
Abstract
Background
Hermansky-Pudlak syndrome (HPS) is a rare inherited platelet disorder characterized by bleeding diathesis, oculocutaneous albinism (OCA) and a myriad of often-serious clinical complications.
Methods
We established the clinical and laboratory phenotype and genotype of six unrelated pedigrees comprising ten patients with clinical suspicion of HPS; including platelet aggregation, flow cytometry, platelet dense granule content, electron microscopy and high-throughput sequencing (HTS).
Results
The clinical presentation showed significant heterogeneity and no clear phenotype-genotype correlations. HTS revealed two known and three novel disease-causing variants. The Spanish patients carried a homozygous p.Pro685Leufs17* deletion (n = 2) in HPS4, or the novel p.Arg822* homozygous variant (n = 1) in HPS3. In the case of two Turkish sisters, a novel missense homozygous HPS4 variant (p.Leu91Pro) was found. In two Portuguese families, genetic studies confirmed a previously reported nonsense variant (p.Gln103*) in DTNBP1 in three patients and a novel duplication (p.Leu22Argfs*33) in HPS6 in two unrelated patients.
Conclusions
Our findings expand the mutational spectrum of HPS, which may help in investigating phenotype-genotype relationships and assist genetic counselling for affected individuals. This approach is a proof of principle that HTS can be considered and used in the first-line diagnosis of patients with biological and clinical manifestations suggestive of HPS.
We established the relationships between the clinical and laboratory phenotype and genotype of six unrelated pedigrees comprising ten patients with clinical suspicion of HPS.
Molecular analysis is useful in confirming the diagnosis and may offer some prognostic information that will aid in optimizing monitoring and surveillance for early detection of end-organ damage.
This approach is a proof of principle that HTS can be considered and used in the first-line diagnosis of patients with biological and clinical manifestations suggestive of HPS.
Key messages
1. Introduction
Hermansky-Pudlak syndrome (HPS) is a rare multisystemic recessive congenital disorder characterized by bleeding diathesis and oculocutaneous albinism (OCA) that is sometimes associated with serious complications, such as immunodeficiency, granulomatous colitis, and/or pulmonary fibrosis [Citation1]. HPS arises from defects in ten genes (HPS1, AP3B1, AP3D1, HPS3, HPS4, HPS5, HPS6, DTNBP1, BLOC1S3, BLOC1S6) that encode proteins involved in the biogenesis and/or function of intracellular organelles known as lysosome-related organelles (LROs), which interact with each other in complexes called BLOCS (biogenesis of lysosome-related organelle complexes) [Citation1]. Each gene has been associated with a clinical subtype [Citation2]. Genetic variants of the HPS1 and HPS4 genes, which define the HPS-1 and HPS-4 subtypes, respectively, cause the most common and most severe clinical forms of HPS [Citation2,Citation3]. As the HPS1 and HPS4 proteins are a part of BLOC3 complexes, which regulate the biogenesis and/or function of pneumocyte lamellar bodies, they are responsible for the pulmonary complications observed in these subtypes [Citation3]. The HPS-2 subtype is caused by genetic variants in the AP3B1 gene and is the only one to cause immunodeficiency [Citation2]. The HPS3 protein associates with HPS5 and HPS6 proteins to form the BLOC2 complex and their deficiencies manifest as a milder phenotype (variable hypopigmentation and sporadic granulomatous colitis, without pulmonary fibrosis) [Citation4]. Molecular defects affecting subunits of the BLOC1 complex: dysbindin-1 (encoded by DTNBP1 gene), BLOC1S3 (encoded by BLOC1S3 gene) and BLOC1S6 (pallidin) (encoded by BLOC1S6 gene) are very infrequent; they result, respectively, in the HPS-7, HPS-8 and HPS-9 subtypes that represent mild forms of HPS with limited clinical manifestations [Citation2]. The AP3D1 gene has recently been proposed as being an HPS-10 subtype and genetic variants in this gene associate to severe neurological disorders with immunodeficiency and albinism [Citation5].
The suspicion of HPS is based on clinical features. When bleeding diathesis is present, the diagnosis relies on the identification of clinical manifestations, and ideally on the combination of platelet function tests as well as on the verification of reduced platelet δ-granules content [Citation2,Citation6]. In this disorder, a large number of candidate genes (140 exons) and the lack of meaningful genotype-phenotype correlations complicate the accurate molecular diagnosis of patients. High-throughput sequencing (HTS) enables the molecular testing of candidate genes simultaneously and is currently implemented in this setting [Citation7–11].
In this study, we investigated six unrelated pedigrees from Spain, Turkey and Portugal who presented with OCA and bleeding diathesis. We illustrate the proof of principle of the utility of HTS sequencing to complement the phenotypic and genotypic characterization of patients with suspected HPS and expand the mutational spectrum of this syndrome.
2. Methods
2.1. Patients
We studied ten patients from six families (two Spanish, one Turkish and three Portuguese) ().
Table 1. Clinical features, laboratory findings and molecular characterization of the six HPS families studied.
2.2. Material and methods
Venous blood was drawn from each patient into commercial 7.5% K3 EDTA tubes for complete blood count, flow cytometry [FC] and DNA isolation, and into buffered 3.2% sodium citrate for platelet studies. The release and expression on the platelet surface of CD63 present in dense granules and lysosomal membranes were evaluated by FC (Becton Dickinson, San Jose, CA) after the activation of platelets with 25 μM TRAP or vehicle, using an anti CD63*PE antibody [Citation12,Citation13]. The ability of platelets to take up and store radioactive serotonin ([14C]‐5‐hydroxytryptamine; [14C]‐5HT) in dense granules was tested according to standard procedures. The number of dense granules per platelet was estimated using electron microscopy (EM) on whole mounts. These methods have been described in detail elsewhere [Citation12,Citation13]. Aggregation studies were performed using a Lumi-aggregometer (Menarini Diagnostics, Florence, Italy) to measure ATP secretion by luminescence while aggregation was measured by light transmission (LTA) as reported elsewhere [Citation13,Citation14]. Disease-causing variants in these six families were identified using HTS technology. The Spanish and Turkish patients underwent molecular screening for variants in 72 candidate genes related to inherited platelet disorders (IPDs), using an Illumina HTS platform (MiSeq, Illumina, San Diego, CA) according to Nextera sequencing design [Citation9,Citation15–17]. This panel not only include genes involved in transcription factors (RUNX1, GATA1, MPL), agonist platelet receptors (GP1BA, ITGB3 or P2RY12), cytoskeletal assembly and structural proteins (MYH9, WAS, TUBB1) or related in the signal transduction (RASGRP2 or TBXAS1), but also in the genes, which codified proteins related to platelet granules (LYST, NBEAL2), even, 9 genes which cause HPS” [Citation9]. In the Portuguese patients, HTS analysis was performed with an Ion Torrent platform. DNA libraries were prepared based on an amplicon-based approach using Ion AmpliSeq (Hematology Research Panel, Unidade de Genética Molecular, Centro de Genética Médica Dr Jacinto Magalhães, Centro Hospitalar Universitário do Porto, Porto, Portugal), followed by sequencing in the Ion S5 sequencer (Thermo Fisher Scientific, Life Technologies, Madrid, Spain), and conventional analysis of data was performed in all cases [Citation9,Citation15–17]. Briefly, data were filtered according to the sequence quality and coverage, severity of the consequence, minor allelic frequencies less than 0.05. Synonymous and intronic variants were disregarded. Public databases and in silico platforms provides extensive information. Finally, variants were classified according to the American College of Medical Genetics and Genomics recommendations [Citation9,Citation15–18]. The causative genetic variants were also confirmed in the patients and screened in the available family members by Sanger sequencing (SS). All samples were obtained after subjects, parents or family members provided informed consent.
3. Results
3.1. Clinical presentation
Patients 1 and 2 (P1 and P2) are two sisters (with 13- and 16-years of age, respectively), born to non-related healthy parents (Family 1). At birth, they presented with OCA, which was confirmed by skin biopsy. The peripheral blood smears did not show morphological alterations, ruling out other diagnoses such as Chediak-Higashi syndrome (CHS). Their lifelong bleeding tendency consisted of epistaxis, ecchymosis and menorrhagia since menarche; ISTH-BAT bleeding scores (BSs) were 11 and 3, respectively (). P1 had several episodes of gastrointestinal (GI) bleeding, which was attributed to Crohn’s disease, requiring surgery and red blood cell transfusion.
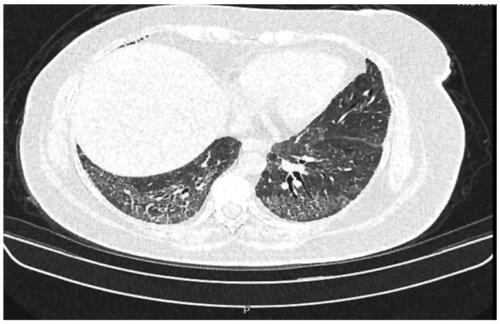
Patients 3 and 4 (P3 and P4) are 40- and 48-year-old Turkish sisters born to healthy consanguineous parents (Family 2). They are the only affected individuals among their family of 7 siblings, and there is no known family history of the disease (). Since early childhood, they presented with easy bruising and recurrent epistaxis. Their BSs were 7 and 6, respectively. Examinations revealed OCA and nystagmus. At the age of 22, P3 had severe rectal bleeding. A total colonoscopy revealed signs of colitis. She underwent colectomy combined with ileostomy due to persistent serious GI haemorrhage. Pathological examination of the colon was reported as non-granulomatous colitis. In contrast, her older sister (P4) exhibited dyspnea and shortness of breath since 30 years of age. High-resolution chest computerized axial tomography (CAT) scans showed diffuse bilateral pulmonary fibrosis (). No platelet functional assays could be performed.
Figure 1. High-resolution chest computerized axial tomography (CAT), showing diffuse bilateral pulmonary fibrosis in P4.
Patient 5 (P5) is a 25-year-old Spanish male born to non-related parents (Family 3). He had a history of OCA, with ocular findings revealing mild translucent irides and patched hypopigmentation choroids related to ocular albinism, strabismus and a BS of 5 (). At 24-years of age, he presented with self-limited GI bleeding and required hospital admission due to acute haemorrhage. Colonoscopy revealed only angiodysplasia.
Patients 6 and 7 (P6 and P7) were born to non-related parents from Portugal. Their parents and nine siblings were healthy and their family (Family 4) had no significant history of the disease. P6 was a regular blood donor; he referred frequent epistaxis, easy bruising, bleeding from minor wounds and after teeth extractions (BS of 6). He experienced no significant hemorrhagic events following two inguinal hernia surgeries with prophylactic administration of 1-desamino-8-D-arginine vasopressin (DDAVP). He has blond hair (pale straw colour), retinal hypopigmentation, iris transillumination, horizontal nystagmus, low visual acuity (VOD 0.2; VOS 0.2) and photophobia. His older sister (P7) was also diagnosed with HPS when she was 56 years old. She also had low visual acuity (VOD 0.16; VOS 0.16), fundus hypopigmentation, foveal hypoplasia and photophobia, but her rotary nystagmus was more modest. Her OCA was masked because she used dark brown hair-colouring products. She also complained from cutaneous bruises, epistaxis and spontaneous bleeding from gums and following tooth extraction (BS of 8). She gave birth to 5 children, some of them at home, without medical assistance; one died during delivery and another shortly thereafter. Two daughters suffered from bilateral acoustic neurinoma and neurofibromatosis (paternal inheritance) and died at the ages of 23 and 29. At present, only one of her descendants, who is 35 years old, remains alive.
Patient 8 (P8) is an 18-year-old Portuguese woman, an only child born to non-related healthy parents (Family 5). At 4 months of age, she was suspected to be blind and was investigated by Pediatric Ophthalmology. She had OCA, with blond hair, retinal hypopigmentation and iris transillumination; in addition, she showed horizontal nystagmus, photophobia, deep visual deficiency (VOD: 20/200; VOE: 20/160) and exotropia with compensatory torticollis. Strabismus surgery was performed with prophylactic administration of DDAVP, without any bleeding complications. Her parents noticed frequent cutaneous bruises since the age of 18 months. She also bled from minor wounds and had excessive bleeding on tooth extractions. The most relevant hemorrhagic events occurred since menarche with severe menorrhagia, requiring oral hormonal therapy (BS of 7).
Patients 9 and 10 (P9 and P10) were born to first-cousin parents from Portugal (Family 6). The youngest son (P9) was studied at 5 months of age due to OCA (fair hair and skin). Ophthalmic findings included iris hypopigmentation and transillumination, foveal hypoplasia, horizontal and torsional nystagmus and reduced visual activity. In addition, he had easy cutaneous bruises, bleeding from minor wounds and episodes of epistaxis controlled with packing and antifibrinolytic drugs (BS of 6). The oldest daughter (P10) was studied at age four, due to easy cutaneous bruises, bleeding from minor wounds, and episodic epistaxis and petechiae. She also presented skin, hair and iris hypopigmentation, iris transillumination, impaired binocular visual acuity and torsional nystagmus. She was diagnosed with epilepsy and mild developmental delay. A clinical suspicion of HPS was made at 13 years of age, due to menorrhagia requiring hormonal therapy and bleeding gums (BS of 8).
3.2. Platelet studies
Except for the Turkish family, in which HPS was suspected only from the clinical features, the platelets of other patients were functionally analyzed ().
In the Spanish patients (P1, P2, and P5) 14C-5HT uptake was reduced by approximately 25% in P1 and P2 and by 70% in P5. Similarly, TRAP-induced CD63 release was severely impaired (≈70%) in patients from Family 1 and was undetectable in P5 (). Moreover, whole-mount EM experiments showed a severe reduction of δ-granules in platelets from P1 and P2 (Supplementary Figure 1), and in P5 (not shown). In the Portuguese patients (P6-P10) the ATP release was absent upon stimulation with thrombin (1 U), ADP (10 μM), collagen (2 μg/mL) and arachidonic acid (1 mM), in accordance with a defect in the platelet content of δ-granules. In addition, TRAP-induced CD63 release was severely impaired (58% and 37%) in patients from Family 4 ().
3.3. Genotyping
HTS was carried out in all families. In Family 1 (P1 and P2), a previously reported 1-bp deletion (c.2054delC) in exon 13 of HPS4 (NM_022081), which produces a frameshift with a premature stop codon (p.Pro685Leufs*17) [Citation3], was detected in a homozygous state, while their parents were asymptomatic heterozygotes for this genetic variant. In Family 2 (P3 and P4), the molecular analysis demonstrated a novel missense variant in exon 4 of the HPS4 gene (c.272T > C; p.Leu91Pro), in a homozygous state, while the parents and the other five family members were heterozygous carriers of this variant. In Family 3, a novel nonsense variant (c.2464C > T; p.Arg822*) in exon 13 of HPS3 (NM_032383) was identified in the propositus (P5) and in his asymptomatic mother in a heterozygous state. In Family 4 (P6 and P7) and Family 5 (P8), a reported nonsense homozygous variant (c.307C > T; p.Gln103*) in exon 5 of DTNBP1 (NM_183040) was detected. In the last family (P9 and P10), a novel homozygous duplication (c.60_64dup) resulting for a frameshift variant (p.Leu22Argfs*33) was detected in exon 1 of the HPS6 (NM_024747).
4. Discussion
HPS belongs to a genetically heterogeneous group of rare autosomal recessive disorders with an estimated worldwide prevalence of 1–9 per 1,000,000 individuals (www.orpha.net) that share oculocutaneous albinism and platelet storage disease. Ocular findings include reduced iris pigment with iris transillumination, reduced retinal pigment, foveal hypoplasia with significant reduction in visual acuity, nystagmus; additionally, some individuals develop pulmonary fibrosis, granulomatous colitis, or immunodeficiency. In humans, ten causative genes involved in the formation, transport or fusion of intracellular vesicles of lysosomal lineage can be affected. The diagnosis of HPS is established by clinical features and platelet phenotyping that show a reduction in dense granules [Citation1–5]. The identification of biallelic pathogenic variants in AP3B1, AP3D1, BLOC1S3, BLOC1S6, DTNBP1, HPS1, HPS3, HPS4, HPS5, or HPS6 confirms the diagnosis and distinguish this disease from other genetic disorders characterized by hypopigmentation, immunodeficiency and bleeding, including CHS or Griscelli Syndrome (GS) [Citation1–5]. However, HPS is a heterogeneous genetic disorder and a molecular diagnosis is difficult to reach [Citation5,Citation7,Citation13]. The most commonly reported subtypes are HPS-1, HPS-3, and HPS-4. The HPS1 gene was first screened by SS in patients with severe phenotypes and in Puerto Rican patients [Citation4,Citation18], while founder mutations in HPS3 were analyzed in patients with mild phenotypes and in Ashkenazi Jewish patients [Citation2,Citation19]. In addition, the prevalence of HPS-4, one of the severe forms of the disease, is relatively high in different populations, such as Japanese, or inhabitants of Mediterranean regions, among others [Citation1,Citation4,Citation19,Citation20]. Few variants have been reported in HPS6 and DTNBP1 resulting in HPS-6 and HPS-7, respectively, most of them being diagnosed in European Caucasian patients [Citation21–27]. Although some studies have suggested a genotype-phenotype association in this disease [Citation19,Citation22,Citation23], to date, correlations between specific HPS-causing variants in any one gene and specific clinical presentations are not convincing [Citation23]. When using serial single-gene testing strategy, it has been suggested a sequence analysis of HPS3, HPS5 and/or HPS6 in mildly affected individuals, of HPS1 and HPS4 in severely affected individuals, or AP3B1 in affected individuals with neutropenia or infections [Citation28]. In the era of HTS, several genes can be analyzed simultaneously, which is very helpful for detecting genetic variants in heterogeneous disorders such as HPS [Citation7–9].
The HTS approach allowed us to identify the causative variants in these six families (). In Family 1, we found the same single-base-pair deletion (c.2054delC) that had been previously detected in HPS4 in an Asian patient with pulmonary fibrosis and no history of albinism or of excessive bleeding [Citation3]. However, our HPS-4 phenotype was different and although no respiratory manifestations were present in these two patients, we cannot rule out pulmonary fibrosis to develop at later age, since symptoms usually begin in the thirties and are fatal within a decade (). Pulmonary fibrosis is most frequently found in the HPS-1 type (78% of HPS-1 patients vs. 14% of HPS-4 patients) [Citation29], and annual pulmonary function testing and a CAT examination of the chest is recommended for young adults over 20 years of age with pathogenic variants in HPS1, HPS4 and AP3B1 [Citation20,Citation28].
In Family 2, a novel missense variant (c.272T > C; p.Leu91Pro) was found in the HPS4 gene. To date only 15 variants in 21 patients with HPS-4 had been reported in the literature [Citation29]; these are mainly nonsense (eight), and other variants are frameshift (four), missense (one), splice site (one), and a large in-frame duplication (one). In this family, one of the affected members suffered from interstitial pneumonia, while the other presented colitis, highlighting the extremely heterogeneous presentation of clinical manifestations even within the same family members. In this case, it was not possible to perform functional platelet tests, and HTS analyses confirmed the clinical suspicion of HPS.
In Family 3, a novel nonsense variant (c.2464C > T; pArg822*) in the HPS3 gene was found only in the heterozygous state both in P5 and in his asymptomatic mother. In this context, other mechanisms such as haploinsufficiency of chromosomes, epigenetics, and also other regulatory genes, may contribute to the patient’s phenotype, and may explain how the heterozygous variant was the causative variant of this HPS. Additionally, the inability to detect a second variant in the propositus may be a consequence of the limitations of HTS due to the incapacity to detect variants that are in deep introns, gross abnormalities, rearrangements, large heterozygous deletions or duplications that are not usually detected by sequencing-based technology and routinely-used bioinformatic approaches [Citation30]. In this case, the HPS3 expression analysis at the mRNA level, or the use of other genomic approaches such as multiplex ligation-dependent probe amplification (MLPA) or array comparative genome hybridization (aCGH), might enable the identification of these HTS-resilient variants [Citation31]. Interestingly, the genetic analysis carried out in the two Portuguese families (Family 4 and Family 5), who were not related but from the same geographical area, revealed a nonsense variant (c.307C > T, p.Gln103*) in exon 5 of the DTNBP1 that was described for the first time in a Portuguese woman [Citation21], suggesting a possible founder effect in the Portuguese population. Genetic variants affecting DTNBP1 are usually present in families with consanguineous parents [Citation21,Citation28]. In contrast, in these two families and in a Paraguayan boy, no consanguinity was apparent [Citation26]. In addition, the previously reported patients diagnosed with HPS-7 showed mild shortness of breath on exertion and reduced lung compliance but otherwise normal pulmonary function and CAT chest scans, and delayed motor and language development [Citation21,Citation26]. In contrast, although there was no history of pulmonary fibrosis in affected members from the two families carrying this mutation, both P6 and P7 had pulmonary hypertension, with pulmonary arterial pressure estimated to be of 26–27 mmHg through transthoracic echocardiography (mean value of 12–16 mmHg).
Individuals with pathogenic variants in HPS3, HPS5, or HPS6 are BLOC-2 deficient and generally have mild symptoms: patients can present with minimal hypopigmentation, better visual acuity than in other forms, bleeding is also mild, and pulmonary involvement has not been observed. Therefore, individuals with HPS6 variants may go undiagnosed for decades [Citation24,Citation25]. However, one of the affected siblings with variants in HPS6 exhibited neurodevelopmental delay and generalized seizures, features not commonly seen in individuals with BLOC2 deficient-HPS. Previous reports of other cases have shown a variety of variants in the single exon of the HPS6 gene, including frameshift, missense, and nonsense variants as well as a deletion spanning the entire HPS6 genomic region [Citation24,Citation25]. In Family 6, a novel duplication that led to a frameshift was detected (p.Leu22Argfs*33).
5. Conclusions
Hermansky-Pudlak syndrome is a rare genetic disorder presenting with hypopigmentation, bleeding diathesis, and organ dysfunction secondary to the accumulation of ceroid-like material and resultant tissue damage. Variants in one of the ten currently known genes can result in this autosomal recessive disorder. Molecular analysis is useful in confirming the diagnosis and may offer some prognostic information that will aid in optimizing monitoring and surveillance for early detection of end-organ damage. Given the poor genotype-phenotype correlation, a sequential assessment of the most likely genes can be lengthy and expensive. A target gene panel analyzed by HTS can be considered and could be used in the first-line diagnosis of patients with biological and clinical manifestations suggestive of HPS. Our findings also expand the mutational spectrum of HPS, which may help in investigating phenotype-genotype relationships and assist genetic counselling for affected individuals.
Supplemental Material
Download MS Word (259.2 KB)Acknowledgements
We are grateful to Irene Rodríguez and Sara González for their help isolating DNA, and Dr Phil Mason for his help with other technical aspects.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Masliah-Planchon J, Darnige L, Bellucci S. Molecular determinants of platelet delta storage pool deficiencies: an update. Br J Haematol. 2013;160:5–11.
- Sánchez-Guiu I, Torregrosa JM, Velasco F, et al. Hermansky-Pudlak syndrome. Overview of clinical and molecular features and case report of a new HPS-1 variant. Hamostaseologie. 2014;34:301–309.
- Bachli EB, Brack T, Eppler E, et al. Hermansky-Pudlak syndrome type 4 in a patient from Sri-Lanka with pulmonary fibrosis. Am J Med Genet. 2004;127:201–207.
- Carmona-Rivera C, Golas G, Hess RA, et al. Clinical, molecular and cellular features of non-Puerto Rican Hermansky-Pudlak syndrome patients of Hispanic descent. J Invest Dermatol. 2011;131:2394–2400.
- Ammann S, Schulz A, Krägeloh-Mann I, et al. Mutations in AP3D1 associated with immunodeficiency and seizures define a new type of Hermansky-Pudlak syndrome. Blood. 2016;127:997–1006.
- Bastida JM, Hernández-Rivas JM, González-Porras JR. Novel approaches for diagnosing inherited platelet disorders. Med Clin. 2017;148:71–77.
- Simeoni I, Stephens JC, Hu F, et al. A high-throughput sequencing test for diagnosing inherited bleeding, thrombotic, and platelet disorders. Blood. 2016;127:2791–2803.
- Bastida JM, Palma-Barqueros V, Lozano ML, et al. A modern approach to the molecular diagnosis of inherited bleeding disorders. J Mol Genet Med. 2018;12:322.
- Bastida JM, Lozano ML, Benito R, et al. Introducing high-throughput sequencing into mainstream genetic diagnosis practice in inherited platelet disorders. Haematologica. 2018;103:148–162.
- Bastida JM, Del Rey M, Lozano ML, et al. Design and application of a 23-gene panel by next-generation sequencing for inherited coagulation bleeding disorders. Haemophilia. 2016;22:590–597.
- Bastida JM, González-Porras JR, Jiménez C, et al. Application of a molecular diagnostic algorithm for haemophilia A and B using next-generation sequencing of entire F8, F9 and VWF genes. Thromb Haemost. 2017;117:66–74.
- González-Conejero R, Rivera J, Escolar G, et al. Molecular, ultrastructural and functional characterization of a Spanish family with Hermansky-Pudlak syndrome: role of insC974 in platelet function and clinical relevance. Br J Haematol. 2003;123:132–138.
- Sánchez-Guiu I, Antón AI, Padilla J, et al. Functional and molecular characterization of inherited platelet disorders in the Iberian Peninsula: results from a collaborative study. Orphanet J Rare Dis. 2014;9:213.
- King SM, McNamee RA, Houng AK, et al. Platelet dense-granule secretion plays a critical role in thrombosis and subsequent vascular remodeling in atherosclerotic mice. Circulation. 2009;120:785–791.
- Lozano ML, Cook A, Bastida JM, et al. Novel mutations in RASGRP2 encoding for CalDAG-GEFI abrogate Rap1 activation causing platelet dysfunction. Blood. 2016;128:1282–1289.
- Bastida JM, Del Rey M, Revilla N, et al. Wiskott-Aldrich syndrome in a child presenting with macrothrombocytopenia. Platelets. 2017;28:417–420.
- Bastida JM, Benito R, Janusz K, et al. Two novel variants of the ABCG5 gene cause xanthelasmas and macrothrombocytopenia: a brief review of hematologic abnormalities of sitosterolemia. J Thromb Haemost. 2017;15:1859–1866.
- Bastida JM, Benito R, Lozano ML, et al. Molecular diagnosis of inherited coagulation and bleeding disorders. Semin Thromb Hemost. 2019;1–17.
- Huizing M, Anikster Y, Fitzpatrick DL, et al. Hermansky-Pudlak syndrome type 3 in Ashkenazi Jews and other non-Puerto Rican patients with hypopigmentation and platelet storage-pool deficiency. Am J Hum Genet. 2001;69:1022–1032.
- Seward SL, Jr, Gahl WA. Hermansky-Pudlak syndrome: health care throughout life. Pediatrics. 2013;132:153–160.
- Li W, Zhang Q, Oiso N, et al. Hermansky-Pudlak syndrome type 7 (HPS-7) results from mutant dysbindin, a member of the biogenesis of lysosome-related organelles complex 1 (BLOC-1). Nat Genet. 2003;35:84–89.
- Loredana Asztalos M, Schafernak KT, Gray J, et al. Hermansky-Pudlak syndrome: report of two patients with updated genetic classification and management recommendations. Pediatr Dermatol. 2017;34:638–646.
- Miyamichi D, Asahina M, Nakajima J, et al. Novel HPS6 mutations identified by whole-exome sequencing in two Japanese sisters with suspected ocular albinism. J Hum Genet. 2016;61:839–842.
- Andres O, Wiegering V, König EM, et al. A novel two-nucleotide deletion in HPS6 affects mepacrine uptake and platelet dense granule secretion in a family with Hermansky-Pudlak syndrome. Pediatr Blood Cancer. 2017;64. DOI:https://doi.org/10.1002/pbc.26320
- Huizing M, Pederson B, Hess RA, et al. Clinical and cellular characterisation of Hermansky-Pudlak syndrome type 6. J Med Genet. 2009;46:803–810.
- Bryan MM, Tolman NJ, Simon KL, et al. Clinical and molecular phenotyping of a child with Hermansky-Pudlak syndrome-7, an uncommon genetic type of HPS. Mol Genet Metab. 2017;120:378–383.
- Lowe GC, Guiu I, Chapman O, UK GAPP collaborative, et al. Microsatellite markers as a rapid approach for autozygosity mapping in Hermansky-Pudlak syndrome: identification of the second HPS7 mutation in a patient presenting late in life. Thromb Haemost. 2013;109:766–768.
- Huizing M, Malicdan MCV, Gochuico BR, et al. Hermansky-Pudlak Syndrome. [updated 2017 Oct 26]. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2019.
- Sandrock-Lang K, Böckelmann D, Eberl W, et al. A novel nonsense mutation in a patient with Hermansky-Pudlak syndrome type 4. Blood Cells Mol Dis. 2018;69:113–116.
- Weaver JM, Edwards PA. Targeted next-generation sequencing for routine clinical screening of mutations. Genome Med. 2011;3:58.
- Daber R, Sukhadia S, Morrissette JJ. Understanding the limitations of next generation sequencing informatics, an approach to clinical pipeline validation using artificial data sets. Cancer Genet. 2013;206:441–448.