
Abstract
Background
Amyloid light chain (AL) amyloidosis is the most common systemic amyloidosis. The objective of this scoping review was to map the available literature on the diagnosis of AL amyloidosis in China.
Materials and Methods
The published academic papers related to the diagnosis of AL amyloidosis were screened from 1 January 2000 to 15 September 2021. Chinese patients who have suspected AL amyloidosis were included. The included studies were categorized into accuracy studies and descriptive studies based on if the studies supplied the diagnostic accuracy data or not. The information on the diagnostic methods reported by included studies was synthesized.
Results
Forty-three articles were included for the final scoping review, with 31 belonging to descriptive studies and 12 having information on diagnostic accuracy. Although cardiac involvement was second top in Chinese patients with AL amyloidosis, a cardiac biopsy was rare. Next, we found light chain classification and monoclonal (M-) protein identification were essential methods for the diagnosis of AL amyloidosis in China. In addition, some combined tests (e.g. immunohistochemistry and serum free light chain, immunohistochemistry and immunofixation electrophoresis, and serum free light chain and immunofixation electrophoresis) can increase the sensitivity of the diagnosis. Finally, several adjuvant methods (e.g. Imaging, N-terminal-pro hormone BNP, and brain natriuretic peptide test) were important for AL amyloidosis diagnosis.
Conclusion
This scoping review details the characteristics and results of the recently published studies on diagnosing AL Amyloidosis in China. Biopsy is the most important method for AL Amyloidosis diagnosis in China. In addition, combined tests and some adjuvant methods played essential roles in the diagnosis. Further research is required to determine an acceptable and feasible diagnostic algorithm after symptom onset. REGISTRATION: INPLASY2022100096
This scoping review details the characteristics and results of the recently published studies on diagnosing Amyloid light chain (AL) Amyloidosis in China.
Biopsy is the most important method for AL Amyloidosis diagnosis in China.
Combined tests and some adjuvant methods played essential roles in the diagnosis.
KEY MESSAGES
Introduction
Amyloid light chain (AL) amyloidosis is the most common systemic amyloidosis with transthyretin amyloidosis being the next most common. The disease usually presents with impairment of multiorgan function due to extracellular aggregates generated through the deposition of misfolded light chains produced by an indolent plasma cell clone [Citation1]. The estimated incidence of AL amyloidosis was between 5.73 to 12.80 per million persons per year [Citation2–4]. Furthermore, the prevalence of amyloidosis has a geographical distribution difference, and an increasing trend was observed during the last decades [Citation5,Citation6]. Although AL amyloidosis is rare, its prognosis can be poor: the median survival after diagnosis is reported to be between six months to three years after diagnosis [Citation6]. The current treatments of AL amyloidosis focus on eliminating the clonal plasma cell that produces amyloid [Citation7]. Recently, several clinical trials suggested that CD38 monoclonal antibodies (e.g. Daratumumab) appeared to be well tolerated and could improve clinical response to this disease [Citation8–11]. However, although the prognosis of AL amyloidosis improved due to the optimized treatment during the past forty years, the overall survival remains poor [Citation12,Citation13].
Early diagnosis is another strategy to improve the prognosis for AL amyloidosis besides optimal treatment [Citation14]. It was reported that patients with diagnostic delays were found to have a significantly worse prognosis than those without a delay [Citation15]. AL amyloidosis could be involved in almost all organs other than the brain, and the clinical manifestations are diverse, with common symptoms such as feeling fainting, numbness feeling in the hands and feet (peripheral neuropathy), nausea, diarrhea, or constipation, tingling and pain in the wrist, hand and fingers (carpal tunnel syndrome), and easy bruising [Citation16,Citation17]. In addition, as the clinical features of AL amyloidosis are mainly nonspecific, the disease is easily overlooked or misdiagnosed [Citation18,Citation19]. According to an observational study in the USA, a median delay from symptom onset to AL amyloidosis diagnosis was 2.7 years [Citation20]. Hence, most of the patients with AL amyloidosis were advanced when the diagnosis was made [Citation21,Citation22]. Cardiac involvement is the most important prognosis factor in AL amyloidosis patients, Mayo staging system, combining cardiac biomarkers, troponin T and NT-proBNP, is commonly used to evaluate the severity of cardiac dysfunction and the prognosis of patients with AL amyloidosis [Citation23]. Therefore, early diagnosis is the most important factor in avoiding irreversible organ damage and prolonging survival [Citation20,Citation24].
Both Chinese and International guidelines stipulate that three conditions must be met for a proper diagnosis of AL amyloidosis. These include 1. A combination of clinical signs, lab values and imaging that confirm organ involvement. 2. Identification of amyloid tissue by biopsy of involved tissue followed by Congo red staining. It should also be confirmed that the amyloid deposits are composed of light chains by immunohistochemical methods, electron microscopy, or mass spectrometry. 3. Evidence of monoclonal antibodies or identification of light chains in the serum or urine, or bone marrow examination which reveals monoclonal plasma cells or B cells [Citation25]. However, diagnosing AL amyloidosis is difficult due to the lack of solid pathognomonic blood test or imaging test for it [Citation26]. The definite diagnosis now depends on the tissue biopsy of involved organs stained with Congo red dye and immunoassays for further typing remain indispensable to establish the diagnosis [Citation17]. Some clinical characteristics, such as symptoms, serum biomarkers, immunohistochemistry (IHC), and methods for M-protein identification, may raise the clinical suspicion of the disease and improve its diagnostic dilemma [Citation19]. However, to our best knowledge, there is a lack of systematic synthesis on the diagnosis of AL amyloidosis in China. A scoping review is appropriate for this purpose [Citation27].
The objective of this scoping review was to map the available literature on the diagnosis of AL amyloidosis and to consolidate recommendations for practice and future research in China. With this review, we also aimed to provide insights into AL amyloidosis that is specific to the current state of the Chinese medical system. This will both give medical professionals a frame of reference when testing patients, as well as allow policymakers to make better decisions when creating and updating relevant regulations.
Methods
The protocol of the present review was provided in Supplementary Appendix I. Besides, this scoping review was reported in accordance with the Preferred Reporting Items for Systematic Reviews and Meta-Analyses extension for Scoping Reviews (PRISMA-ScR) guidelines [Citation28].
Eligibility criteria
The published academic papers related to the diagnosis of AL amyloidosis were screened from 1 January 2000 to 15 September 2021. The inclusion criteria were 1) Chinese patients who have suspected AL amyloidosis without age restriction, 2) published in English or Chinese, 3) having a sample size of at least 25 patients, 4) original studies included both experimental studies and observational studies, and 5) having information on the diagnosis of AL amyloidosis. Exclusion criteria were as follows: case reports, review, consensus, thesis, questionnaire, and correspondence.
Literature search
References were searched through MEDLINE, EMBASE, and CNKI databases from 1 January 2000 to September 15 September 2021. The key search terms included ‘amyloidosis’, ‘amyloidos*’ ‘amyloido*’, ‘AL amyloidosis’ and ‘China’. The details of the search strategies can be found in the protocol (Supplementary Appendix I).
Grey literature search of Google Scholar was also included in the search. We also explored sources identified by searching the reference list of all the included full-text papers. Where more information related to a potentially included literature is lacking, we contacted the literature authors and request further information.
Study selection
Search results were entered to Endnote version X9 and duplicates were removed before screening for relevance. Two authors (PJ & LW) reviewed the titles and abstracts of publications retrieved by the search to identify potentially eligible papers. All potentially relevant citations were requested and inspected in detail using the full-text version. In case of doubt on whether a study could be eligible, a third author (XL) reviewed the publication to make a collegial decision.
Data extraction
Data items were reviewed as follows: 1) clinical characteristics, such as age, sex, stage, type, organ involvement (as defined by the studies), and diagnostic delay; 2) diagnostic yield of Congo red staining, using different kinds of tissues; 3) light chains classification method; 4) radiological tools, flow cytometry, genetic tests, and other routine biomarkers, such as 24 h urinary protein, NT-proBNP or BNP and alkaline phosphatase. The data extraction form can be found in the protocol (Supplementary Appendix I).
A pretested data extraction form based on the PICOT (population, intervention, control, outcome, and time) structure was used to perform the data extraction. Data from each literature review were extracted by one reviewer and double-checked by another using a standard data extraction form. Any disagreements were resolved by discussion, with assistance from a third party if necessary. Data were extracted as follows: first author, title, year of publication, publication journal, country of study, study design, diagnostic delay, Congo red staining (biopsies, performance, and others), amyloid typing method (proportion and performance), AL Amyloidosis typing method (proportion and performance), organ involvement (diagnostic methods and corresponding performance).
Synthesis of results
The included studies were categorized into accuracy studies and descriptive studies based on if the studies supplied the diagnostic accuracy data or not. The information on the diagnostic methods reported in included studies was synthesized according to different organ involvement and expressed using the count and proportion of the studies. For each diagnostic method, the sample size of related patients was summed up and expressed as count and percentage. For the available accuracy data for any diagnostic method, a descriptive analysis was performed.
Results
Characteristics of the included studies and patients
A total of 891 papers were identified via reference searching. After removing duplicate articles and screening, 43 articles were included in the final scoping review [Citation29–71]. A PRISMA flow diagram was constructed to show the whole literature selection process with reasons for exclusions at each stage (). Of all included studies, 31 belong to descriptive studies [Citation29,Citation30,Citation32–39,Citation43,Citation46–51,Citation53–58,Citation60,Citation62,Citation63,Citation65,Citation66,Citation69–71], and 12 have information of diagnostic accuracy [Citation31,Citation40–42,Citation44,Citation45,Citation52,Citation59,Citation61,Citation64,Citation67,Citation68] (the characteristics of each included study can be found in Supplementary Appendix II).
Figure 1. PRISMA flow diagram for article selection for analysis.
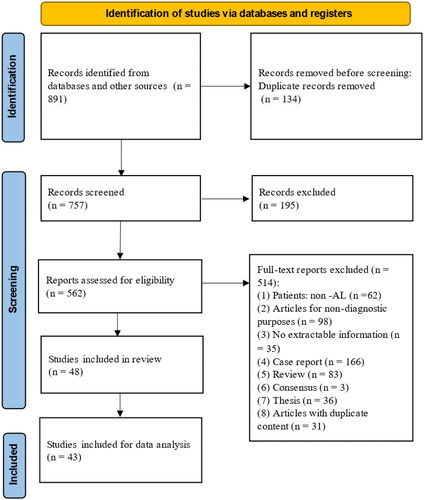
A total of 2447 AL patients with amyloidosis were included in the final analysis. Among patients with amyloidosis, AL amyloidosis comprised a majority proportion, which depends on the kind of organ involved. The classification of AL amyloidosis was reported in 686 patients, most of whom were primary AL amyloidosis (656/686, 95.6%; 13 studies). The mean age of the included patients ranged from 49 to 61 years. In 2060 patients who reported gender, 63.1% (1299) were male. In terms of Mayo stage categorization, 170 patients were categorized according to Mayo 2004 cardiac staging system, and 537 patients were categorized according to Mayo 2012 cardiac staging system. In addition, there were 5% (32/695) patients who reported clonal plasma cells in bone marrow ≥ 10%. The details of the characteristics of patients collected from the 43 included studies can be found in .
Table 1. Summary of patients’ characteristics based on 43 included studies.
AL amyloidosis diagnosis in China
Biopsy sites
The most common organ involvement reported in the 43 papers was the kidney (1979/2143, 92.3%), followed by the heart (899/1566, 57.4%), and the skin & soft tissue (150/300, 50.0%). The details of the organ involved in the included studies can be found in Supplementary Appendix III. Meanwhile, forty studies (40/43, 93.0%) reported the sites of biopsy in total 2101 patients with the top three of the kidneys (27/40, 67.5%) in 1367 (65.1%) patients, bone marrow (16/40, 40.0%) in 656 (31.2%) patients, and rectal mucosa (5/40, 12.5%) in 334 (15.9%) patients ().
Table 2. Summary of biopsy sites and related sample size of the patients based on 40 included studies (n = 2101).
Light chain classification and monoclonal (M-) protein identification
A total of 23 studies supplied the details of the methods of classification of light chains. The most popular diagnostic method for light chain classification reported in China was IHC (15/43, 34.9%), immunofluorescence (IF) (12/43, 27.9%), and electron microscopy (10/43, 23.3%). Meanwhile, a total of 29 studies described the methods of detection of M-protein with the top three reported tests being serum-free light chain (20/43, 46.5%), blood immunofixation electrophoresis (8/43, 18.6%), urine-free light chain (8/43, 18.6%). The number of studies and patients that reported light chain classification methods and M protein identification tests in the 43 papers were described in .
Table 3. Summary of diagnostic methods for light chains classification and different M protein identification in 43 included studies.
Adjuvant diagnostic methods
As an important adjuvant diagnostic method for cardiac involvement, imaging has been reported in 19 (44.2%) studies, including echocardiogram (15/19, 78.9%), electrocardiogram (14/19, 73.7%), and cardiac MRI (10/19, 52.6%). In addition, N-terminal (NT)-pro hormone BNP (NT-proBNP) and brain natriuretic peptide (BNP) test were reported in 12 (12/43, 27.9%) and 3 (3/43, 7.0%) studies, respectively. Next, for renal involvement, 24 h urinary protein test was reported in 22 (51.2%) studies. Third, for bone marrow involvement, the flow cytometry (FCM) and genetic tests (CD138 MACS-FISH magnetic bead sorting combined with interphase fluorescence in situ hybridization) were stated valuable for the diagnosis of AL in 4 (4/43, 9.3%) and 3 (3/43, 7.0%) studies, respectively. Finally, for liver involvement, the liver percussion and alkaline phosphatase test were reported in 2 (2/43, 4.7%) and 1 (1/43, 2.3%) study, respectively (Supplementary Appendix II).
Diagnostic delay
Diagnostic delay was reported in two studies [Citation44,Citation55] (Citations), the median and mean delays were reported as 9.5 months (median) and 11.3 ± 10.4 months (mean ± SD), respectively.
Accuracy of the AL amyloidosis diagnosis in China
Amyloidosis testing accuracy
When stained with Congo red, the amyloidosis tissue will demonstrate apple-green birefringence under a polarizing microscope [Citation72]. In the forty studies having information on biopsy, Congo red stain was the most popular method for detecting amyloid (32/40, 80%). The details of the sensitivity of Congo red staining ranged from 15.0% to 100.0%, as calculated using data reported by the studies. The lowest sensitivity (15.0%) was found in the marrow biopsy and the highest sensitivity (100%) was found in lung, renal, and rectal mucosal tissue. One of the included studies Li et al. [Citation61] supplied the sensitivity of the deposit sites of amyloid and the IF. According to Li et al. combining skin fat biopsy with rectal mucosal biopsy can significantly increase the test sensitivity [Citation61] ().
Table 4. The performance of diagnostic methods for detecting amyloidosis in China.
Light chain classification
Only four studies reported the accuracy of data on the light chain classification (). The top method for light classification was IHC, which was reported in 15 (34.9%) studies. However, the high sensitivity was more likely related to the combined method of immunofixation electrophoresis and serum light chain (88%) or IHC and serum free light chain (96%).
M Protein identification
For M protein identification, the reported sensitivity of different methods varied in a wide range. However, the highest sensitivity (88%) was reported for the Serum free light chain and immunofixation electrophoresis by Zhai et al. On the contrary, using blood immunofixation electrophoresis was reported the lowest (43.8%) sensitivity by Zhou et al. One study by Fan et al. reported the sensitivity of urine immunofixation electrophoresis (Bence-Jones protein). The sensitivity of the M protein identification was 64.0% by Bence-Jones protein and 79.5% by Bence-Jones protein and blood M protein, respectively ().
Three-dimensional speckle tracking imaging (3D-STI)
One of the studies [Citation43] reported the accuracy of the 3D-STI assessed global circumferential strains (see details data in ). The highest sensitivity (94.2%) and specificity (87.5%) were found in the 3D-STI diagnosis of GCS.
Multiparametric flow cytometry (MFC)
One study [Citation59] reported that the positive rate of MFC to measure the light chain restriction was 80.7% ().
Discussion
In the present study, we studied the condition of AL amyloidosis diagnosis in China. According to our findings, although cardiac involvement was second top in Chinese patients with AL amyloidosis, cardiac biopsy was rare. Next, we found light chain classification and monoclonal (M-) protein identification were essential methods for the diagnosis of AL amyloidosis in China. In addition, some combined tests can increase the sensitivity of the diagnosis. Finally, some adjuvant methods were essential for AL amyloidosis diagnosis.
Congo red staining is the gold standard method for amyloid diagnosis and has been used for decades [Citation73]. However, using Congo red also has several disadvantages. For instance, false-positive or false-negative results can occur due to inexperienced examiners [Citation74]. In addition, the usual site of choice for biopsy is the involved organ. However, there are sometimes alternative sites chosen for biopsy due to the organ or technical limitations. In this study, we observed that the most biopsied sites in Chinese hospitals were for kidney, bone marrow, and skin/fat in that order, with varying favorable rates. In addition, we found that cardiac biopsy reports were rare, which may be due to the low acceptance of biopsy itself and the small number of healthcare providers with the relevant skills. Besides, we found that only 31.2% related patients were performed bone marrow biopsy. The reason may be due to the low percentage of bone marrow involvement and the percentage of myeloma plasma cells. Therefore, bone marrow biopsy may not be a good method for AL amyloidosis detection [Citation75]. Hence, besides Congo Red staining, IF, IHC, electron microscope, mass spectrometry, and potassium permanganate staining are also recommended to improve the diagnostic accuracy of pathological examinations [Citation29,Citation36,Citation76].
IF and IHC can distinguish multiple amyloid types but are prone to false negatives with a sensitivity of about 60% [Citation77]. Therefore, IF or other sensitive immunoassays in combination with immunofixation electrophoresis of serum and urine are required to check serum k and λ light chain to determine the presence of monoclonal free light chains in serum or urine [Citation78].
As nonquantitative techniques, serum and urine IFE (SIFE/UIFE) are routinely performed for the detection of monoclonal Ig among patients with plasma cell dyscrasia. As shown in the previous studies [Citation40,Citation41,Citation44,Citation45,Citation59], blood IFE has a moderate sensitivity of 50–60% for AL Amyloidosis. However, the sensitivity could be improved if blood IFE is combined with urine IFE [Citation44]. Recently, sFLC showed the sensitivities for diagnosis of AL Amyloidosis were 52.7% [Citation41] and 76.0% [Citation68], respectively. Prokaeva et al. found that the combination of each method (SIFE/UIFE and HLC, SIFE/UIFE and sFLC, and HLC and sFLC) have a sensitivity of 94%, 100%, and 93.5% for the AL amyloidosis diagnosis [Citation79]. Similar findings were also observed in other reports. The sensitivity of combined sFLC and SIFE/UIFE testing for the diagnosis of AL amyloidosis ranges from 97 to 100% [Citation80,Citation81]. Therefore, baseline sFLC measurements and SIFE/UIFE are recommended by consensus guidelines for screening underlying clonal disease in suspected AL amyloidosis cases [Citation79,Citation82–84].
Two of the included studies reported on diagnosis delay. The reported diagnosis delay was considerably shorter than the delay reported by other international studies [Citation20]. There are two possible hypotheses that may explain this disparity: 1. The Chinese studies that we included were conducted in relatively large medical institutions with amyloidosis subspeciality, which may have had access to a lot of healthcare resources to allocate to the patients, thus allowing the diagnosis to be conducted faster. 2. The staff at these institutions are experienced and familiar with the process of AL amyloidosis diagnosis and were able to make use of multi-disciplinary teams to speed up the process of diagnosis. It is important to keep in mind that the data we have obtained on this specific topic is limited and likely not representative of the situation in China as a whole.
In this article, we found that the knowledge gap as follows: (1) the lack of awareness of AL amyloidosis, which leads to delayed diagnosis of patients, and it takes nearly a year from the appearance of symptoms to the diagnosis [Citation85], and it is a problem to raise the awareness of early suspicion of amyloidosis among doctors in related departments; (2) There is a lack of research on practical and applicable tests unless some hospitals are experienced in AL diagnosis, AL patients are easily misdiagnosed as other diseases in the clinic for treatment; (3) there is an urgent need to study other detection techniques that can identify AL amyloidosis as a disease at an early stage, including imaging assessment of the affected organs, early detection of abnormal biochemical indicators such as NT-proBNP, cardiac troponin T, and proteinuria.
This is the first attempt to systematize existing evidence on diagnosing AL Amyloidosis in China. The study has well addressed recent findings of AL Amyloidosis in China, which may improve the current diagnostic dilemma of AL Amyloidosis. However, some limitations should be considered when interpreting and applying those results. First, as known, the assessment of individual literature methodological quality is not required for scoping reviews. The authors could not identify gaps in the literature related to low-quality research. Second, this review is descriptive and would be limited by the low quality and heterogeneity of the included studies. Third, the rare nature of AL amyloidosis resulted in many studies with small sample sizes, limiting definitive conclusions. Fourth, this review was based exclusively on information from published reports. There has been no registered study and no data from the ClinicalTrials.gov website. Therefore, the accuracy and completeness of the data depend upon these previously reported studies. In the future, further research is required to draw more reliable conclusions.
Conclusions
This scoping review details the characteristics and results of the recently published studies on diagnosing AL Amyloidosis in China. Biopsy was still the most critical method for AL Amyloidosis diagnosis in China. In addition, combined tests and some adjuvant methods played essential roles in the diagnosis. However, further research and deliberation are required to amend the current Chinese guidelines in order to make them more practical and feasible.
Author contributions
Search strategy, X.C. and X.W.; draft protocol, M.C. and J.R.L.; study selection, J.R.L. and X.W.; data extraction, L.X., J.P. and W.L.; data analysis, M.C. and X.G.; writing review and editing, J.L. and J.R.L.; All authors have read and agreed to the published version of the manuscript.
Supplemental Material
Download MS Word (20.7 KB)Supplemental Material
Download MS Word (53.4 KB)Supplemental Material
Download PDF (562 KB)Acknowledgments
We thank Medjaden Inc. for the editorial assistance and language editing and all other contributors who provided assistance and support but have not been mentioned. The collection and assembly of data and statistical expertise were provided by Yang Zhang and Sai Zhao, from Systematic Review Solutions, Ltd.
Disclosure statement
The authors declared that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The authors are responsible for all content and editorial decisions and received no honoraria related to the development of this publication.
Data availability statement
The original contributions presented in the study are included in the article, further inquiries can be directed to the corresponding author.
Additional information
Funding
References
- Merlini G. AL amyloidosis: from molecular mechanisms to targeted therapies. Hematology Am Soc Hematol Educ Program. 2017;2017(1):1–12. doi: 10.1182/asheducation-2017.1.1.
- Nienhuis HL, Bijzet J, Hazenberg BP. The prevalence and management of systemic amyloidosis in Western countries. Kidney Dis . 2016;2(1):10–19. doi: 10.1159/000444206.
- Maurer MS, Elliott P, Comenzo R, et al. Addressing common questions encountered in the diagnosis and management of cardiac amyloidosis. Circulation. 2017;135(14):1357–1377. doi: 10.1161/CIRCULATIONAHA.116.024438.
- Hou H-A, Tang C-H, Goh CH, et al. A population-based cohort study of the epidemiology of light-chain amyloidosis in Taiwan. Sci Rep. 2022;12(1):15736. doi: 10.1038/s41598-022-18990-3.
- Seo SR, Jang SY, Lee GY, et al. Prevalence of amyloidosis in korea. Orphanet J Rare Dis. 2017;12(1):152. doi: 10.1186/s13023-017-0705-2.
- Quock TP, Yan T, Chang E, et al. Epidemiology of AL amyloidosis: a real-world study using US claims data. Blood Adv. 2018;2(10):1046–1053. doi: 10.1182/bloodadvances.2018016402.
- Aimo A, Buda G, Fontana M, et al. Therapies for cardiac light chain amyloidosis: an update. Int J Cardiol. 2018;271:152–160. doi: 10.1016/j.ijcard.2018.05.018.
- Kastritis E, Palladini G, Minnema MC, et al. Daratumumab-based treatment for immunoglobulin light-chain amyloidosis. N Engl J Med. 2021;385(1):46–58. doi: 10.1056/NEJMoa2028631.
- Kaufman GP, Cerchione C. Beyond andromeda: improving therapy for light chain amyloidosis. Front Oncol. 2020;10:624573. doi: 10.3389/fonc.2020.624573.
- Palladini G, Kastritis E, Maurer MS, et al. Daratumumab plus CyBorD for patients with newly diagnosed AL amyloidosis: safety run-in results of ANDROMEDA. Blood. 2020;136(1):71–80. doi: 10.1182/blood.2019004460.
- Kastritis E, Rousakis P, Kostopoulos IV, et al. Consolidation with a short course of daratumumab in patients with AL amyloidosis or light chain deposition disease. Amyloid. 2021;28(4):259–266. doi: 10.1080/13506129.2021.1971192.
- Staron A, Zheng L, Doros G, et al. A 40-Year natural history study of overall survival and primary causes of death in systemic light chain (AL) amyloidosis. Blood. 2021;138(Supplement 1):155–155. doi: 10.1182/blood-2021-151804.
- Parkin S, Seidman MA, Davis M, et al. Clinical characteristics and outcome of patients with wild-type transthyretin and AL cardiac amyloidosis confirmed by mass spectrometry. Blood. 2018;132(Supplement 1):3126–3126. doi: 10.1182/blood-2018-99-115317.
- McCausland KL, White MK, Guthrie SD, et al. Light chain (AL) amyloidosis: the journey to diagnosis. Patient. 2018;11(2):207–216. doi: 10.1007/s40271-017-0273-5.
- Schulman A, Connors LH, Weinberg J, et al. Patient outcomes in light chain (AL) amyloidosis: the clock is ticking from symptoms to diagnosis. Eur J Haematol. 2020;105(4):495–501. doi: 10.1111/ejh.13472.
- Dispenzieri A, Merlini G. Immunoglobulin light chain systemic amyloidosis. Cancer Treat Res. 2016;169:273–318.
- Fotiou D, Dimopoulos MA, Kastritis E. Systemic AL amyloidosis: current approaches to diagnosis and management. Hemasphere. 2020;4(4):e454. doi: 10.1097/HS9.0000000000000454.
- Gertz MA, Dispenzieri A. Systemic amyloidosis recognition, prognosis, and therapy: a systematic review. Jama. 2020;324(1):79–89. doi: 10.1001/jama.2020.5493.
- Vaxman I, Gertz M. When to suspect a diagnosis of amyloidosis. Acta Haematol. 2020;143(4):304–311. doi: 10.1159/000506617.
- Hester LL, Gifkins DM, K MB, et al. Diagnostic delay and characterization of the clinical prodrome in AL amyloidosis among 1523 US adults diagnosed between 2001 and 2019. Eur J Haematol. 2021;107(4):428–435. doi: 10.1111/ejh.13679.
- Comenzo RL, Reece D, Palladini G, et al. Consensus guidelines for the conduct and reporting of clinical trials in systemic light-chain amyloidosis. Leukemia. 2012;26(11):2317–2325. doi: 10.1038/leu.2012.100.
- Merlini G, Wechalekar AD, Palladini G. Systemic light chain amyloidosis: an update for treating physicians. Blood. 2013;121(26):5124–5130. doi: 10.1182/blood-2013-01-453001.
- Dispenzieri A, Buadi F, Kumar SK, et al. Treatment of immunoglobulin light chain amyloidosis: mayo stratification of myeloma and risk-adapted therapy (mSMART) consensus statement. Mayo Clin Proc. 2015;90(8):1054–1081. doi: 10.1016/j.mayocp.2015.06.009.
- Muchtar E, Gertz MA, Kumar SK, et al. Improved outcomes for newly diagnosed AL amyloidosis between 2000 and 2014: cracking the glass ceiling of early death. Blood. 2017;129(15):2111–2119. doi: 10.1182/blood-2016-11-751628.
- Kumar SK, Callander NS, Adekola K, et al. Systemic light chain amyloidosis, version 2.2023, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2023;21:67–81.
- Vaxman I, Dispenzieri A, Muchtar E, et al. New developments in diagnosis, risk assessment and management in systemic amyloidosis. Blood Rev. 2020;40:100636. doi: 10.1016/j.blre.2019.100636.
- Peters MDJ, Marnie C, Tricco AC, et al. Updated methodological guidance for the conduct of scoping reviews. JBI Evid Implement. 2021;19(1):3–10. doi: 10.1097/XEB.0000000000000277.
- Tricco AC, Lillie E, Zarin W, et al. PRISMA extension for scoping reviews (PRISMA-ScR): checklist and explanation. Ann Intern Med. 2018;169(7):467–473. doi: 10.7326/M18-0850.
- Xiang L, Yuan S, Zhang X, et al. [Clinicopathological features of renal impairment in 76 cases with amyloidosis]. J Clin Pathol. 2018;38:1675–1681.
- Mao X, Deng S, Sui W, et al. Clinical analysis of patients with MGUS, primary light chain amyloidosis, multiple myeloma or multiple myeloma with concurrent amyloidosis. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2021;29(3):812–818. doi: 10.19746/j.cnki.issn.1009-2137.2021.03.025.
- Xu Y, Cui X, Li M, et al. Cardiac manifestations of 21 patients with immunoglobulin light chain cardiac amyloidosis. Chin J Hyperten. 2021;29:371–375.
- Cheng P, Zhang Y, Li Y, et al. Clinicopathological analysis of 321 patients with renal amyloidosis. J Chin Pract Diagn Ther. 2021;35:588–591.
- Guo L, Tian Z. Clinical characteristics of patients with cardiac light-chain amyloidosis and normal left ventricular wall thickness. J Clin Cardiol. 2018;34:239–243.
- Li X, Huang S, Han P, et al. Nonenhanced chemical exchange saturation transfer cardiac magnetic resonance imaging in patients with amyloid Light-Chain amyloidosis. J Magn Reson Imaging. 2022;55(2):567–576. doi: 10.1002/jmri.27859.
- Zhang L, Tang H, Cheng L, et al. Analysis of clinical and imaging features of cardiac amyloidosis: a multicenter study. J Southern Med Univ. 2014;34:295–302.
- Zhu X, Liu F, Liu Y, et al. Analysis of clinical and pathological characteristics of 28 cases with renal amyloidosis. Clin Lab. 2011;57:947–952.
- Xia R, Gao F, Sun J, et al. Cardiac magnetic resonance imaging of systemic amyloidosis patients with normal left ventricular ejection fraction: an initial study. Pak J Med Sci. 2013;29(6):1300–1305. doi: 10.12669/pjms.296.3775.
- Shen K, Sun W, Sun J, et al. Classification of amyloidosis by laser micro- dissection and mass spectrometry based proteomic analysis. Chin J Hematol. 2015;36:99.
- Diao X, Li J, Ouyang J, et al. Flow cytometry-based immunophenotypic analysis of primary systemic light chain amyloidosis. Oncol Lett. 2017;13(4):2691–2697. doi: 10.3892/ol.2017.5767.
- Fan Y, Wu G, Zhang S. Clinical characteristics and screening indexes of light chain amyloidosis nephropathy. Chin J Pract Med. 2019;46:6–10.
- Li T, Huang X, Chen W. The significance of skin fat and rectal mucosal biopsy in the diagnosis of systemic light chain myloidosis. Chinese J Nephrol Dialysis Transplant. 2015;24:426–428.
- Zhang J, Sun J, Wu M, et al. Clinicopathological study of renal amyloidosis. Chin J Laborat Diag. 2012;16:131–133.
- Lei C, Zuo L, Wang Y, et al. The role of three-dimensional speckle tracking imaging in the diagnosis of immunoglobulin light-chain cardiac amyloidosis with normal left ventricular ejection fraction. Chin J Ultrasonogr. 2020;29:213–218.
- Zhou FD, Zhang LX, Yao Y, et al. Immunofixation electrophoresis was highly specific for the diagnosis of renal light-chain amyloidosis. Am J Med Sci. 2013;345(1):18–21. doi: 10.1097/MAJ.0b013e31824e0ec5.
- Yao Y, Wang S, Zhang Y, et al. Clinicopathological correlation analysis of AL type renal amyloidosis. Chin J Nephrol. 2013;29:216–218.
- Mao X, Zhao D, Fang Q, et al. Clinicopathology of myocardial amyloidosis. Med J Peking Union Medical College Hosp. 2012;3:89–93.
- Yang J, Shi H, Xu J, et al. Clinicopathological analysis of 33 cases of primary renal amyloidosis. Modern Pract Med. 2016;28:998–999.
- Xiao H, Liu R, Yuan S, et al. Clinical and pathological features of 20 cases of renal amyloidosis. Inter J Pathol Clin Med. 2010;1:43–46.
- Wang X, Dong K, Sun L, et al. Hepatic light chain deposition disease and light chain amyloidosis clinicopathological features. Beijing Med J. 2016;38:877–879.
- Yang Y. Clinical study of renal amyloidosis and cutaneous amyloidosis in elderly and presenile patients. China Continuing Med Educ. 2015;7:78–79.
- Mei J, Shao J, Cao H, et al. Analysis of cytogenetic abnormalities in systemic light chain amyloidosis. Laborat Med Clinic. 2016;13:3463–3466.
- Zhang F, Yu X, Wang S, et al. The role of commonly used immunopathological methods in the typing of light chain renal amyloidosis. Labeled Immunoassays Clin Med. 2020;27:818–822.
- Ren L, Liu H, Xu X, et al. Diagnosis and pathological type analysis of renal amyloidosis. Chin J Nephrol. 2011;27:730–734.
- Zhang L, Wang Y, Cheng L, et al. The value of peak left ventricular long-axis systolic strain in the diagnosis of primary cardiac amyloidosis and hypertrophic cardiomyopathy. J Southern Med University. 2014;34:609–616.
- Liu D, Liu G. Analysis of clinical characteristics of 20 cases of AL amyloidosis. J Capital Med Univ. 2010;31:416–419.
- Sui Y, Jiang N, Xie L, et al. Clinicopathological analysis of 31 cases of amyloid nephropathy. Chin J Clin Exper Pathol. 2014;30:1379–1382.
- Mu X, Xiong Y, Chen J, et al. Clinical analysis of 11 cases of respiratory amyloidosis. Chin J Tubercul Respir Dis. 2013;36:88–93.
- Ren H, Lu M, Cai Q, et al. Clinicopathological analysis of 10 cases of renal amyloidosis. Military Med J Southeast China. 2011;13:348–349.
- Sa Q, Ren G, Xu X, et al. Characterization of abnormal plasma cells in systemic light chain amyloidosis. Chin J Nephrol Dialysis Transplant. 2019;28:401–405.
- Li R, Yang ZG, Xu HY, et al. Myocardial deformation in cardiac amyloid light-chain amyloidosis: assessed with 3T cardiovascular magnetic resonance feature tracking. Sci Rep. 2017;7(1):3794. doi: 10.1038/s41598-017-03699-5.
- Li T, Huang X, Cheng S, et al. Utility of abdominal skin plus subcutaneous fat and rectal mucosal biopsy in the diagnosis of AL amyloidosis with renal involvement. PLOS One. 2017;12(9):e0185078. doi: 10.1371/journal.pone.0185078.
- Cui Q, Yu J, Shen W. The value of delayed gadolinium enhancement combined with quantitative longitudinal relaxation time imaging in the assessment of myocardial amyloidosis. Chinese Crit Care Med. 2019;31:1538–1541.
- Zhang L, Zhou X, Wang J, et al. Differentiation of light-chain cardiac amyloidosis from hypertrophic cardiomyopathy using myocardial mechanical parameters by velocity vector imaging echocardiography. Int J Cardiovasc Imaging. 2017;33(4):499–507. doi: 10.1007/s10554-016-1027-5.
- Wang L, Tian Y, Lei Z, et al. Clinical analysis of immunoglobulin light chain myocardial amyloidosis. Chinese General Pract. 2020;23:3474–3478.
- Li L, Duan X, Sun Y, et al. Application value of immunohistochemical indexes in cardiac amyloidosis typing. Chin J Pathol. 2018;47:105–108.
- Sa Q, Ren G, Xu X, et al. Bone marrow plasmacytogenetic characteristics of 102 patients with systemic light chain amyloidosis. Chin J Nephrol Dialysis Transplant. 2020;29:413–419.
- Xu T, Wei Z, Chen C, et al. The value of combined abdominal wall fat and labial gland biopsy in the diagnosis of light chain amyloidosis. Chin J Clin Med. 2019;26:37–42.
- Zhai y, Song P, Li F, et al. Clinical significance of serum free light chain detection in primary systemic amyloidosis. Chin J Internal Med. 2011;50:404–407.
- Hu Y, Wang M, Chen Y, et al. Immunophenotypic analysis of abnormal plasma cell clones in bone marrow of primary systemic light chain amyloidosis patients. Chin Med J. 2014;127:2765–2770.
- Li R, Yang ZG, Wen LY, et al. Regional myocardial microvascular dysfunction in cardiac amyloid light-chain amyloidosis: assessment with 3T cardiovascular magnetic resonance. J Cardiovasc Magn Reson. 2016;18:16. doi: 10.1186/s12968-016-0240-7.
- Liu Y, Lai Y, Ma L, et al. Fluorescence in situ hybridisation combined with CD138 immunomagnetic sorting is effective to identify cytogenetic abnormalities whplosich play significant prognostic roles in chinese AL amyloidosis patients. Amyloid. 2020;27(3):208–209. doi: 10.1080/13506129.2020.1723538.
- Howie AJ, Brewer DB, Howell D, et al. Physical basis of colors seen in Congo red-stained amyloid in polarized light. Lab Invest. 2008;88(3):232–242. doi: 10.1038/labinvest.3700714.
- Guselnikova VV, Antimonova OI, Fedorova EA, et al. A novel method for amyloid detection in human tissue load using a fluorescent dye - Congo red analogue. Sovrem Tekhnologii Med. 2020;12(1):65–70. doi: 10.17691/stm2020.12.1.08.
- Sjolander D, Rocken C, Westermark P, et al. Establishing the fluorescent amyloid ligand h-FTAA for studying human tissues with systemic and localized amyloid. Amyloid. 2016;23(2):98–108. doi: 10.3109/13506129.2016.1158159.
- Salameh OK, Darok MC, Kane JA, et al. Unusual case of nephrotic syndrome from light chain amyloidosis in a 37-year-old patient. Cureus. 2021;13(9):e18120. doi: 10.7759/cureus.18120.
- Murray DL, Dasari S. Clinical mass spectrometry approaches to myeloma and amyloidosis. Clin Lab Med. 2021;41(2):203–219. doi: 10.1016/j.cll.2021.03.003.
- Verine J, Mourad N, Desseaux K, et al. Clinical and histological characteristics of renal AA amyloidosis: a retrospective study of 68 cases with a special interest to amyloid-associated inflammatory response. Hum Pathol. 2007;38(12):1798–1809. doi: 10.1016/j.humpath.2007.04.013.
- Novak L, Cook WJ, Herrera GA, et al. AL-amyloidosis is underdiagnosed in renal biopsies. Nephrol Dial Transplant. 2004;19(12):3050–3053. doi: 10.1093/ndt/gfh503.
- Prokaeva T, Spencer B, Sun F, et al. Immunoglobulin heavy light chain test quantifies clonal disease in patients with AL amyloidosis and normal serum free light chain ratio. Amyloid. 2016;23(4):214–220. doi: 10.1080/13506129.2016.1219715.
- Palladini G, Russo P, Bosoni T, et al. Identification of amyloidogenic light chains requires the combination of serum-free light chain assay with immunofixation of serum and urine. Clin Chem. 2009;55(3):499–504. doi: 10.1373/clinchem.2008.117143.
- Katzmann JA, Abraham RS, Dispenzieri A, et al. Diagnostic performance of quantitative kappa and lambda free light chain assays in clinical practice. Clin Chem. 2005;51(5):878–881. doi: 10.1373/clinchem.2004.046870.
- Gertz MA, Comenzo R, Falk RH, et al. Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): a consensus opinion from the 10th international symposium on amyloid and amyloidosis, tours, France, 18–22 april 2004. Am J Hematol. 2005;79(4):319–328. doi: 10.1002/ajh.20381.
- Dispenzieri A, Kyle R, Merlini G, et al. International myeloma working group guidelines for serum-free light chain analysis in multiple myeloma and related disorders. Leukemia. 2009;23(2):215–224. doi: 10.1038/leu.2008.307.
- Gillmore JD, Wechalekar A, Bird J, et al. Guidelines on the diagnosis and investigation of AL amyloidosis. Br J Haematol. 2015;168(2):207–218. doi: 10.1111/bjh.13156.
- Hwa YL, Fogaren T, Sams A, et al. Immunoglobulin light-chain amyloidosis: clinical presentations and diagnostic approach. J Adv Pract Oncol. 2019;10:470–481.