
Abstract
Background: Copy number variants of uncertain significance (VUS) has brought much distress for patients and great counselling challenges for clinicians. Of these, a special type of VUS (HT-VUS), harbouring one or both breakpoints within the established haploinsufficient or triplosensitive genes, were considered to be more likely to cause clinical effects compared with other types of VUS.
Methods: We retrospectively evaluated the properties and clinical significance of those HT-VUS samples in clinical testing for chromosome microarray analysis (CMA).
Results: A total of 7150 samples were selected for HT-VUS screening, and 75 (1.05%) subjects with 75 HT-VUS were found. The majority of these HT-VUS were heterozygous duplications and chromosome X had the most HT-VUS. The prevalence of HT-VUS was 0.90% (28/3116) for prenatal low-risk samples, 1.18% (26/2196) for prenatal high-risk samples, 1.37% (10/728) for postnatal samples and 0.99% (11/1110) for early pregnancy loss samples. However, the incidence of HT-VUS was not statistically different between different groups.
Conclusions: HT-VUS (deletions or duplications) involving introns and HT-VUS (duplications) including terminal coding exons (either the first or last exons) might be clinically neutral. Our study will be helpful for both interpretation and genetic counselling in the future.
KEY MESSAGES
This study assessed the clinical impact and features of a special type of copy number variants of uncertain significance (HT-VUS) in samples from CMA retrospectively.
Out of 7150 samples screened, 75 (1.05%) subjects had HT-VUS. Most HT-VUS were heterozygous duplications and chromosome X had the highest frequency of HT-VUS.
HT-VUS (deletions or duplications) involving introns and HT-VUS (duplications) including terminal coding exons might be clinically neutral. This study would be helpful for future interpretation and genetic counselling.
Introduction
Copy number variants (CNVs) have been implicated in the genetic aetiology of multiple disorders [Citation1–3]. With the rapid development of high-resolution CNVs detection platforms, a large series of CNVs have been reported [Citation4,Citation5]. Of these, the uncertain significance of CNVs (VUS) brought much anxiety for those who were tested and great challenges for genetic counselling [Citation5–7].
According to the 2019 ACMG classification guidelines, the clinical significance of CNVs was categorized into five classes: Benign, Likely benign, Variants of uncertain significance, Likely pathogenic, and Pathogenic [Citation8]. The VUS category is the broadest CNVs with scores between −0.89 and 0.89 points. During the process of analysis and interpretation of VUS, it is important to note one special type of VUS (termed as HT-VUS in the present study) with one or both breakpoints within the established haploinsufficient (HI) or triplosensitive (TS) genes (a score of 3 in Dosage Sensitivity Map [Citation9,Citation10]). It is reported that these VUS might be more likely to cause clinical effects due to overlapping with known pathogenic genes, although the final categories need to be further evaluated combined with more evidence such as breakpoint locations, involvement of coding sequence and literature evidences [Citation8].
In recent years, several studies have indicated the crucial roles of HT-VUS in a variety of diseases including intelligence disability (ID), developmental delay (DD), autistic spectrum disorder (ASD) and schizophrenia [Citation11–14]. These HT-VUS involving HI/TS genes included NRXN1 (partial deletions or duplications) [Citation11–14], MYT1L (partial duplications) [Citation15] and MBD5 (partial deletions or duplications) [Citation16–18]. However, all the above patients with HT-VUS were postnatal, and the prevalence and distribution of these CNVs in prenatal settings and miscarriage remain unknown.
In this study, we retrospectively analyzed CNVs data from 9910 subjects undergoing CMA clinical testing and systematically evaluated the incidence and distribution of HT-VUS in prenatal, postnatal and miscarriage samples. Furthermore, we evaluated their properties and clinical significance to provide more evidence for the assessment of CNV pathogenicity and genetic counselling.
Materials and methods
Study population
A retrospective study was conducted in Nanjing Maternity and Child Health Care Hospital (Jiangsu, China) during the period from 1 June 2011 to 30 September 2020. A total of 9910 subjects requesting for CMA testing in the Department of Prenatal Diagnosis were enrolled in the present study. By clinical indication, all samples were divided into four groups described below: (1) prenatal low-risk group: pregnancies with advanced maternal age (age ≥ 35, AMA), abnormal aneuploidy screening, history of a genetic disorder in the family (HGD), parental request, and ultrasound soft markers (foetal pyelectasis, echogenic intracardiac focus, thickened nuchal fold, choroid plexus cysts, single umbilical artery, enlarged cisterna magna (≥ 10 mm), short femur length, echogenic foci in bowel, absent or hypoplasia of nasal bone, aberrant right subclavian artery); (2) prenatal high-risk group: pregnancies with foetal structural anomalies, increased nuchal translucency (NT ≥ 3.0 mm), mild ventriculomegaly (10–15mm), intrauterine growth restriction (IUGR) and poly/oligohydramnios; (3) postnatal group: cases with DD/ID, multiple congenital anomalies (MCA) and ASD; (4) early pregnancy loss group: women who had pregnancy loss before 13 weeks gestation. After excluding those subjects (n = 2760) with pathogenic or likely pathogenic chromosomal abnormalities (numerical chromosomal abnormalities, partial aneuploidy, pathogenic or likely pathogenic microdeletions/microduplications and uniparental disomy), 7150 samples with non-pathogenic chromosomal abnormalities (with benign, likely benign, VUS CNVs or without any CNVs) were included for further HT-VUS screening (). All subjects provided written informed consent and agreed to publish their personal case details. All the procedures in our study were approved by Nanjing Maternity and Child Health Care Hospital Ethics Committee (2021KY004).
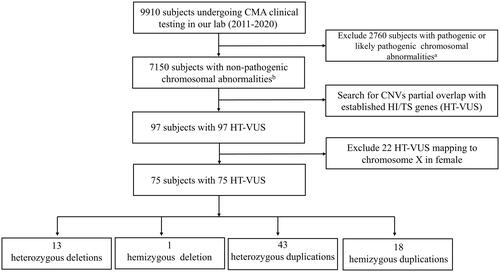
Figure 1. Flow diagram of the HT-VUS selection. HT-VUS, those copy number variants of uncertain significance partially overlapping with the haploinsufficient or triplosensitive genes; CMA, chromosomal microarray analysis; CNVs, copy number variants; HI, haploinsufficiency; TS, triplosensitivity; a, pathogenic or likely pathogenic chromosomal abnormalities include numerical chromosomal abnormalities, partial aneuploidy, pathogenic or likely pathogenic microdeletion/microduplication and uniparental disomy; b, non-pathogenic chromosomal abnormalities include benign or likely benign CNVs or without any CNVs.
Chromosomal microarray analysis
The Affymetrix CytoScan 750K array (Affymetrix, Santa Clara, CA), containing about 750,000 markers for copy number analysis, was performed to detect CNVs during the study period. The 750K array methodology has been described in detail previously [Citation19]. Cytogenetic analysis was performed using Chromosome Analysis Suite Software (Affymetrix, Santa Clara, CA) and CNVs coordinates were determined according to the human genome GRCh37/hg19 assembly. The CNVs size reporting criteria were called at a minimal resolution of 100 kb within the backbone coverage and 50 kb within those well-established HI or TS genes which were covered by higher probe density. Those CNVs with sizes small than 100 kb were confirmed by MLPA (Multiplex Ligation-dependent Probe Amplification) or Affymetrix CytoScan HD array (Affymetrix, Santa Clara, CA) containing about 2.7 million markers for copy number analysis. The clinical significance of CNVs was re-evaluated according to the latest CNVs interpretation standards [Citation8]. HT-VUS were defined as those VUS with one or both breakpoints within the HI or TS genes in the ClinGen database (https://www.ncbi.nlm.nih.gov/projects/dbvar/clingen/) [Citation10]. In this study, those genes (score = 2) were also included considering their scores might be upgraded to 3 as the continuous updating of the ClinGen database. In total, 374 HI genes (316 score for 3, 58 score for 2) and 7 TS genes (3 score for 3, 4 score for 2) were selected in this study (Supplementary Table 1). To comprehensively investigate the features and implications of HT-VUS in various clinical scenarios, we focused on both copy number loss and gain for HI genes and TS genes in this study.
HT-VUS were divided into three types: (1) One-5′: CNVs with one breakpoint within the 5′ end of an established HI or TS gene (3′ end of the gene not involved); (2) One-3′: CNVs with one breakpoint within the 3′ end of an established HI or TS gene (5′ end of the gene not involved); (3) Two: CNVs with two breakpoints within an established HI or TS gene.
Postnatal follow-up
For prenatal samples undergoing invasive prenatal diagnosis, we conducted postnatal follow-up by telephone interviews from birth to six years old. The outcomes of interest included normal postnatal phenotype, pregnancy termination, and other obvious abnormal phenotypes.
Statistical analysis
Statistical comparisons between groups were performed using the χ2 test or Fisher exact test. p-values < 0.05 were considered significant.
Results
The selection process of HT-VUS in our study
A total of 9910 subjects requesting CMA testing were included in our study. After excluding 2760 with pathogenic or likely pathogenic chromosomal abnormalities, 7150 subjects were included for further analysis. By searching for those CNVs overlapping with established HI/TI genes, 97 subjects with 97 HT-VUS were found. Among these, 22 heterozygous HT-VUS mapping to chromosome X in females were excluded from further analysis due to the complexity of deciphering the clinical significance. Finally, we found 75 subjects with 75 HT-VUS including 13 (17.3%, 13/75) heterozygous deletions, 1 (1.3%, 1/75) hemizygous deletion, 43 (57.3%, 43/75) heterozygous duplications and 18 (24.0%, 18/75) hemizygous duplications (). Detailed HT-VUS-related information is described in Supplementary Table 2.
Characteristic analysis of the HT-VUS
Among 75 subjects with HT-VUS, there were 54 (72.0%, 54/75) subjects with only HT-VUS and 21 (28.0%, 21/75) subjects with secondary VUS (Supplementary Table 2). These HT-VUS involved 35 HI genes and one TS gene (APP) (). Among the 35 HI genes, NRXN1 was the most commonly observed HI gene (n = 10). To investigate these HT-VUS better, we mapped these HI genes into human chromosomes and found they were enriched in 14 chromosomes (chr 1, 2, 7, 8, 9, 10, 11, 12, 14, 15, 16, 17, 21 and X), among which, chromosome X had the most HI genes (n = 9) and the most HT-VUS (n = 19) ().
Table 1. List of established HI or TS genes partial overlap with HT-VUS.
Through analysis of gene-associated phenotypes, we found these genes were most associated with developmental neuropsychiatric disorders such as autistic spectrum disorder (ASD) (e.g., NRXN1, SHANK2), schizophrenia (e.g., NRXN1) and mental retardation (e.g., MYT1L, MBD5, TRIP12, GLI3, KMT2C, DYRK1A, CNKSR2, IL1RAPL1).
Evaluating the incidence and clinical significance of HT-VUS in clinical population requesting for CMA testing
To identify the potential impact of HT-VUS in the clinical population, we evaluated the incidence and distribution of HT-VUS in prenatal, postnatal and miscarriage samples. Overall, the incidence of HT-VUS was 0.90% (28/3116) in prenatal low-risk samples, 1.18% (26/2196) in prenatal high-risk samples, 1.37% (10/728) in postnatal samples and 0.99% (11/1110) in early pregnancy loss samples, respectively (). By selecting the prenatal low-risk group as a control, we found the incidence of HT-VUS was not statistically different between different groups.
Table 2. Distribution of HT-VUS in clinical population requesting for CMA testing.
To provide a better understanding of the potential clinical significance of HT-VUS, deletions and duplications were analyzed separately. All 14 deletions of HT-VUS belonged to the type “Two” (CNVs with two breakpoints within an established HI or TS gene) and involved only introns of HI genes ( and Supplementary Table 2). The incidence of HT-VUS (deletions) was not statistically different between different groups. Among 61 subjects with HT-VUS (duplications), 21 with the type One-5′, 36 with the type One-3′ and 4 with the type “Two” (involving only introns of HI genes) were identified. Similarly, there were no significant differences in the detection rates of HT-VUS (duplications) between the four groups.
Notably, five DMD-related HT-VUS were identified in our study with two deletions and three duplications (Supplementary Table 2). Among these, two male foetuses carried the DMD (NM_004006.3) exon 64–79 (case 83) and 62–79 (case 84) duplications respectively. Both of them were inherited from their unaffected mothers and maternal grandfathers. A female foetus (case 85) carried the DMD exon 2 deletion which was inherited from her asymptomatic father. The other two foetuses carried the DMD intron 60 deletion (case 86) and exon 51–79 duplications (case 87) respectively, however, there was no further information about their parental results.
Postnatal follow-up assessments for prenatal samples undergoing invasive prenatal diagnosis
We performed postnatal follow-up for 46 amniotic fluid samples and obtained the following results: (1) For the 18 prenatal high-risk samples: 16 had no obvious abnormality at birth or in subsequent follow up, while two foetuses’ parents elected termination of pregnancy (Case 5 and Case 32). In case 5, a third-trimester ultrasound revealed a gastrointestinal malformation, along with the previously diagnosed ventricular septal defect. Case 32 had no other abnormalities detected by third-trimester ultrasound, except for the previously found ventricular septal defect. However, the parents decided to terminate the pregnancy due to the concern that the de novo HT-VUS could lead to other abnormal postnatal phenotypes; (2) For the 28 prenatal low-risk samples: 25 had no obvious abnormality after follow-up, while three were lost to follow-up (Supplementary Table 2).
Discussion
Interpretation and genetic counselling of VUS have long been difficult [Citation2,Citation20]. Through expanding the laboratory’s threshold size for reporting (e.g., 500 Kb for deletions, 1 Mb for duplications), the number of VUS has decreased markedly [Citation21,Citation22]. However, many reported VUS remains challenging for genetic counsellors. Of these, the special VUS (HT-VUS), with one or both breakpoints within the established HI or TS genes, may be more likely to cause clinical effects owing to overlapping important genes. Therefore, these types of VUS may be significant and valuable to be further studied.
In this study, 75 subjects with 75 HT-VUS were found (females with heterozygous HT-VUS in chromosome X were excluded), of which, the majority were heterozygous duplications and chromosome X had the most HI genes and the most HT-VUS. Furthermore, we found 0.90% of the prenatal low-risk population, 1.18% of prenatal high-risk population, 1.37% of postnatal population and 0.99% of early pregnancy loss population carried HT-VUS, however, the incidence was not statistically different between different groups. To further investigate the significance of HT-VUS, we evaluated deletions and duplications separately, and found the incidences of deletions and duplications were also not statistically different between different groups. One possible explanation was that that deletions and the type “Two” duplications of HT-VUS in our study only involved introns and might be non-pathogenic. Indeed, it was reported that intronic CNVs were common, often recurrent and might have no clinical significance [Citation11,Citation23,Citation24]. Besides, the type “one” duplications might also be not deleterious because functional gene structure may be preserved [Citation8]. In addition, the postnatal follow-up results also showed that most of the foetuses carrying HT-VUS had no obvious abnormal phenotype after birth. These results suggested that HT-VUS (deletions or duplications) involving introns and HT-VUS (duplications) including terminal coding exons (either the first or last exons) might have no apparent clinical effects.
Particularly, our study suggested that the duplication including the last exon and the deletion involving only the second exon of the DMD gene might be clinically benign. The DMD gene, consisting of 79 exons, is one of the largest human genes, and DMD mutations cause Duchenne and Becker muscular dystrophies (DMD/BMD). Previous literature reported a male BMD patient carrying a DMD exon 64–79 duplication with unknown inheritance information [Citation25]. Besides, a maternal DMD exon 64–79 duplication was detected in a 3-year-old boy manifesting autism and mild neuromotor delays [Citation26]. However, there was no family pedigree analysis especially without other male carriers in both above reports. In our study, the DMD exon 64–79 and 62–79 duplications were identified in two male foetuses and both duplications were inherited from their unaffected maternal grandfathers. Considering that the functional gene structure of DMD might be preserved, it is suggested that DMD duplications including the last exon might be clinically neutral. Wein et al. reported a deletion of DMD exon 2 in an Italian male with entirely normal neurological examination at 15 years of age. Besides, no muscular dystrophy history was ever reported in his family. The authors raised the point that it might be due to the translation from a DMD exon 5 internal ribosome entry site resulting in a functional dystrophin isoform that attenuates dystrophinopathy [Citation27]. This is consistent with our data showing a asymptomatic father carrying the DMD exon 2 deletion. Taken together, we speculated that subjects carrying the duplication including the last exon and the deletion involving only the second exon of the DMD gene might be asymptomatic or exceedingly mild.
Our study also has some limitations. The first limitation of our study is its retrospective nature; thus some detailed information of subjects were not available. Second, our CMA results did not exclude other genetic aetiologies such as nucleotide base-pair changes, balanced rearrangements and epigenetic events, which may be the real genetic aetiology for corresponding phenotypic features. Another limitation of our study is that we did not investigate the orientation of the duplication involving HI genes, which might affect their expression and regulation. Future studies might be helpful to elucidate this aspect.
In general, our study is the first to systematically evaluate the characteristics, distribution and clinical significance of HT-VUS in prenatal, postnatal and miscarriage samples, which will be helpful for both CNVs interpretation and genetic counselling in the future.
Authors’ contributions
ZR designed the study and drafted the manuscript. JJ performed data analysis and comparison, and revised the manuscript. WY drafted and revised the manuscript. MLL and LYM performed data collection. XYY analyzed the data. HP and XZF provided guiding suggestion. All the authors read and approved the final manuscript.
Supplemental Material
Download Zip (56.8 KB)Acknowledgements
We greatly appreciate all the participants for their cooperation in this study.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Additional information
Funding
References
- Manning M, Hudgins L, Professional P, et al. Array-based technology and recommendations for utilization in medical genetics practice for detection of chromosomal abnormalities. Genet Med. 2010;12(11):1–7. doi: 10.1097/GIM.0b013e3181f8baad.
- Miller DT, Adam MP, Aradhya S, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86(5):749–764. doi: 10.1016/j.ajhg.2010.04.006.
- American College of Obstetricians and Gynecologists Committee on Genetics. Committee Opinion No. 581: the use of chromosomal microarray analysis in prenatal diagnosis. Obstet Gynecol. 2013;122(6):1374–1377. doi:10.1097/01.AOG.0000438962.16108.d1
- Hollenbeck D, Williams CL, Drazba K, et al. Clinical relevance of small copy-number variants in chromosomal microarray clinical testing. Genet Med. 2017;19(4):377–385. doi: 10.1038/gim.2016.132.
- Kearney HM, Thorland EC, Brown KK, et al. American college of medical genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet Med. 2011;13(7):680–685. doi: 10.1097/GIM.0b013e3182217a3a.
- Bernhardt BA, Soucier D, Hanson K, et al. Women’s experiences receiving abnormal prenatal chromosomal microarray testing results. Genet Med. 2013;15(2):139–145. doi: 10.1038/gim.2012.113.
- Lou S, Lomborg K, Lewis C, et al. It’s probably nothing, but… ‘couples’ experiences of pregnancy following an uncertain prenatal genetic result. Acta Obstet Gynecol Scand. 2020;99(6):791–801. doi: 10.1111/aogs.13813.
- Riggs ER, Andersen EF, Cherry AM, et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American college of medical genetics and genomics (ACMG) and the clinical genome resource (ClinGen). Genet Med. 2020;22(2):245–257. doi: 10.1038/s41436-019-0686-8.
- Riggs ER, Church DM, Hanson K, et al. Towards an evidence-based process for the clinical interpretation of copy number variation. Clin Genet. 2012;81(5):403–412. doi: 10.1111/j.1399-0004.2011.01818.x.
- Rehm HL, Berg JS, Brooks LD, et al. –The clinical genome resource. N Engl J Med. 2015;372(23):2235–2242. doi: 10.1056/NEJMsr1406261.
- Cosemans N, Vandenhove L, Vogels A, et al. The clinical relevance of intragenic NRXN1 deletions. J Med Genet. 2020;57(5):347–355. doi: 10.1136/jmedgenet-2019-106448.
- Lowther C, Speevak M, Armour CM, et al. Molecular characterization of NRXN1 deletions from 19,263 clinical microarray cases identifies exons important for neurodevelopmental disease expression. Genet Med. 2017;19(1):53–61. doi: 10.1038/gim.2016.54.
- Wiśniowiecka-Kowalnik B, Nesteruk M, Peters SU, et al. Intragenic rearrangements in NRXN1 in three families with autism spectrum disorder, developmental delay, and speech delay. Am J Med Genet B Neuropsychiatr Genet. 2010;153B(5):983–993. doi: 10.1002/ajmg.b.31064.
- Rujescu D, Ingason A, Cichon S, et al. Disruption of the neurexin 1 gene is associated with schizophrenia. Hum Mol Genet. 2009;18(5):988–996. doi: 10.1093/hmg/ddn351.
- Mansfield P, Constantino JN, Baldridge D. MYT1L: a systematic review of genetic variation encompassing schizophrenia and autism. Am J Med Genet B Neuropsychiatr Genet. 2020;183(4):227–233. doi: 10.1002/ajmg.b.32781.
- Tadros S, Wang R, Waters JJ, et al. Inherited 2q23.1 microdeletions involving the MBD5 locus. Mol Genet Genomic Med. 2017;5(5):608–613. doi: 10.1002/mgg3.316.
- Kushima I, Aleksic B, Nakatochi M, et al. High-resolution copy number variation analysis of schizophrenia in Japan. Mol Psychiatry. 2017;22(3):430–440. doi: 10.1038/mp.2016.88.
- Mullegama SV, Rosenfeld JA, Orellana C, et al. Reciprocal deletion and duplication at 2q23.1 indicates a role for MBD5 in autism spectrum disorder. Eur J Hum Genet. 2014;22(1):57–63. doi: 10.1038/ejhg.2013.67.
- Wang Y, Li Y, Chen Y, et al. Systematic analysis of copy-number variations associated with early pregnancy loss. Ultrasound Obstet Gynecol. 2020;55(1):96–104. doi: 10.1002/uog.20412.
- Tsuchiya KD, Shaffer LG, Aradhya S, et al. Variability in interpreting and reporting copy number changes detected by array-based technology in clinical laboratories. Genet Med. 2009;11(12):866–873. doi: 10.1097/GIM.0b013e3181c0c3b0.
- Buchanan JA, Chitayat D, Kolomietz E, et al. Prenatal genomic microarray and sequencing in Canadian medical practice: towards consensus. J Med Genet. 2015;52(9):585–586. doi: 10.1136/jmedgenet-2015-103223.
- Marcou CA, Pitel B, Hagen CE, et al. Limited diagnostic impact of duplications <1 Mb of uncertain clinical significance: a 10-year retrospective analysis of reporting practices at the Mayo clinic. Genet Med. 2020;22(12):2120–2124. doi: 10.1038/s41436-020-0932-0.
- Mullegama SV, Elsea SH. Intragenic MBD5 familial deletion variant does not negatively impact MBD5 mRNA expression. Mol Cytogenet. 2014;7(1):80. doi: 10.1186/s13039-014-0080-9.
- Yatsenko SA, Wood-Trageser M, Chu T, et al. A high-resolution X chromosome copy-number variation map in fertile females and women with primary ovarian insufficiency. Genet Med. 2019;21(10):2275–2284. doi: 10.1038/s41436-019-0505-2.
- De Wel B, Willaert S, Nadaj-Pakleza A, et al. Respiratory decline in adult patients with Becker muscular dystrophy: a longitudinal study. Neuromuscul Disord. 2021;31(3):174–182. doi: 10.1016/j.nmd.2020.12.010.
- Qiao Y, Tyson C, Hrynchak M, et al. Clinical application of 2.7M cytogenetics array for CNV detection in subjects with idiopathic autism and/or intellectual disability. Clin Genet. 2013;83(2):145–154. doi: 10.1111/j.1399-0004.2012.01860.x.
- Wein N, Vulin A, Falzarano MS, et al. Translation from a DMD exon 5 IRES results in a functional dystrophin isoform that attenuates dystrophinopathy in humans and mice. Nat Med. 2014;20(9):992–1000. doi: 10.1038/nm.3628.