e1005889–23. https://doi.org/10.1371/journal.pcbi.1005889
- Mitchell, A. L., Attwood, T. K., Babbitt, P. C., Blum, M., Bork, P., Bridge, A., Brown, S. D., Chang, H.-Y., El-Gebali, S., Fraser, M. I., Gough, J., Haft, D. R., Huang, H., Letunic, I., Lopez, R., Luciani, A., Madeira, F., Marchler-Bauer, A., Mi, H., … Finn, R. D. (2019). InterPro in 2019: Improving coverage, classification and access to protein sequence annotations. Nucleic Acids Research, 47(D1), D351–D360. https://doi.org/10.1093/nar/gky1100
- Panda, A. K., Bisht, S. P. S., Panigrahi, A. K., De Mandal, S., & Senthil Kumar, N. (2016). Cloning and In Silico Analysis of a High-Temperature Inducible Lipase from Brevibacillus. Arabian Journal for Science and Engineering, 41(6), 2159–2170. https://doi.org/10.1007/s13369-015-1975-4
- Pleiss, J., Fischer, M., Peiker, M., Thiele, C., & Schmid, R. D. (2000). Lipase engineering database: Understanding and exploiting sequence-structure-function relationships. Journal of Molecular Catalysis - B Enzymatic, 10(5), 491-508. https://doi.org/10.1016/S1381-1177(00)00092-8
- Rajeswari, T., Palaniswamy, M., Venil, C. K., Nathiya, K., & Joyruth, P. (2010). Production, partial purification and characterization of lipase from Aspergillus flavus KUF108. Pakistan Journal of Scientific and Industrial Research, 53(5), 258–264.
- Raveendran, S., Parameswaran, B., Ummalyma, S. B., Abraham, A., Mathew, A. K., Madhavan, A., Rebello, S., & Pandey, A. (2018). Applications of microbial enzymes in food industry. Food Technology and Biotechnology, 56(1), 16–30. https://doi.org/10.17113/ftb.56.01.18.5491
- Rosano, G. L., & Ceccarelli, E. A. (2014). Recombinant protein expression in Escherichia coli: Advances and challenges. Frontiers in Microbiology, 5, 1-17. https://doi.org/10.3389/fmicb.2014.00172
- Rubio, M. V., Terrasan, C. R. F., Contesini, F. J., Zubieta, M. P., Gerhardt, J. A., Oliveira, L. C., de Souza Schmidt Gonçalves, A. E., Almeida, F., Smith, B. J., de Souza, G. H. M. F., Dias, A. H. S., Skaf, M., & Damasio, A. (2019). Redesigning N-glycosylation sites in a GH3 β-xylosidase improves the enzymatic efficiency. Biotechnology for Biofuels, 12(1), 1–14. https://doi.org/10.1186/s13068-019-1609-2
- S., Kumar, D. (2015). Fungal lipase production by solid state fermentation – An overview. Journal of Analytical & Bioanalytical Techniques, 06(01), 1-6. https://doi.org/10.4172/2155-9872.1000230
- Sanchez, S., & Demain, A. (2012). Special issue on the production of recombinant proteins. Biotechnology Advances, 30(5), 1100–1101. https://doi.org/10.1016/j.biotechadv.2011.12.004
- Sandi, J., Mata-Araya, I., & Aguilar, F. (2020). Diversity of Lipase-Producing Microorganisms from Tropical Oilseeds Elaeis guineensis, Ricinus communis, and Jatropha curcas L. from Costa Rica. Current Microbiology, 77(6), 943–952. https://doi.org/10.1007/s00284-020-01886-8
- Santhoshkumar, R., & Yusuf, A. (2020). In silico structural modeling and analysis of physicochemical properties of curcumin synthase (CURS1, CURS2, and CURS3) proteins of Curcuma longa. Journal of Genetic Engineering and Biotechnology, 18(1), 1-9. https://doi.org/10.1186/s43141-020-00041-x
- Schmid, R. D. (1998). Lipases: Interfacial enzymes with attractive applications. Angewandte Chemie International.
- Schrödinger, L. (2010). The PyMOL molecular graphics system, version 1.3r1. Schrödinger LLC, 37, 362–387. https://doi.org/10.1038/hr.2014.17
- Sharma, S., & Kanwar, S. S. (2014). Organic solvent tolerant lipases and applications. The Scientific World Journal, 2014, 1–15. https://doi.org/10.1155/2014/625258
- Sharma, A., Meena, K. R., & Kanwar, S. S. (2018). Molecular characterization and bioinformatics studies of a lipase from Bacillus thermoamylovorans BHK67. International Journal of Biological Macromolecules, 107(Pt B), 2131–2140. https://doi.org/10.1016/j.ijbiomac.2017.10.092
- Sharma, A. K., Sharma, V., & Saxena, J (2016). A review on applications of microbial lipases. International Journal of Biotech Trends and Technology, 19(1), 1–5. https://doi.org/10.14445/22490183/IJBTT-V19P601
- Shreya, Sharma, A. K., Sharma, V., & Saxena, J. (2018). Optimization of culture conditions for extracellular fungal lipase production by submerged fermentation process. Plant Science Today, 5(3), 135–141. https://doi.org/10.14719/pst.2018.5.3.399
- Singh, R., Gupta, N., Goswami, V. K., & Gupta, R. (2006). A simple activity staining protocol for lipases and esterases. Applied Microbiology and Biotechnology, 70(6), 679–682. https://doi.org/10.1007/s00253-005-0138-z
- Singh, R., Kumar, M., Mittal, A., & Mehta, P. K. (2016). Microbial enzymes: Industrial progress in 21st century. 3 Biotech, 6(2), 1–15. https://doi.org/10.1007/s13205-016-0485-8
- Steentoft, C., Vakhrushev, S. Y., Joshi, H. J., Kong, Y., Vester-Christensen, M. B., Schjoldager, K. T.-B G., Lavrsen, K., Dabelsteen, S., Pedersen, N. B., Marcos-Silva, L., Gupta, R., Bennett, E. P., Mandel, U., Brunak, S., Wandall, H. H., Levery, S. B., & Clausen, H. (2013). Precision mapping of the human O-GalNAc glycoproteome through SimpleCell technology. The EMBO Journal, 32(10), 1478–1488. https://doi.org/10.1038/emboj.2013.79
- Suplatov, D., Voevodin, V., & Švedas, V. (2015). Robust enzyme design: Bioinformatic tools for improved protein stability. Biotechnology Journal, 10(3), 344–355. https://doi.org/10.1002/biot.201400150
- Tan, Y., & Miller, K. J. (1992). Cloning, expression, and nucleotide sequence of a lipase gene from Pseudomonas fluorescens B52. Applied and Environmental Microbiology, 58(4), 1402–1407. https://doi.org/10.1128/aem.58.4.1402-1407.1992
- Tang, L., Su, M., Yan, J., Xie, S., & Zhang, W. (2015). Lid hinge region of Penicillium expansum lipase affects enzyme activity and interfacial activation. Process Biochemistry, 50(8), 1218–1223. https://doi.org/10.1016/j.procbio.2015.04.022
- Toida, J., Fukuzawa, M., Kobayashi, G., Ito, K., & Sekiguchi, J. (2000). Cloning and sequencing of the triacylglycerol lipase gene of Aspergillus oryzae and its expression in Escherichia coli. FEMS Microbiology Letters, 189(2), 159–164. https://doi.org/10.1016/S0378-1097(00)00271-8
- Toscano, L., Montero, G., Stoytcheva, M., Gochev, V., Cervantes, L., Campbell, H., Zlatev, R., Valdez, B., Pérez, C., & Gil-Samaniego, M. (2013). Lipase production through solid-state fermentation using agro-industrial residues as substrates and newly isolated fungal strains. Biotechnology & Biotechnological Equipment, 27(5), 4074–4077. https://doi.org/10.5504/BBEQ.2012.0145
- Tran, D. T., & Ten Hagen, K. G. (2013). Mucin-type o-glycosylation during development. Journal of Biological Chemistry, 288(10), 6921–6929. https://doi.org/10.1074/jbc.R112.418558
- Umesha, S., Manukumar, H. M., & Raghava, S. (2016). A rapid method for isolation of genomic DNA from food-borne fungal pathogens. 3 Biotech, 6(2), 1–9. https://doi.org/10.1007/s13205-016-0436-4
- Waterhouse, A., Bertoni, M., Bienert, S., Studer, G., Tauriello, G., Gumienny, R., Heer, F. T., de Beer, T. A. P., Rempfer, C., Bordoli, L., Lepore, R., & Schwede, T. (2018). SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Research, 46(W1), W296–W303. https://doi.org/10.1093/nar/gky427
- Weerapana, E., & Imperiali, B. (2006). Asparagine-linked protein glycosylation: From eukaryotic to prokaryotic systems. Glycobiology, 16(6), 91R–101. https://doi.org/10.1093/glycob/cwj099
- Wohlfarth, S., Hoesche, C., Strunk, C., & Winkler, U. K. (1992). Molecular genetics of the extracellular lipase of pseudomonas aeruginosa PAO1. Journal of General Microbiology, 138(7), 1325–1335. https://doi.org/10.1099/00221287-138-7-1325
- Yang, M., Yu, X. W., Zheng, H., Sha, C., Zhao, C., Qian, M., & Xu, Y. (2015). Role of N-linked glycosylation in the secretion and enzymatic properties of Rhizopus chinensis lipase expressed in Pichia pastoris. Microbial Cell Factories, 14(1), 1–14. https://doi.org/10.1186/s12934-015-0225-5
- Yuan, B. H., Cai, Y. J., Liao, X. R., Yun, L. H., Zhang, F., & Zhang, D. B. (2010). Isolation and identification of a cold-adapted lipase producing strain from decayed seeds of Ginkgo biloba L. and characterization of the lipase. African Journal of Biotechnology, 9(18), 2661–2667. https://doi.org/10.5897/AJB2010.000-3085
- Yu, X. W., Tan, N. J., Xiao, R., & Xu, Y. (2012). Engineering a disulfide bond in the lid hinge region of Rhizopus chinensis lipase: Increased thermostability and altered acyl chain length specificity. PLoS One, 7(10), e46388–7. https://doi.org/10.1371/journal.pone.0046388
- Yu, X. W., Zhu, S. S., Xiao, R., & Xu, Y. (2014). Conversion of a Rhizopus chinensis lipase into an esterase by lid swapping. Journal of Lipid Research, 55(6), 1044–1051. https://doi.org/10.1194/jlr.M043950
- Zhan, N., & An, Z. (2010). Manual of Industrial Microbiology and Biotechnology. Heterologous Protein Expression in Yeasts and Filamentous Fungi, 3(11), 145–156.
- Zhang, B., Li, J., & Lü, Q. (2018). Prediction of 8-state protein secondary structures by a novel deep learning architecture. BMC Bioinformatics, 19(1), 1–13. https://doi.org/10.1186/s12859-018-2280-5
- Zou, X., Pham, T. K., Wright, P. C., & Noirel, J. (2012). Bioinformatic study of the relationship between protein regulation and sequence properties. Genomics, 100(4), 240–244. https://doi.org/10.1016/j.ygeno.2012.07.003