
Abstract
Background
The multi-country mpox outbreak across the globe has led to the systematic surveillance of mpox cases in India. During the surveillance of mpox, we encountered cases of Varicella Zoster Virus (VZV) in suspected mpox cases amongst children & adults. This study focused on the genomic characterization of VZV in India.
Methods
A total of 331 mpox suspected cases were tested for VZV through real-time PCR, and the positive samples were subjected to next-generation sequencing to retrieve the whole genome of VZV using CLC genomics software. Phylogenetic analysis has been done in MEGA 11.0 software to identify circulating clades.
Result
Of the 331 suspected cases, 28 cases with vesicular rashes were found to be positive for VZV. The maximum genome could be retrieved from the clinical specimens of 16 cases with coverage greater than 98% when mapped with reference strain Dumas (NC 001348). The phylogenetic analyses of these sequences determined the circulation of clades 1, 5, and 9 in India. Further, the sequence analysis demonstrated non-synonymous single nucleotide polymorphism (SNPs) among specific ORF of VZV including ORF 14, ORF 22, ORF 36, ORF 37 and ORF 51. Although clade 1 and 5 has been reported earlier, the circulation of clade 9 of VZV has been determined for the first time in India.
Conclusion
Although the circulation of different clades of VZV was reported from India, the presence of clade 9 was detected for the first time during the mpox surveillance.
1. Introduction
Varicella (chickenpox) and Herpes Zoster (Shingles) are caused by a ubiquitous neurotropic Varicella Zoster Virus (VZV), which belongs to the human Herpesviridae family, human alphaherpesvirinae subfamily, genus Varicellovirus. Varicella is a common childhood disease characterized by fever, and vesicular rashes. In rare cases, VZV infections can progress to central nervous system involvement and severe complications [Citation1]. Reactivation of the virus from neurons of a sensory ganglion can occur and result in herpes zoster [Citation2]. VZV spreads with ease amongst contact through the vesicular fluid and nasopharyngeal secretions with an infectivity rate of more than 85–90% [Citation3]. It is critical to distinguish chickenpox from measles, bacterial skin infections, scabies, syphilis, medication-associated allergies, and mpox disease. Recently, a multicentric mpox outbreak has been observed across 110 countries, total 86,500 cases and 111 deaths observed till 16.03.2023 including 22 cases and 1 death from India (https://www.cdc.gov/poxvirus/monkeypox/response/2022/world-map.html). Mpox is an emerging disease caused by the mpox virus (mpoxv) belonging to the genus Orthopoxvirus. The incubation period of mpox ranges from 5 to 21 days whereas, in the case of Chickenpox, it is from 10 to 21 days. The skin rashes in mpox are often monomorphic while in Chickenpox it is usually pleiomorphic. The evolution of rashes occurs quite slowly over a few days in mpox but this occurs rapidly in chickenpox. The pattern of rash distribution is centrifugal in mpox whereas it is centripetal in chickenpox. Lymphadenopathy is a prominent feature of mpox, but it is usually absent in chickenpox [Citation4].
Atypical varicella-zoster virus infections involving palms and soles were reported in African countries [Citation5] underscoring the importance of laboratory diagnosis for the differentiation of mpox and chickenpox. The current scenario of the mpox outbreak, waning population immunity to orthopoxviruses, and non-inclusion of chickenpox vaccination in the national immunization program emphasize more on the laboratory diagnosis and identification of different clades of circulating VZV in India. The additional details of mpox and VZV differences has been provided ().
VZV is an enveloped virus with a genome consisting of linear double-stranded DNA of approximately 125 kb packaged into a nucleocapsid core surrounded by tegument proteins. Davison and Scott determined the complete genome sequence in 1986. The genome is divided into unique long (UL) and unique short (US) segments flanked by two inverted repeats. Five repeat regions, R1-R5 are identified in different ORF regions and the length of these repeat regions plays an important role in distinguishing the different strains of VZV [Citation6]. It has approximately 72 distinct predicted/known open reading frames (ORF), 10 of which translate to produce glycoproteins: ORFS/L (ORF0), gK (ORF5), gN (ORF9a), gC (ORF14), gB (ORF31), gH (ORF37), gM (ORF50), gL (ORF60), gI (ORF67) and gE (ORF68) [Citation7–9]. There are different methods employed for the classification of VZV into different clades. It includes Restriction Fragment Length Polymorphism (RFLP) in ORF38, ORF 54, and ORF62 and Single Nucleotide Polymorphism (SNPs) in ORF1, ORF21, ORF50, and ORF54 [Citation10,Citation11].
Glycoproteins and IE62 sequences of VZV also form the basis of the classification of VZV as A, B, C, and D clades [Citation12]. Based on the genotyping of the ORF22, ORF21, and ORF50, five confirmed clades (E1, E2, J, M1, M2) and two provisional clades (M3, M4) have been designated. Based on the complete genome sequences of VZV, the VZV nomenclature recognizes 7 established clades (clades 1-6 and 9) and two provisional clades (VII and VIII) are circulating worldwide [Citation13–16]. Two whole genome sequences are required for the re-designation of a provisional clade as an established clade. The advent of next-generation sequencing (NGS) technologies enabled the sequencing of VZV from clinical samples, which had previously been limited due to its nonviable nature and also the limited quantity of viral DNA. The present study aimed to determine the circulating clades of VZV from clinical samples that were initially suspected of mpox virus in India.
2. Material & methods
2.1. Sample collection
A total of 331 suspected mpox cases were screened from May 2022 to November 2022 at Indian Council of Medical Research-National Institute of Virology (ICMR-NIV), Pune. The clinical specimens included EDTA blood, serum, nasopharyngeal swab/oropharyngeal swab, urine, lesion scraping, lesion fluid, lesion crust/scab, lesion roof, and lesion floor. The samples were received from different geographical regions of India including Chandigarh, New Delhi, Gujarat, Jharkhand, Karnataka, Kerala, Maharashtra, Meghalaya, Telangana, Uttar Pradesh, Uttarakhand, West Bengal, and Tamil Nadu.
2.2. Screening of samples by Real-time PCR
The total nucleic acid was extracted from the clinical specimens using the Magmax Viral Pathogen Total Nucleic acid extraction kit (Applied Biosystems) following an automated DNA extraction method (KingFisher, USA). The extracted nucleic acid was used to screen mpox virus and other non-variola Orthopoxviruses using specific Real-Time PCR. The negative samples were further screened for Herpes Simplex Virus (HSV-1/2) and VZV using real-time PCR.
2.3. Next-generation sequencing
2.3.1. DNA Library preparation
The viral DNA was extracted from 200 µl of the clinical specimen using QIAamp DNA Blood Mini Kit [Qiagen, USA] following a manual extraction method. The extracted DNA was quantified using the Qubit dsDNA High Sensitivity kit (Invitrogen, USA), and the concentration of the input DNA from each specimen was measured and diluted to obtain approx. 0.2 ng/µl [Input DNA conc. max 1 ng/5 µl]. Nextera XT DNA Library preparation kit (Illumina, USA) was used for preparing DNA libraries from concentration optimized/diluted viral DNA. The prepared DNA libraries were quantified using a Qubit dsDNA HS kit. Libraries with a concentration ≥2 nM were pooled to obtain a final pool of libraries of 4 nM. Libraries were denatured and diluted as per the MiniSeq guide (Illumina, USA) and the run was loaded using the Miniseq Highoutput 150 × 2 cycle chemistry on the Illumina Miniseq at a loading concentration of 1.4 picomoles for paired-end sequencing.
2.3.2. Analysis of NGS data
The FastQ files generated from the Illumina machine were imported to CLC Genomics Workbench v22.0.2. (Qiagen, Aarhus, Denmark) for analysis. The raw files (FastQ) generated through the NGS platform were mapped with the reference sequence of VZV, Dumas (Accession Id: NC_001348) in CLC genomics software to retrieve the maximum genome of VZV.
2.3.3. Phylogenetic tree and single nucleotide polymorphism analysis
The phylogenetic analysis was done using maximum retrieved sequences with >98% genome length (n = 16) with few representative VZV sequences of each clade [(clade 1 (n = 8), clade 2 (n = 8), clade 3 (n = 6), clade 4 (n = 5), clade 5 (n = 15), clade 6 (n = 3), clade VIII (n = 1), and clade 9 (n = 5)] that were downloaded from NCBI. All the sequences were aligned with the reference sequence of VZV-Dumas in an online tool MAFFT v 7. The Neighbor-Joining (NJ) tree was generated using the substitution Model as Maximum Composite Likelihood model/method in MEGA 11.0 software with 1000 bootstrap replications to assess the significance of the phylogenetic tree (). The variants were calculated at a 20% frequency level to explore the SNP in the whole genome ().
Figure 1. The maximum likelihood method was utilized to generate the NJ phylogenetic tree of the VZV genome, exhibiting ≥98% genomic coverage from India. Out of 16 sequences retrieved, 11 sequences belonged to clade 5; 2 belonged to clade 1; while 3 belonged to novel clade 9. The sequenceshighlighted in red colour are foreign travellers, while those marked in blue do not have a travel history.
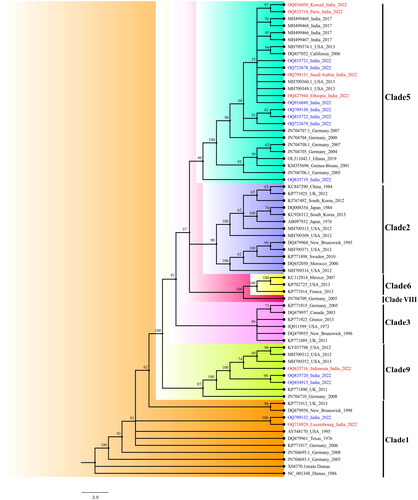
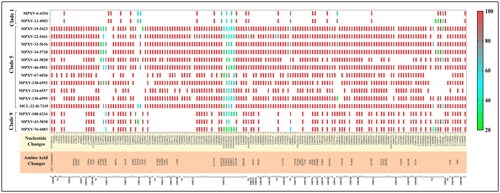
Figure 2. The graph depicts the variants observed throughout the entire VZV genome for clade 1, clade 9, and clade 5. The robustness of variation observed at the amino-acid and nucleotide levels for each orf were predicted by the frequency of residue changes from least (green) to highest (red).
3. Results
3.1. Clinical profile and Real-time PCR of VZV-positive cases
Of the total 331 suspected cases, 22 cases were positive for mpox virus infection (15 from New Delhi and 7 from Kerala), while 17 were positive for Enteroviruses and 1 case was confirmed as Buffalopox virus. Of these 331 suspects, 28 were positive for VZV, with primary presentation of vesicular rashes all over the body. The other clinical manifestations included fever (82%), myalgia (46%), headache (36%), fatigue (29%), loss of appetite (14%), and lymphadenopathy (11%) (). The real-time PCR of all 28 cases showed positive result for VZV infection with the cyclic threshold (Ct) ≤ 35 ().
Figure 3. The graphical representation depicted the real-time PCR Ct values of all 28 VZV positive cases.
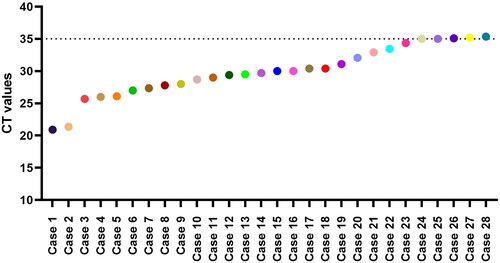
Table 1. Clinical features specific to VZV-positive cases from various geographical locations of India.
3.2. Next-generation sequencing
The extracted genomic DNA from positive VZV clinical specimens were used for NGS. The reference-based mapping of FastQ raw files using the VZV reference genome strain Dumas (NC_001348) led to the retrieval of the maximum genome of VZV. Out of 28 cases, the genome of 16 cases was retrieved with coverage of 98% − 100% (122,631 bp –124,881 bp).
3.3. Phylogenetic analysis
The phylogenetic tree of VZV complete sequences with genomic coverage >98% to reference sequence, Dumas is depicted in . The phylogenetic tree using the NJ method with a bootstrap value of 1000 revealed two sequences belonged to clade 1, three to clade 9, and eleven to clade 5. Six out of the 16 cases selected for analysis had international travel history with one sequence in Clade 1 (Luxembourg), one in clade 9 (Indonesia), and four sequences in clade 5 (Kuwait, Paris, Saudi Arabia & Ethiopia). The phylogenetic analysis revealed the circulation of multiple clades of VZV in India. One case with travel history from Luxembourg, a West European country where clade 1 is predominant, showed 99.99% similarity to the Indian sequence (MCL-22-H-6-4354 India 2022) and 99.82% similarity to USA strain AY548170. The VZV sequences published from India in previous years show similarity to the new sequences and are clustered in the same clade 5.
3.4. Single nucleotide Polymorphism (SNP) analysis
The whole genome sequencing and phylogenetic analysis revealed VZV sequences under study belong to three clades; Clade 1 (n = 02), Clade 9 (n = 03), and Clade 5 (n = 11). The genotyping scheme proposed by Jensen et al. after incorporating the less common discriminating SNP can differentiate the clades from 1-6, VIII, and 9). All 16 sequences were analyzed using the revised strategy by Jensen et al. () for the confirmation of the clades obtained from the phylogenetic tree constructed by the neighbor-joining statistical method using MEGA11.0. Other than the clade-specific less discriminating SNPs, more non-synonymous mutations were also observed in different ORF in comparison with the Dumas strain (NC_001348).
Table 2. Identification of the specific clades based on less common discriminating SNP in different ORF of VZV sequences using the revised strategy proposed by Jensen et al.
Table 3. The table showing the differentiation of features of mpox and VZV aids in clinical diagnosis.
The common mutational changes were analyzed in each sequence of specific clades. Clade 1 (n = 02), calde 9 (n=03) and clade 5 (n=11) sequences showed a total of 10, 29 and 59 non-synonymous SNPs respectively. (Supplementary Table 1).
The SNPs were most commonly observed in ORF 14 (envelope glycoprotein C), ORF 22 (tegument protein), ORF 36 (thymidine kinase), ORF 37 (envelope glycoprotein H), and ORF 51 (DNA replication origin binding-Helicase) for all three clades [clade 1,5 and 9]. The SNP analysis was also performed on the Indian sequences, with the most recent common ancestor (MRCA) observed in the phylogenetic tree serving as a reference for each clade. The reference sequences for clade 5 (DQ457052_California 2006), clade 9 (KP771890_UK 2011), and clade 1 (AY548170_USA 1995) were used to identify the SNPs in Indian sequences. A total of 27 SNPs were observed in clade 5, and 20 SNPs each in clade 9 and clade1 (Supplementary Table 2).
4. Discussion
We describe the identification and genomic characterization of VZV from the clinical specimens of mpox suspected cases. Amongst the 16 whole genome sequences retrieved, eleven belonged to clade 5, three to clade 9, and two to clade 1. Normally, VZV is diagnosed clinically, but the ongoing mpox outbreak in India enhanced the surveillance of fever with rash, enforcing specimens to be sent to laboratory for ruling out the mpox infections. Even though clinical presentation can distinguish between mpox and VZV, there are reports of atypical varicella infection in some African countries, which involves lesions on the palms and soles [Citation5]. We encountered inavoidable issues with the few set of samples where we were not able to obtain the sufficient genome for future research. This could be attributed to a multiple factors, including big genome size of VZV, challenge in collection of clinical samples, period of collection from disease onset, method of collection, adequacy of tissues or fluids collected during sampling, and immediate cold chain maintenance following collection.
The present study focused on the circulation of different clades of VZV and the major SNPs. This is the first study reporting the circulations of VZV clade 9 in India, whereas the clade 9 is the most common strain in circulation in countries such as Germany, UK, and USA. In a study of patients with central nervous system (CNS) involvement in VZV infection from the metropolitan region of New York City, the clade 9 strains were found in areas with a higher concentration of immigrants of Middle Eastern and Indian ethnicity, indicating the presence of clade 9 circulation in India [Citation14]. The specific geographic distribution of clades of VZV is plummeting now due to more immigration and travel.
In this study, two sequences from India belonged to clade 1 which were considered to be the European and North American strains. Clades 1 to 5 are most prevalent in the world, with clade 1 and 3 circulating in Oceania, Europe and America while clade 2 in Asia and clade 4-5 in South Asia and Africa. [Citation17]. The majority of VZV infections observed in the USA belong to clade 1 and 3 , but with the introduction of varicella vaccine and changes in current immigration patterns, the current distribution of clades & relative involvement of each clade in CNS and non-CNS diseases is still unknown and opens the arena for research. The above study also concluded that the same viral clades were identified in cerebrospinal fluid (CSF) and non-CSF specimens [Citation18]. The partial genome sequencing data from the clinical samples from the VZV outbreak in Dadra and Nagar Haveli, suggested the circulation of clade 1 VZV in India [Citation19].
The study conducted by Rima et al. revealed the circulation of multiple clades (1/3 and 4/5) of VZV in three Indian states [Citation20]. A clinico epidemiological investigation undertaken in the Indian state of Maharashtra revealed mixed clade circulation and the existence of VZV clade 1 in India. One of the patients who tested positive for clade 1 VZV came from Luxembourg, a Western European country. It was the first report of the circulation of clade 1 VZV based on the complete sequence.
The current research revealed 11 VZV sequences that belonged to clade 5, a finding corroborated by Nyayanit et al.’s comprehensive exploration of VZV genome sequences, wherein clade 5 VZV emerged as the predominant circulating clade in India [Citation21]. The updated genotyping approach proposed by Jensen et al. was employed to validate clade assignments using less common differentiating SNPs. Analyzing all 16 sequences aligned with the reference sequence unveiled additional SNPs within significant ORFs such as ORF14, ORF22, ORF 36, ORF 37, and ORF51. The majority of the SNPs present in the major capsid protein of clade 5 sequences are consistent with previous studies conducted by Nyayanit et al. [Citation21]. In an investigation conducted in China between 2020 and 2021, viral genotyping revealed that VZV Clade 2 had the highest prevalence (94%) within Shandong province [Citation22]. A 2012 UK and European study explored oral fluid as an alternative for VZV genotyping, finding no clade variance between paired oral and vesicular fluids. Clades 1 & 3 dominated UK & Eastern Europe, while clade 5 prevailed in Western Europe. Clade 5 viruses were more common in non-white children, potentially indicating genetic susceptibility to specific clades [Citation23]. The co-circulation of the VZV clades provides the opportunity to investigate the reinfection, coinfection and recombination among VZV strains [Citation24,Citation25]. A recent study demonstrated the genotypic differentiation of wild type (wt) strain from vaccine strain vOKa of varicella-zoster virus based on snp pattern at 4 nucleotide positions on snp 69,349 in ORF38 [Citation26,Citation27] and 106,262, 107,252 & 10,8111in ORF 62 [Citation28–33]. Wild type VZV strain demonstarted snp pattern A-T-T-T (Genbank accession ID: NC_001348, Dumas) at 69,349, 106,262, 107,252 & 108,111 nucleotide positions, whereas vOKa vaccine strain shown G-C-C-C (Genbank accession ID: AB097932) snp pattern. The efficacy and safety of the VZV vaccine have already been established [Citation34–36], however there is a need for continuous monitoring of the vaccinated population to differentiate between wt & vOKa strains for further evaluation of recombination patterns.
In our study, genotypic characterization of VZV was performed to identify the different clades based on snp pattern in VZV ORF. All the 16 sequences were also analysed to differentiate between wt and vOKa strains. Based on the snp pattern in ORF38 and ORF62, all 16 sequences revealed the wt snp pattern A-T-T-T, and none of the cases were vaccinated with the vOKa strain. Out of the total of 28 reported cases of VZV infection, all individuals recovered successfully, and no instances of mortality were observed.
Mpox disease symptoms are frequently mistaken for VZV, as their clinical presentations often closely resemble each other. There is a need for clinical differentiation between mpox & VZV for accurate diagnosis. In a region with mpox cases, earlier research revealed a significant count of VZV cases that tested negative for mpox [Citation37–39]. Similarly, we have also reported the VZV positive cases among the cases suspected of mpox virus infection. The key difference between mpox & VZV during disease progression and symptoms onset can help to presumptive diagnosis [Citation40]. The mpox-infected patients often experienced febrile prodrome with high fever before the development of rash onset, while in VZV low-grade fever was observed at the rash onset. Lesions were observed on the palm of hands & soles of feet and lymphadenopathy are common characteristics feature of mpox but not observed in VZV cases [Citation5]. The pox further characterised lesions with deep-seated, well-circumscribed, evolved slowly with each stage while VZV evolved rapidly from macules to the crust, lesions were superficial with irregular shape [Citation40–42]. According to data availability and outcomes, lesions (scraping/fluid/swab/floor) are the most superior and priority samples for diagnosis and sequencing of VZV, followed by oropharyngeal swab/nasopharyngeal swab, urine, and blood.
In summary, based on our findings, this is the first report revealing the presence of the VZV clade 9 variant in India. Among the sequences examined, three are associated with clade 9. Despite infection with the VZV clade 9 strain, there were no significant indications of heightened disease severity in the patients. Further studies warrant investigating the recombination patterns among wild-type and vaccinated populations to explore the snp evolution to help in disease monitoring and surveillance of VZV infections in India.
Author’s contribution
PDY and RRS made contributions to a critical evaluation, study design, data analysis, interpretation, and writing. The following individuals AK, LSR, KSPS, RRS, DYP, AMS, NI, KK, A, SGB, JS, SP, YJ, SP, TM, AV, JY, NS, VK, PV, SS and PG made contributions to the sample collection, data collecting, laboratory experiments, interpretation, writing, and critical review. PDY, RRS, AMS, DYP helped with the final editing and review.
Ethical statement
The study was approved by the Institutional Human Ethics Committee of ICMR-NIV, Pune, India under the project ‘Providing diagnostic support for referred samples of viral hemorrhagic fever and other unknown aetiology and outbreak investigation’. The clinical data collected were anonymized.
Supplemental Material
Download MS Word (183 KB)Acknowledgement
The authors extend their gratitude towards Dr. Sheela Godbole, Director-In Charge of ICMR-NIV, Pune for her continuous administrative support during this study.
Disclosure statement
There is no potential conflict of interest declared by the author(s).
Data availability statement
The sequence data generated during this study is available on the open-access database.
Additional information
Funding
References
- Arvin AM. Varicella-zoster virus. Clin Microbiol Rev. 1996;9(3):1–10. doi: 10.1128/CMR.9.3.361.
- Harpaz R, Ortega-Sanchez IR, Seward JF . Prevention of herpes zoster: recommendations of the advisory committee on immunization practices (ACIP). MMWR Recomm Rep. 2008;57(RR-5):1–30.
- Singh MP, Singh G, Kumar A, et al. Epidemiologic lessons: chickenpox outbreak investigation in a rural community around Chandigarh, North India. Indian J Pathol Microbiol. 2011;54(4):772–774.
- Barry M, Akkielah L, Al-Tawfiq AJ, et al. Not every skin rash in a returning adult male traveler is monkeypox. Travel Med Infect Dis. 2022; 50:102432. doi: 10.1016/j.tmaid.2022.102432.
- MacNeil A, Reynolds MG, Braden Z, et al. Transmission of atypical Varicella-Zoster virus infections involving palm and sole manifestations in an area with monkeypox endemicity. Clin Infect Dis. 2009;48(1):e6–e8. Jandoi: 10.1086/595552.
- Davison AJ, Scott JE. The complete DNA sequence of Varicella-Zoster virus. J Gen Virol. 1986;67 (Pt 9)(9):1759–1816. doi: 10.1099/0022-1317-67-9-1759.
- Cohen JI. The varicella-zoster virus genome. Curr. Top. Microbiol. Immunol. 2010;342:1–14.
- Oliver SL, Yang E, Arvin AM. Varicella-zoster virus glycoproteins: entry, replication, and pathogenesis. Curr Clin Microbiol Rep. 2016;3(4):204–215. doi: 10.1007/s40588-016-0044-4.
- Zhang Z, Selariu A, Warden C, et al. Genome-wide mutagenesis reveals that ORF7 is a novel VZV skin-tropic factor. PLoS Pathog. 2010;6(7):e1000971. doi: 10.1371/journal.ppat.1000971.
- LaRussa P, Lungu O, Hardy I, et al. Restriction fragment length polymorphism of polymerase chain reaction products from vaccine and wild-type varicella-zoster virus isolates. J Virol. 1992;66(2):1016–1020. doi: 10.1128/JVI.66.2.1016-1020.1992.
- Chanasit JS, Sauerbrei A. Evolution and world-wide distribution of Varicella–Zoster virus clades. Infect Genet Evol. 2011;11(1):1–10. doi: 10.1016/j.meegid.2010.08.014.
- Loparev VN, Gonzalez A, Deleon CM, et al. Global identification of three major genotypes of Varicella-Zoster virus: longitudinal clustering and strategies for genotyping. J Virol. 2004;78(15):8349–8358. doi: 10.1128/JVI.78.15.8349-8358.2004.
- Breuer J, Grose C, Norberg P, et al. A proposal for a common nomenclature for viral clades that form the species varicella-zoster virus: summary of VZV nomenclature meeting 2008, Barts and the London School of Medicine and Dentistry, 24-25 July 2008. J Gen Virol. 2010; 91(Pt 4):821–828. doi: 10.1099/vir.0.017814-0.
- Zell R, Taudien S, Pfaff F, et al. Sequencing of 21 varicella-zoster virus genomes reveals two novel genotypes and evidence of recombination. J Virol. 2012;86(3):1608–1622. doi: 10.1128/JVI.06233-11.
- Norberg P, Depledge DP, Kundu S, et al. Recombination of globally circulating varicella-zoster virus. J Virol. 2015;89(14):7133–7146. doi: 10.1128/JVI.00437-15.
- Jensen NJ, Rivailler P, Tseng FH, et al. Revisiting the genotyping scheme for varicella-zoster viruses based on whole-genome comparisons. J Gen Virol. 2017;98(6):1434–1438. doi: 10.1099/jgv.0.000772.
- Loparev VN, Rubtcova EN, Bostik V, et al. Identification of five major and two minor genotypes of varicella-zoster virus strains: a practical two-amplicon approach used to genotype clinical isolates in Australia and New Zealand. J Virol. 2007;81(23):12758–12765. doi: 10.1128/JVI.01145-07.
- Bryant P, Yildirim T, Griesemer SB, et al. Vaccine strain and wild-type clades of Varicella-Zoster virus in Central nervous system and Non-CNS disease, New York state, 2004–2019. J Clin Microbiol. 2022;60(4):e0238121. doi: 10.1128/jcm.02381-21.
- Vaidya SR, Tilavat SM, Kumbhar NS, et al. Chickenpox outbreak in a tribal and industrial zone from the union territory of Dadra and Nagar Haveli, India. Epidemiol Infect. 2018;146(4):476–480. doi: 10.1017/S0950268818000201.
- Sahay RR, Yadav PD, Majumdar T, et al. Clinico-epidemiological investigation on Varicella Zoster virus indicates multiple clade circulation in Maharashtra state, India. Heliyon. 2018;4(8):e00757. doi: 10.1016/j.heliyon.2018.e00757.
- Nyayanit DA, Chaubal G, Sahay R, et al. Molecular characterization of varicella zoster virus isolated from clinical samples in India. Indian J Med Res. 2021;154(4):592–597. doi: 10.4103/ijmr.IJMR_434_19.
- Chen M, Wang ST, Liu Y, et al. Genetic characteristics of varicella zoster virus in Shandong province from 2020 to 2021. Zhonghua Yu Fang Yi Xue Za Zhi. 2022;56(8):1080–1086.
- Quinlivan M, Sengupta N, Papaevangelou V, et al. Use of oral fluid to examine the molecular epidemiology of varicella zoster virus in the United Kingdom and continental Europe. J Infect Dis. 2013;207(4):588–593. doi: 10.1093/infdis/jis649.
- Quinlivan M, Sengupta N, Breuer J. A case of varicella caused by coinfection with two different genotypes of Varicella-Zoster virus. J Clin Virol. 2009;44(1):66–69. doi: 10.1016/j.jcv.2008.09.004.
- Quinlivan M, Hawrami K, Barrett-Muir W, et al. The molecular epidemiology of Varicella-Zoster virus: evidence for geographic segregation. J Infect Dis. 2002;186(7):888–894. doi: 10.1086/344228.
- LaRussa P, Lungu O, Hardy I, et al. Restriction fragment length polymorphism of polymerase chain reaction products from vaccine and wild-type varicella-zoster virus isolates. J Virol. 1992;66(2):1016–1020.
- Hondo R, Yogo Y, Yoshida M, et al. Distribution of varicella-zoster virus strains carrying a Pst1 site-less mutation in Japan and DNA change responsible for the mutation. Jpn J Exp Med. 1989;59(6):233–237.
- Harbecke R, Oxman MN, Arnold BA, et al. A real time PCR assay to identify and discriminate among wild-type and vaccine strains of varicella zoster virus and herpes simplex virus in clinical specimens, and comparison with the clinical diagnoses. J Med Virol. 2009;81(7):1310–1322. doi: 10.1002/jmv.21506.
- Higashimoto Y, Ihira M, Ohta A, et al. Discriminating between varicella-zoster virus vaccine and wild-type strains by loop-mediated isothermal amplification. J Clin Microbiol. 2008;46(8):2665–2670. doi: 10.1128/JCM.00216-08.
- Ihira M, Higashimoto Y, Kawamura Y, et al. Cycling probe technology to quantify and discriminate between wild-type Varicella-Zoster virus and Oka vaccine strains. J Virol Methods. 2013;193(2):308–313. doi: 10.1016/j.jviromet.2013.06.031.
- Loparev VN, Argaw T, Krause PR, et al. Improved identification and differentiation of varicella-zoster virus (VZV) wild-type strains and an attenuated varicella vaccine strain using a VZV open reading frame 62-based PCR. J Clin Microbiol. 2000;38(9):3156–3160. doi: 10.1128/JCM.38.9.3156-3160.2000.
- Kaushik KS, Lahiri KK, Kapila K, et al. Differentiation of wild-type varicella-zoster strains from India and the oka vaccine strain using a VZV open reading frame-62 based PCR-RFLP technique. Brazi J Infect Dis. 2008;12(4):313–315.
- Thiele S, Borschewski A, Küchler J, et al. Molecular analysis of varicella vaccines and varicella-zoster virus from vaccine-related skin lesions. Clin Vaccine Immunol. 2011;18(7):1058–1066. doi: 10.1128/CVI.05021-11.
- Schmader KE, Levin MJ, Gnann Jr JW, et al. Efficacy, safety, and tolerability of herpes zoster vaccine in persons aged 50–59 years. Clin Infect Dis. 2012;54(7):922–928. doi: 10.1093/cid/cir970.
- Vazquez M, Larussa PS, Gershon AA, et al. The effectiveness of the varicella vaccine in clinical practice. N Engl J Med. 2001;344(13):955–960. doi: 10.1056/NEJM200103293441302.
- LaRussa P, Steinberg SP, Shapiro E, et al. Viral strain identification in varicella vaccines with disseminated rashes. Pediatr Infect Dis J. 2000;19(11):1037–1039. doi: 10.1097/00006454-200011000-00002.
- Jezek Z, Szczeniowski M, Paluku KM, et al. Human monkeypox: confusion with chickenpox. Acta Trop. 1988;45(4):297–307.
- Rimoin AW, Kisalu N, Kebela-Ilunga B, et al. Endemic human monkeypox, democratic republic of Congo, 2001–2004. Emerg Infect Dis. 2007;13(6):934–937. doi: 10.3201/eid1306.061540.
- Leung J, McCollum AM, Radford K, et al. Varicella in Tshuapa province, democratic republic of Congo, 2009–2014. Trop Med Int Health. 2019;24(7):839–848. doi: 10.1111/tmi.13243.
- Breman JG, Henderson DA. Diagnosis and management of smallpox. N Engl J Med. 2002;346(17):1300–1308. doi: 10.1056/NEJMra020025.
- Jezek Z, Szczeniowski M, Paluku KM, et al. Human monkeypox: clinical features of 282 patients. J Infect Dis. 1987;156(2):293–298. doi: 10.1093/infdis/156.2.293.
- McCollum AM, Damon IK. Human monkeypox. Clin Infect Dis. 2014;58(2):260–267. doi: 10.1093/cid/cit703.