Abstract
Von Willebrand disease (VWD) is the most frequent inherited bleeding disorder and is due to quantitative (types 1 and 3) or qualitative (type 2) defects of von Willebrand factor (VWF). VWD is inherited by autosomal dominant or recessive patterns, but women with mild forms are more symptomatic. VWD is classified in six VWD types (1, 2A, 2B, 2M, 2N, 3) with peculiar phenotype and genotype. The ristocetin cofactor activity (VWF:RCo) is the most useful test for VWD diagnosis, because it can mimic the interactions of VWF with its platelet receptor. Knowledge of the segments of VWF involved in the binding to its receptor and to factor VIII prompted the search for mutations in specific exons of the VWF gene, with mutations causing VWD types 2A, 2B, 2M, 2N localized in exons 18–28. In case of VWD types 1 and 3 the mutations are spread within the entire gene. Desmopressin (DDAVP) is the treatment of choice for type 1 VWD because it can induce release of normal VWF from cellular compartments. In type 3 and in severe forms of types 1 and 2 VWD, DDAVP is not effective and plasma virally inactivated VWF concentrates should be used in bleedings, surgery, and secondary long‐term prophylaxis.
| Abbreviations | ||
| VWD | = | von Willebrand disease |
| VWF | = | von Willebrand factor |
| FVIII | = | factor VIII |
| VWF:RCo | = | ristocetin cofactor activity of VWF |
| VWF:Ag | = | antigen of VWF |
| GPIbα | = | glycoprotein Ib α |
| FVIII/VWF | = | the complex of FVIII and VWF |
| RGD | = | arginine, glycine, aspartic acid |
| DDAVP | = | 1‐deamino‐8‐D‐arginine vasopressin, or desmopressin |
| PK | = | pharmacokinetics |
Introduction
Von Willebrand disease (VWD) is, together with hemophilia A, the most frequent bleeding disorder, with a prevalence of 66–100 cases per million in the general population, taking patients referred for clinical manifestations of bleeding as a basis of the estimate. Much higher prevalences (1 per 100) are reported in population‐based studies, but the clinical relevance of many of these cases is uncertain Citation1. Last year (2006) marked the 80th anniversary of the first description of the disease by Erik von Willebrand. He was an internist at the Deaconess Hospital in Helsinki, Finland. In April 1924, a 5‐year‐old girl from Föglo on the islands of Åland in the Gulf of Bothnia (between Finland and Sweden) was admitted to the hospital of Helsinki for investigation of severe bleedings from nose and gums. Her parents were cousins, and there was a bleeding history within the family: 11 siblings had a bleeding history, and 3 had died from massive menorrhagia and gastrointestinal bleeds. In contrast to hemophilia, the epitome of inherited bleeding disorders, both sexes were affected, and mucosal bleedings were the predominant symptoms. A prolonged bleeding time (BT) with a normal platelet count was the most important laboratory abnormality, and a functional disorder of the platelets associated with a systemic lesion of the vessel wall was suggested as a possible cause of the disease. Erik von Willebrand called this novel clinical disorder ‘hereditary pseudo‐hemophilia’, and this disease was named after him since then Citation2. However, it was not until 1971 that it was understood that the deficiency of a new factor, different from factor VIII (FVIII), was actually responsible for the disease. Theodore S. Zimmerman et al. produced an antiserum against a highly purified FVIII preparation and used it to determine a new plasma protein by an immunochemical technique. This protein, called at that time FVIII‐related antigen (actually VWF antigen), was present in normal individuals and those with hemophilia A but was lacking in VWD patients Citation3. Throughout nearly a century, a lot of progress was made in our understanding on von Willebrand factor (VWF), the protein deficient or defective in VWD, as well as on the molecular basis, natural history, and treatment of the disease Citation4.
Key messages
Von Willebrand disease (VWD) is the most frequent inherited disorder of hemostasis and is due to quantitative (VWD types 1 and 3) or qualitative (VWD type 2) defects of von Willebrand factor (VWF). Three criteria should be always satisfied for a correct VWD diagnosis: 1) a positive bleeding history in the patients since childhood; 2) reduced levels of VWF activity in plasma; and 3) a positive family history (inheritance pattern usually autosomal dominant; recessive in type 3).
The current classification in six VWD types (1, 2A, 2B, 2M, 2N, 3) is important to understand the basic mechanisms of VWF defects, to determine the risk of bleeding, and to select the best therapeutic approach. Molecular screening can be important to confirm phenotypic diagnosis.
Prenatal diagnosis is required only in the case of parents already known to be carriers of VWD type 3. Since young children with VWD type 3 might carry deletions of VWF gene that predispose to the alloantibodies to VWF, deletions should be searched for before starting extensive therapy with exogenous VWF concentrates.
Desmopressin (DDAVP) is the first‐line treatment in most VWD types 1 and 2, except for VWD type 2B because of the transient reduction in platelet counts. After excluding VWD 2B at diagnosis, all VWD types 1 and 2 should be exposed to an infusion trial with DDAVP to determine their biologic response to DDAVP.
Plasma‐derived VWF concentrates are safe and effective in the management of most bleeding episodes and in preventing bleeding during surgeries. They are indicated in VWD types 3 and 2B, in all VWD unresponsive to DDAVP, and in cases of tachyphylaxis after repeated injections of DDAVP. In case of repeated injections of concentrates, especially during surgery, factor VIII levels should be carefully observed before any decision on an additional dose.
Structure–function of von Willebrand factor
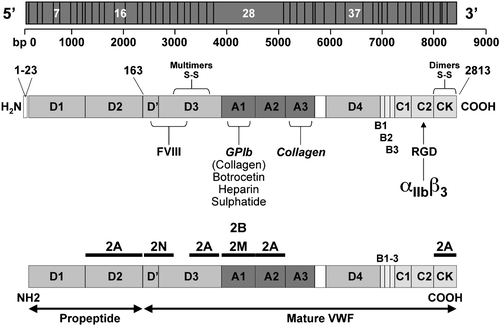
Von Willebrand factor (VWF) is a large circulating glycoprotein synthesized by endothelial cells and megakaryocytes Citation5. The gene encoding VWF, located on chromosome 12p13.2, is a large gene that spans 178 kilobases of DNA and contains 52 exons. A noncoding, highly homologous pseudogene was identified in chromosome 22, spanning the gene sequence from exon 23 to 34 Citation6. The primary product of the VWF gene is a 2,813 amino acid protein made of a signal peptide of 22 amino acids, an unusually large propeptide of 741 amino acids, and a mature subunit of 2,050 amino acids. In keeping with a recently proposed nomenclature Citation7, numbering of VWF starts from the first amino acid of the signal peptide, so that number 764 is the first amino acid of the mature protein. Different regions, corresponding to four types of repeated protein domains (D1, D2, D′, D3, A1, A2, A3, D4, B, C1, C2), are responsible for the different functions of VWF (). Mature VWF is the result of ordered maturation steps as it moves along the secretory pathway of endothelial cells, leading to the storage in Weibel‐Palade bodies and then to the constitutive or regulated secretion of a huge multimeric glycoprotein. Circulating VWF, which is mainly derived from the endothelium, has two major functions in hemostasis. It is essential for platelet adhesion to the subendothelium, platelet‐to‐platelet interactions, and platelet aggregation in vessels like small arteries and large stenotic arteries in which rapid blood flow results in high shear stress. Adhesion is promoted by the interaction of a region of the A1 domain with the platelet membrane glycoprotein Ibα (GPIbα) Citation5. High shear stress activates the A1 domain of VWF bound to subendothelial collagen by stretching the largest multimers into filamentous forms. The interaction between GPIbα and VWF can be mimicked in platelet‐rich plasma by ristocetin, which promotes binding of VWF to GPIbα. Aggregation of platelets within the growing hemostatic plug is promoted by the interaction of VWF with another platelet receptor, glycoprotein IIb–IIIa (or integrin αIIbβ3), which once activated binds to VWF and fibrinogen to recruit more platelets into a stable plug. Both these binding activities are highly expressed in the largest VWF multimers Citation5. VWF is also the carrier of factor VIII (FVIII) in plasma. VWF protects FVIII from proteolytic degradation, prolonging its half‐life in the circulation and efficiently localizing it at the site of vascular injury Citation8. Each VWF monomer has one FVIII‐binding domain located in the first 272 amino acids of the mature subunit (D′ domain). Therefore, any change in plasma VWF level is usually associated with a parallel change in FVIII. In this review article we use the recommended nomenclature and abbreviations of FVIII/VWF activities Citation9.
Figure 1. Schematic representation of the von Willebrand factor(VWF) gene located in chromosome 12: the main exons are indicated with the number of base pairs from 5′ to 3′ (upper panel). The structure of VWF functional domains: the pre‐pro‐VWF is indicated with amino acids numbered from the amino‐ (aa 1) to carboxy‐terminal portions (aa 2813) of VWF. Note the important CK and D3 domains for formation of VWF dimers and multimers. The native mature subunit of VWF, after the cleaving of the pre‐pro‐VWF, is described with its functional domains: the VWF binding sites for factor VIII (D′ and D3), GPIb, botrocetin, heparin, sulfatide, collagen (A1), collagen (A3) and the arginine, glycine, aspartic acid (RGD) sequence for binding to αIIbβ3 (middle panel). Distribution of VWF mutations in patients with VWD types 2: the positions of mutations causing VWD types 2A, 2B, 2M, 2N are indicated with black bars throughout the VWF domains (lower panel).
Classification of VWD
The current classification of VWD was proposed in 1994 Citation10 and was recently updated after a fruitful discussion by a panel of experts participating in the Working Party organized on behalf of the Sub‐Committee on VWF of the Scientific Standardization Committees of the International Society on Thrombosis and Haemostasis (ISTH‐SSC‐SC on VWF) Citation11. Six different VWD types have been proposed: VWD 1, 3, 2A, 2B, 2M, 2N. A partial quantitative defect marks type 1, whereas type 3 is characterized by the nearly total absence of VWF in plasma and platelets. Type 1 is easily distinguished from type 3 by milder VWF deficiency (usually in the range of 10–30 U/dL), the autosomal dominant pattern of inheritance, and the presence of a milder bleeding tendency Citation1. In the past type 1 was reported to be the most frequent form of VWD, accounting for approximately 70% of cases. A study based on the reappraisal of diagnoses of type 1 after 10 years (1994–2004) in 1,234 patients followed by 16 Italian centers has established that only 671/1234 (54%) were VWD type 1, because many cases previously diagnosed as type 1 were re‐diagnosed type 2 due to discrepant VWF measurements (ratio of ristocetin cofactor activity (VWF:RCo) to VWF:Ag <0.7) Citation12,13. The presence of VWF defects in previously diagnosed VWD type 1 has been recently shown in 154 families evaluated prospectively by the European study, as reported below.
Four VWD types 2 are identified, reflecting different pathophysiological mechanisms Citation10–13. Types 2A and 2B are marked by the absence of high‐molecular‐weight VWF multimers in plasma, but in type 2B there is also an increased affinity of VWF for GPIbα. The identification of qualitatively abnormal variants with decreased platelet‐dependent function and a normal multimeric structure marks subtype 2M. Type 2N shows a full array of multimers, the defect being in the N‐terminal region of the VWF where the binding domain for FVIII is located Citation14. This type is phenotypically distinguishable from mild hemophilia A only by the abnormal binding of FVIII to VWF (VWF:FVIIIB).
Patterns of inheritance
The inheritance pattern of VWD type 3 is autosomal recessive. In type 2 VWD patients, the pattern of inheritance is mainly autosomal dominant, even though rare cases with recessive pattern have been reported Citation1. The inheritance of the mild type 1 VWD is usually autosomal dominant, with variable phenotype and penetrance. In type 1 VWD, a number of genetic and nongenetic factors are likely to contribute to the wide variability of the clinical and laboratory phenotype. About 60% of the variation in VWF plasma is due to genetic factors, with ABO group accounting for only about 30%. In type O subjects the VWF level is 25%–35% lower than in non‐O individuals Citation15. Other factors outside the VWF gene, such as platelet polymorphisms, have been proposed to modify the bleeding tendency of type 1 VWD, as reported Citation16.
Clinical manifestations
Clinical manifestations are excessive mucocutaneous bleeding and prolonged oozing after surgical procedures. In women menorrhagia may be the only clinical manifestation. Soft tissue and joint bleeding are rare, except in patients with type 3 VWD and severe deficiencies of VWF and FVIII (prevalence approximately 1 per million in the general population). The clinical expression of the disease is usually mild in most patients with type 1, whereas severity increases in type 2 and particularly in type 3. Generally, the severity of bleeding correlates with the degree of reduction of VWF:RCo and FVIII. To date, only few detailed descriptions of symptoms are available Citation12,Citation13–18. shows the relative frequency of bleeding symptoms in three large series of patients diagnosed at specialized centers Citation12,Citation13–18. Several attempts were recently made to evaluate sensitivity and specificity of bleeding symptoms, specially in the mild cases with type 1 VWD and VWF:RCo levels >20U/dL. In a multicenter study carried out in obligatory carriers of type 1 VWD, menorrhagia and epistaxis were poor predictors of the disease, while cutaneous bleeding and bleeding after dental extractions were more sensitive symptoms for diagnosis Citation19. A bleeding severity score (BSS) has been developed () and validated in affected and nonaffected members of 154 families enrolled prospectively in a large European study, as well as in 200 normal individuals Citation20. This BSS should be derived from a questionnaire collecting detailed information about 12 different types of bleeds to be administered at the time of diagnosis in every new patient. Despite the fact that this BSS was investigated prospectively in VWD type 1 patients, this approach can be useful in all VWD. Preliminary data collected in our Center suggest that BSS can be a useful parameter to predict bleeding in VWD (data not shown).
Table I. Prevalence (%) of bleeding symptoms in patients with VWD from different cohorts and in normal individuals (adapted from Federici Citation12, Silwer Citation17, Lak et al. Citation18).
Table II. Bleeding score used to evaluate the bleeding history (see Tosetto et al. Citation20).
Laboratory diagnosis
The diagnosis of VWD, particularly type 1, may require several laboratory tests to be repeated at different occasions. These tests are usually applied for patients with suspected bleeding disorders, and summarizes the different steps for VWD diagnosis. The bleeding time, the original hallmark of the disease, is not always prolonged and may be normal in patients with mild forms, such as those with type 1 and normal platelet VWF content Citation21. Hence, it is not particularly useful for diagnosis. Additional tests for VWD diagnosis include the closure time (CT) and assays of VWF activity based on binding to collagen (VWF:CB). Evaluation of CT with the Platelet Function Analyzer (PFA‐100) gives a rapid and simple measure of VWF dependent platelet function at high shear stress: it can be performed in whole blood and therefore can be employed instead of the BT in children or when the BT is not feasible. This system is sensitive and reproducible for VWD screening, even though the CT is normal in type 2N and cannot be modified in VWD type 3 after the administration of VWF/FVIII concentrates Citation22. Assays are also available for VWF:CB and the ratio of VWF:CB to VWF:Ag appears useful for distinguishing types 1 and 2 Citation23,24. Therefore, VWF:CB should be performed always in association with VWF:RCo. Neither assay has been well standardized yet, and thus is not officially approved by the Scientific Standardization Committee on VWF of the International Society of Thrombosis and Haemostasis.
Table III. Clinical and laboratory parameters used for VWD diagnosis.
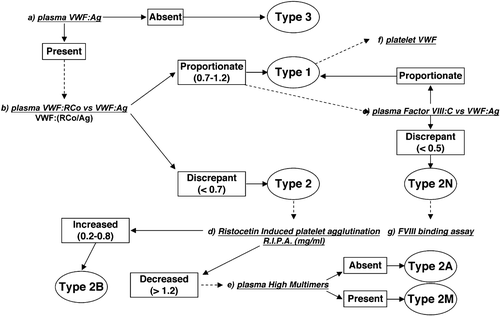
Based on these observations, the differential diagnosis of VWD types can be performed following the flow chart shown in . Type 3 VWD is diagnosed when VWF:Ag is undetectable or less than 1 U/dL. A proportionate reduction of both VWF:Ag and VWF:RCo, with a RCo/Ag ratio >0.7, suggests type 1 VWD. If the VWF:RCo/Ag ratio is <0.7, type 2 is diagnosed. Type 2B VWD is diagnosed when ristocetin‐induced platelet aggregation (RIPA) is heightened (< 0.8 mg/mL), whereas types 2A and 2M are usually associated with low RIPA (> 1.2 mg/mL). Multimeric analysis of plasma VWF is necessary to distinguish type 2A (largest and intermediate multimers lacking) from type 2M (all multimers present). Type 2N can be suspected when FVIII and VWF:Ag levels are discrepant (ratio <0.5), and the differential diagnosis with mild hemophilia A warrants the results of the specific test VWF:FVIIIB Citation14. In type 1 VWD the ratio between Factor VIII and VWF:Ag is always ⩾1, and the severity of the type 1 VWD phenotype can usually be evaluated from platelet VWF measurements Citation21.
Figure 2. Flowchart proposed for the diagnosis of different von Willebrand disease(VWD) types. Type 3 VWD can be diagnosed in case of unmeasurable VWF:Ag (a). A proportionate reduction of both VWF:Ag and VWF:RCo with a RCo/Ag ratio >0.7 suggests type 1 VWD (b). If the VWF:RCo/Ag ratio is <0.7 type 2 is diagnosed. Type 2B VWD (d) can be identified in case of heightened ristocetin‐induced platelet aggregation (RIPA) (<0.8 mg/mL), whereas types 2A and 2M cause low RIPA (>1.2 mg/mL). Multimeric analysis in plasma (e) is necessary to distinguish between type 2A VWD (lack of the largest and intermediate multimers) and type 2M VWD (all the multimers present). Type 2N VWD can be suspected in case of discrepant values for FVIII (c) and VWF:Ag (ratio <0.5), and diagnosis should be confirmed by the specific test (g) of VWF:factor VIII binding capacity (VWF:FVIIIB). In type 1 VWD the ratio between Factor VIII and VWF:Ag is always ⩾1 and the severity of the type 1 VWD phenotype can usually be evaluated from platelet VWF (f) measurements. (This figure was derived from that originally reported previously Citation12.)
No general consensus has been reached for measuring anti‐VWF antibodies in patients with VWD type 3. The assays are currently available in only a few specialized labs and they mimic the Bethesda assays for hemophilia inhibitors by performing VWF and FVIII activities in patient‐normal pool plasma mixtures after 2 hours' incubation at 37°C. The titer of anti‐VWF inhibitor is calculated by the current dilution of VWD plasma inhibiting 50% of normal plasma pool diluted 1:2 compared to control mixture. Several authors did use ristocetin‐induced platelet agglutination (RIPA) in normal platelet‐rich plasma (PRP) to measure anti‐VWF inhibitors Citation25. However, all VWF activities should be performed, such as anti‐VWF:antigen (anti‐VWF:Ag), anti‐VWF ristocetin cofactor (anti‐VWF:RCo), anti‐VWF collagen binding (anti‐VWF:CB) and anti‐factor VIII (anti‐FVIII). These techniques will be standardized by a working party organized on behalf of the ISTH‐SSC‐SC on VWF.
Molecular and prenatal diagnosis
Cloning the VWF gene has allowed the identification of several suitable restriction fragment length polymorphisms (RFLP) which demonstrate the cosegregation of VWD phenotype with haplotype‐specific RFLP patterns in family members of different kindred with VWD Citation1. Knowledge of the crucial segments of VWF involved in the interaction with GPIbα initially prompted the fruitful search for mutations in exon 28 of the VWF gene which encodes for the A1 and A2 domains of mature VWF as reported in (for review see Citation1). The search for mutations has been extended to additional VWF exons encoding for the other functional domains of VWF. Nowadays a registry of mutations identified in all VWD types is available in a website (www.shef.ac.uk/vwf).
Originally, the first mutations were found within exon 28 of VWF gene that is responsible for domains A1, A2, and A3. Most type 2A cases are due to missense mutations in the A1 domain, with R1597W or Q or Y and S1506L accounting for about 60% of them Citation26. The majority of type 2B cases are due to missense mutations in the A1 domain, about 90% being caused by R1306W, R1308C, V1316M, and R1341Q Citation26. A few heterogeneous mutations, also located within the A1 domain, underlie type 2M. A recurrent mutation in type 2M Vicenza has been identified in families from Europe (R1205H); another mutation (M740I) is seen exclusively in families from the Vicenza area in the northeast of Italy Citation27,28. Missense mutations in the FVIII‐binding domain at the amino‐terminal portion of VWF are responsible for type 2N (for review see Citation14). The molecular defects are located in specific VWF domains (): the list of the most frequent mutations of VWF associated with VWD types 2 are summarized in .
Table IV. List of the most frequent mutations in type 2A, 2B, 2M, and 2N according to VWF domains and exons of VWF gene.
The genetic causes of type 1 VWD are still elusive in many cases, especially in those with a mild phenotype. More information on the molecular basis of type 1 has been collected by two multicenter international studies. In the European study, recruitment was based on the historical diagnosis of type 1 VWD as made by 12 expert centers, which included 278 affected cases, 312 nonaffected family members, and 1166 controls Citation29. Three broad groups of patients were identified: 53 had a normal multimeric structure, a VWF:RCo/Ag ratio equal to or greater than 0.7, and mutations in the VWF gene; 55 had VWF gene mutations but abnormal multimers and a ratio lower than 0.7; and 43 had normal multimers, a ratio equal to or greater than 0.7, but no detectable mutation.
The Canadian investigators could recruit 123 families for which the index case had bleeding symptoms and VWF levels between 5 and 50 U/dL Citation30. In this study, subjects with abnormal multimeric patterns or other evidence of qualitative defects were excluded. The most important conclusions from both studies are the following: 1) despite the selection of patients based on bleeding history, candidate VWF mutations were not found for 27% (Canadian) and 36% (European) of index cases diagnosed with VWD type 1; and 2) the spectrum of VWD type 1 mutations was different from that found in VWF type 3 (see later), since about 90% of patients in whom mutations were found had at least one missense mutation, often associated with the loss or creation of cysteine residues. Therefore, VWD type 1 is not at all like heterozygous VWD type 3, because VWF defects that occur in VWD type 1 are usually caused by dominant VWF abnormalities that affect VWF secretion or clearance without substantially altering multimeric patterns or platelet binding.
In type 3 VWD, partial or total gene deletions have been initially reported Citation31. Notably, homozygosity for gene deletion may be associated with the appearance of alloantibodies against VWF, which may render replacement therapy ineffective and stimulate anaphylactic reactions to treatment Citation32. In general, mutations may be scattered over the entire gene, but some (e.g. 2680delC or Arg2535X) are particularly recurrent in Northern Europe (for review see Citation33). The coding region of the VWF gene contains 11 CGA codons (Arg). CG‐dinucleotides are hot spots for mutation, and a C to T mutation will result in a stop codon. Stop codons, in either homozygosity or compound heterozygosity, have been reported in exons 9, 28, 32, and 45 (for review see Citation33). Gene defects of type 3 VWD patients from three different populations have now been studied, but there was no founder effect, and mutations were distributed throughout the entire VWF gene Citation34.
Compared to hemophilia, most VWD patients show relatively mild bleeding symptoms. Therefore, prenatal diagnosis is required mainly in case of parents already known to be carriers of VWD type 3, with gene defects identified in their first affected child. Neonatal diagnosis can be performed in case of children born from parents with VWF defects already characterized, but phenotypic diagnosis of VWD should be always confirmed later on in the child and compared with the other affected members within the same family. Since young children with VWD type 3 might carry deletions of VWF gene that predispose to the alloantibodies to VWF, every new child with VWD type 3 should be intensively investigated by searching deletions, before starting extensive therapy with exogenous VWF concentrates. New methods for screening large deletions are now available Citation35.
Treatment
The goal of treatment is to correct the dual defects of hemostasis, i.e. abnormal platelet adhesion due to low or defective VWF, and abnormal intrinsic coagulation due to low FVIII (for review and additional references, see Citation36). Two main therapeutic approaches are available: desmopressin (DDAVP), that releases endogenous VWF from endothelial cells, and exogenous VWF contained in VWF plasma‐derived concentrates.
Desmopressin
Desmopressin (1‐deamino‐8‐D‐arginine vasopressin (DDAVP)) is a synthetic analogue of vasopressin that is relatively inexpensive and carries no risk of transmitting blood‐borne infections agents. DDAVP, infused intravenously at a dose of 0.3 µg/kg diluted in 50 mL saline over 30 minutes, usually increases plasma VWF and FVIII three to five times above baseline levels within 30–60 minutes, and, in general, high VWF and FVIII levels last for 6–8 hours Citation37. Because the responses in a given patient are consistent on different occasions, a test dose of DDAVP at the time of diagnosis is recommended to establish the individual response patterns Citation12. The protocol of infusion test, with the clinical and laboratory parameters to be measured, is reported in details in . DDAVP infusions can be repeated every 12–24 hours depending on the type and severity of the bleeding episode. However, most patients repeatedly treated become less responsive to therapy. The drug is also available in concentrated forms for subcutaneous and intranasal administration, which can be convenient for home treatment. Repeated administrations in the same subject may induce reduced effects, as has been reported Citation38. Despite the widespread use of DDAVP in the treatment of VWD, there are no prospective clinical studies on efficacy and safety aimed to determine benefits and limit of this therapeutic approach. An investigator‐driven prospective study on clinical efficacy of desmopressin in 200 patients with VWD types 1 and 2 has been organized on behalf of the Subcommittee on VWF of the Scientific Standardization Committees of the International Society on Thrombosis and Haemostasis: the clinical response to desmopressin will be evaluated prospectively for 24 months during bleeding episodes and during minor or major surgeries in the VWD patients who were exposed to an infusion trial at enrollment.
Table V. Recommendations for the infusion test with DDAVP
Side effects of DDAVP are usually mild tachycardia, headache, and flushing: these are attributed to the vasomotor effects of the drug and can often be attenuated by slowing the rate of infusion. Hyponatremia and volume overload due to the antidiuretic effects of DDAVP are relatively rare. A few cases of hyponatremia and seizures have been described, mostly in young children who received closely repeated infusions Citation39. Though no thrombotic episodes have been reported in VWD patients treated with DDAVP, this drug should be used with caution in elderly patients with atherosclerotic disease, because a few cases of myocardial infarction and stroke have occurred in hemophiliacs and uremic patients given DDAVP Citation40,41.
Adjunctive therapies for VWD
Antifibrinolytic amino acids are synthetic drugs that interfere with the lysis of newly formed clots by saturating the binding sites on plasminogen, thereby preventing its attachment to fibrin and making plasminogen unavailable within the forming clot. Epsilon aminocaproic acid (50 mg/kg four times a day) and tranexamic acid (15–25 mg/kg three times a day) are the most frequently used antifibrinolytic amino acids. Both can be administered orally, intravenously or topically and are useful alone or as adjuncts in the management of oral cavity bleeding, epistaxis, gastrointestinal bleeding, and menorrhagia. They should be avoided in the management of urinary tract bleeding.
Estrogens raise plasma VWF levels, but the response is variable and unpredictable, so they are not widely used for therapeutic purposes. It is common clinical experience that the continued use of oral contraceptives is very useful in reducing the severity of menorrhagia in women with VWD, even in those with type 3, despite the fact that FVIII/VWF levels are not modified.
Transfusional therapies
VWF/FVIII concentrates are indicated in type 3 VWD, in type 2B because DDAVP can induce transient thrombocytopenia, and in all types 1 and 2 patients who are not responsive to DDAVP or who may have contraindications to its use. Minimal requirements for plasma‐derived VWF/FVIII concentrates in VWD management are the following: 1) they must contain VWF and some FVIII:C; 2) they should be treated by virucidal methods; and 3) before clinical use, they should be tested for pharmacokinetics (PK) and efficacy in retrospective and prospective clinical trials in relatively large numbers of VWD patients. Among several VWF concentrates, only four have been extensively evaluated in pharmacokinetic (PK) trials as well as in retrospective or prospective efficacy studies in VWD (for review and additional references see Citation36,Citation42).
The Alphanate Study Group published results of PK and clinical efficacy studies in 2002. This was the first study to enroll not only type 3 (n = 12), but also type 2A (n = 5) and type 1 (n = 18) VWD patients. An important finding in this study was that, in VWD type 3, the half‐life of FVIII:C was twice that of VWF:Ag due to the endogenous FVIII:C. Efficacy results showed that 75% of bleeding episodes were controlled with one or two infusions, and 71% of patients who received prophylactic treatment for surgeries or invasive procedures had good clinical responses. In another retrospective study, 22 VWD patients in Italy received Fanhdi, a concentrate similar to Alphanate. Excellent–good clinical responses were seen in 92% of bleeding episodes and in 93% of surgical procedures, despite the relative loss of high‐molecular‐weight VWF multimers in the product.
Haemate P/Humate‐P, an intermediate‐purity VWF/FVIII concentrate, has been widely used in VWD and has been considered the gold standard in management of this disorder. This product was introduced into clinical practice in Europe (Haemate P) in 1984 and in the United States (Humate‐P) in 1999. The first PK study of Haemate P, published in 1998, was a single‐center evaluation involving six type 3 VWD patients. Clinical efficacy data were collected retrospectively, and showed excellent–good responses for 99% of surgeries (n = 73) and for 97% of bleeding episodes (n = 3440). Results of a large retrospective study organized by the Canadian Hemophilia Centers were published in 2002 Citation36,Citation42. Other published studies include a retrospective analysis of Haemate P/Humate‐P efficacy and safety in preventing bleeding during surgery or invasive procedures in 26 Italian VWD patients, as well as two prospective, multicenter, open label, nonrandomized studies conducted in the US on Haemate P/Humate‐P used in urgent bleeding and urgent surgical events Citation36,Citation42. Another plasma‐derived VWF concentrate with low FVIII:C levels was introduced in France in 1992, and the first PK study in type 3 VWD was published in 1996 Citation36,Citation42. An improved version of this concentrate, which is almost devoid of FVIII:C, was evaluated in two large French and European studies, and data on PK are now available Citation36,Citation42. Results in type 3 VWD show no major differences in VWF:RCo and VWF:Ag for the concentrates that did or did not contain FVIII:C: as expected, the only difference was an approximate 6‐hour delay in FVIII:C increase with the concentrate devoid of FVIII:C; therefore, administration of exogenous FVIII:C is recommended in type 3 VWD cases of acute life‐threatening bleeding episodes or emergency surgeries Citation36,Citation42. Clinical efficacy results of the French and European studies have been recently reported Citation43.
Data derived from PK and clinical studies have contributed to more appropriate use of VWF/FVIII concentrates. The specific activity of concentrates is important to derive the degree of VWF/FVIII product purity, while VWF:RCo/Ag and VWF:RCo/FVIII ratios can be considered markers of VWF/FVIII protein activity. The accumulation of FVIII:C that is exogenously infused, together with that endogenously synthesized and stabilized by the infused VWF, may cause very high FVIII levels when multiple infusions are given to cover major surgery. There is some concern that sustained high FVIII levels may increase risk of postoperative deep‐vein thrombosis (DVT): however, DVT are rare events that have been reported only in VWD patients receiving repeated FVIII/VWF concentrate infusions to maintain clinical hemostasis after surgery Citation44. Therefore, when using repeated injections of VWF/FVIII concentrates for recurrent bleeding episodes, and especially after major surgery, we suggest daily monitoring of FVIII:C levels and adjusting the VWF/FVIII concentrate dose to keep the patient's FVIII:C levels between 50 and 150 U/dL. The minimal VWF:RCo levels to maintain sufficient hemostasis in VWD has not yet been determined in prospective studies; however, preliminary retrospective data from a large cohort of well characterized Italian VWD patients suggest that VWF:RCo levels of >30 U/dL are associated with a low incidence of spontaneous mucosal bleedings Citation12. The dosages of concentrates with the most correct therapeutic approaches according to VWD types are summarized ().
Table VI. Treatment of different types of VWD.
Treatment of patients with alloantibodies to VWF
For the rare patients with type 3 VWD who develop anti‐VWF alloantibodies after multiple transfusions, the use of VWF concentrates is not only ineffective, but may even cause postinfusion anaphylaxis due to the formation of immune complexes Citation32. These reactions may be life‐threatening. To overcome this drawback, a patient undergoing emergency abdominal surgery was treated with recombinant FVIII, because this product, that contains no VWF, could not cause anaphylactic reactions. In view of the very short half‐life of FVIII without its VWF carrier, recombinant FVIII had to be administered by continuous intravenous infusion, at very large doses, to keep FVIII levels above 50 U/dL for 10 days after surgery Citation45. Another possible therapeutic approach is recombinant activated factor VII (rFVIIa) that can be used in VWD with alloantibodies according to the same dosage and regimens as for hemophilia A patients with inhibitors Citation46,47. Since only little data on efficacy and safety on the use of recombinant FVIII and VIIa are available, prospective cross‐over studies should be designed to determine the best therapeutic approach in these cases.
Secondary long‐term prophylaxis
Patients with severe forms of VWD may have frequent hemarthroses, especially when FVIII levels are below 5 U/dL, so that some of them develop target joints like patients with severe hemophilia A. Some patients have recurrent gastrointestinal (GI) bleeding, often without lesions in the GI tract, and need treatment every day or every other day. Finally, there are children who have epistaxis frequently and severely enough to cause anemia. In these frequent and severe bleeders, the optimal therapy may be regular prophylaxis with VWF concentrates rather than on‐demand treatment on the occasion of bleeding episodes. The largest experience on secondary prophylaxis in VWD has been collected in Sweden in 35 patients with severe forms of VWD Citation48. Secondary prophylaxis was also implemented in a cohort of Italian patients with VWD Citation49. Among 89 patients who needed treatment with VWF concentrates during the last 2 years because of one or more bleeding episodes, 11 (12%) were included in a prophylaxis program because of frequent recurrence of bleeding at the same sites Citation49. Prophylaxis was started because of GI bleeds in seven patients with types 3 (n = 1), 2A (n = 4), 2M (n = 1), and type 1 (n = 1), and for joint bleeds in four patients with type 3 VWD (n = 4). Prophylaxis prevented bleeding completely in eight patients and largely reduced hospitalization for blood transfusions in the remaining three. When prophylaxis was compared with previous on‐demand regimens, in all the 11 cases the annual total consumption of concentrate, the number of transfused blood units, and days spent in hospital were significantly reduced Citation49. FVIII levels were always higher than 180 U/dL, but no side effects, including thrombosis, were observed. These two retrospective studies suggest that cost‐effectiveness of these prophylaxis regimens versus on‐demand therapy should be further evaluated in larger prospective studies.
Future treatments
On the whole, treatments currently available for patients with VWD are quite satisfactory. For patients unresponsive to DDAVP, VWF concentrates are the only form of available treatment. The fact that they are fractionated from plasma is of concern for some, even if more than one viral inactivation method are used for all concentrates in the manufacturing process. Haemate P/Humate P is the only concentrate that uses only one viral inactivation method (pasteurization), but the safety record of this product is impeccable. This favorable situation notwithstanding, there are advanced plans to develop a therapeutic preparation of recombinant VWF. This product containing only VWF will require the concomitant administration of FVIII for the control of acute bleeding episodes and for the prevention of excessive bleeding at the time of major surgery. Attempts to partially correct VWD through gene replacement therapy are in progress.
Acknowledgements
We acknowledge the work of Dr Luigi Flaminio Ghilardini, who prepared the figures reported in this manuscript.
References
- Castaman G., Federici A. B., Rodeghiero F., Mannnucci P. M. Von Willebrand's disease in the year 2003: towards the complete identification of gene defects for correct diagnosis and treatment. Haematologica 2003; 88: 94–108
- Lee C. A., Kessler C. M., editors. Proceedings of a Nordic von Willebrand symposium. Haemophilia 1999; 5(suppl 2)
- Zimmerman T. S., Ratnoff O. D., Powell A. E. Immunologic differentiations of classic hemophilia (Facor VIII Deficiency) and von Willebrand's disease. J Clin Invest 1971; 50: 244–54
- Federici A. B., Berntorp E., Lee C. A. The 80th Anniversary of von Willebrand Disease: History, Management, Research. Haemophilia 2006; 12: 563–572
- Ruggeri Z. M. Structure of von Willebrand factor and its function in platelet adhesion and thrombus formation. Best Pract Res Clin Haematol 2001; 14: 257–79
- Mancuso D. J., Tuley E. A., Westfield L. A., Lester‐Mancuso T. I., Le Beau M. M., Sorace J. M., et al. Human von Willebrand factor gene and pseudogene: structural analysis and differentiation by polymerase chain reaction. Biochemistry 1991; 30: 253–69
- Goodeve A., Eikenboom J. C. J., Ginsburg D., Hilbert L., Mazurier C., Peake I. R., et al. A standard nomenclature for von Willebrand factor gene mutations and polymorphisms. On behalf of the ISTH SSC Subcommittee on von Willebrand factor. Thromb Haemost 2001; 85: 929–31
- Vlot A. J., Koppelman S. J., Bouma B. N., Sixma J. J. Factor VIII and von Willebrand Factor. Thromb Haemost 1998; 79: 456–65
- Mazurier C., Rodeghiero F. Recommended abbreviations for von Willebrand factor and its activities. Thromb Haemost 2001; 85: 929–31
- Sadler J. E. A revised classification of von Willebrand disease. For the Subcommittee on von Willebrand Factor of the Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis. Thromb Haemost 1994; 71: 520–3
- Sadler J. E., Budde U., Eikenboom J. C. J., Favaloro E. J., Hill F. G., Holmberg L., et al. Update on the pathophysiology and classification of von Willebrand disease. A report of the Subcommittee on von Willebrand factor. J Thromb Haemost 2006; 4: 2103–14
- Federici A. B., Castaman G., Mannucci P. M. Guidelines for the diagnosis and management of VWD in Italy. Haemophilia 2002; 8: 607–21
- Federici A. B. Clinical diagnosis of von Willebrand disease. Haemophilia 2004; 10: 169–76
- Mazurier C., Hilbert L. Type 2 N von Willebrand disease. Current Hematol Rep 2005; 4: 350–8
- Gill J. C., Endres‐Brooks J., Bauer P. J., Marks W. J., Montgomery R. R. The effect of ABO blood group on the diagnosis of von Willebrand disease. Blood 1987; 69: 1691–5
- Kunicki T. J., Federici A. B., Salomon D. R., Koziol J. A., Head S. R., Mondala T. S., et al. An association of candidate gene haplotypes and bleeding severity in Von Willebrand Disease (VWD) type 1 pedigrees. Blood 2004; 104: 2359–67
- Silwer J. von Willebrand's disease in Sweden. Acta Paediat Scand 1973; 238: 1–159
- Lak M., Peyvandi F., Mannucci P. M. Clinical manifestations and complications of childbirth and replacement therapy in 348 Iranian patients with type 3 von Willebrand disease. Br J Haematol 2000; 111: 1223–9
- Rodeghiero F., Castaman G., Tosetto A., Battle J., Baudo F., Cappelletti A., et al. The discriminant power of bleeding history for the diagnosis of von Willebrand disease type 1: an international multicenter study. J Thromb Hemost 2005; 3: 2619–26
- Tosetto A., Rodeghiero F., Castaman G., Goodeve A., Federici A. B., Battle J., et al. A quantitative analysis of bleeding symptoms in type 1 of von Willebrand disease: results from a multicenter European Study (MCMDM‐1VWD). J Thromb Hemost 2006; 4: 774–82
- Mannucci P. M., Lombardi R., Bader R., Vianello L., Federici A. B., Solinas S., et al. Heterogeneity of type I von Willebrand's disease: evidence for a subgroup with an abnormal von Willebrand Factor. Blood 1985; 66: 796–802
- Cattaneo M., Federici A. B., Lecchi A., Agati B., Lombardi R., Stabile F., et al. Evaluation of the PFA‐100 System in the diagnosis and therapeutic monitoring of patients with von Willebrand Disease. Thromb Haemost 1999; 82: 35–39
- Favaloro E. J. Collagen binding assay for von Willebrand factor (VWF:CBA): detection of von Willebrands disease (VWD), and discrimination of VWD subtypes, depends on collagen source. Thromb Haemost 2000; 83: 127–35
- Federici A. B., Canciani M. T., Forza I., Cozzi G. Ristocetin cofactor and collagen binding activities normalized to antigen levels for a rapid diagnosis of type 2 von Willebrand disease: single center comparison of four different assays. Thromb Haemost 2000; 84: 1127–8
- Mannucci P. M., Federici A. B. Antibodies to von Willebrand factor in von Willebrand disease. Adv Exp Med Biol 1995; 386: 87–92
- Meyer D., Fressinaud E., Hilbert L., Ribba A. S., Lavergne J. M., Mazurier C. Type 2 von Willebrand disease causing defective von Willebrand factor‐dependent platelet function. Best Pract Res Clin Haematol 2001; 14: 349–64
- Schneppenheim R., Federici A. B., Budde U., Castaman G., Drewke E., Mannucci P. M., et al. Von Willebrand disease type 2 M “Vicenza” in Italian and German patients: identification of the first candidate mutation (G3864A; R1205H) in 8 families. Thromb Haemost 2000; 83: 136–40
- Castaman G., Missiaglia E., Federici A. B., Schneppenheim R., Rodeghiero F. An additional candidate mutation (G2470A; M740I) in the original families with von Willebrand disease type 2 M Vicenza and the G3864A (R1205H) mutation. Thromb Haemost 2000; 84: 350–1
- Goodeve A., Eikenboom J., Castaman G., Rodeghiero F., Federici A. B., Battle J., et al. Phenotype and genotyoe of a cohort of families historically diagnosed with type 1 von Willebrand disease in the European study, Molecular and Clinical Markers for the Diagnosis and Management of Type 1 von Willebrand disease. Blood 2007; 109: 112–121
- James P. D., Notley C., Hegadorn C., Leggo J., Tuttle A., Tinlin S., et al. The mutational spectrum of type 1 von Willebrand disease: results from a Canadian cohort study. Blood 2007; 109: 145–54
- Shelton‐Inloes B. B., Chebab F. F., Mannucci P. M., Federici A. B., Sadler J. E. Gene deletion correlates with the 21 development of alloantibodies in von Willebrand disease. J Clin Invest 1987; 79: 1459–65
- Mannucci P. M., Tamaro G., Narchi G., Candotti G., Federici A., Altieri D., et al. Life‐threatening reaction to factor VIII concentrate in a patient with severe von Willebrand disease and alloantibodies to von Willebrand Factor. Eur J Haematol 1987; 39: 467–70
- Eikenboom J. C. J. Congenital von Willebrand disease type 3: clinical manifestations, pathophysiology and molecular biology. Best Pract Res Clin Haematol 2001; 14: 365–79
- Baronciani L., Cozzi G., Canciani M. T., Peyvandi F., Srivastava A., Federici A. B., et al. Molecular Characterization of a multiethnic Group of 21 Patients with type 3 von Willebrand Disease. Thromb Haemost 2000; 84: 536–40
- Schneppenheim R., Castaman G., Federici A. B., Kreuz W., Marschalek R., Oldenburg J., et al. A commom 253‐kb deletion involving VWF and TMEM16B in German and Italian patients with severe von Willebrand disease type 3. J Thromb Haemost 2007; 5: 722–8
- Mannucci P. M. Treatment of von Willebrand disease. N Eng J Med 2004; 351: 683–94
- Federici A. B., Mazurier C., Berntorp E., Lee C. A., Sharrer I., Goudemand J., et al. Biological response to desmopressin in patients with severe type 1 and type 2 von Willebrand disease: results of a multicenter European study. Blood 2004; 103: 2032–38
- Mannucci P. M., Bettega D., Cattaneo M. Consistency of responses to repeated DDAVP infusions in patients with von Willebrand disease and haemophilia A. Br J Haematol 1992; 82: 87–93
- Smith T. J., Gill J. C., Ambruso D. R. Hathaway WE. Hyponatremia and seizures in young children given DDAVP. Am J Hematol 1989; 31: 199–202
- Bond L., Bevin D. Myocardial infarction in a patient with hemophilia A treated with DDAVP. N Engl J Med 1988; 318: 121
- Byrnes J. J., Larcada A., Moake J. L. Thrombosis following desmopressin for uremic bleeding. Am J Hematol 1988; 28: 63–5
- Federici A. B. Management of von Willebrand disease with factor VIII/von Willebrand factor concentrates: results from current studies and surveys. Blood Coag Fibrin 2005; 16: S17–21
- Borel‐Derlon A., Federici A. B., Roussel‐Robert V., Goudemand J., Lee C. A., Sharrer I., et al. Treatment of severe von Willebrand disease with a high‐purity von Willebrand factor concentrate (Wilfactin): a prospective study on 50 patients. J Thromb Haemost 2007; 5: 1115–24
- Mannucci P. M. Venous thromboembolism in von Willebrand disease. Thromb Haemost 2002; 88: 378–9
- Bergamaschini L., Mannucci P. M., Federici A. B., Coppola R., Guzzoni S., Agostoni A. Posttransfusion anaphylactic reaction in a patient with severe von Willebrand disease: role of complement and alloantibodies to von Willebrand factor. J Lab Clin Med 1995; 125: 348–55
- Ciavarella N., Schiavoni M., Valenzano E., Mangini F., Inchingolo F. Use of recombinant factor VIIa (Novoseven) in the treatment of two patients with type III von Willebrand disease and an inhibitor against von Willebrand factor. Haemostasis 1996; 26: 150–4
- Boyer‐Neumann C., Dreyfus M., Wolf M., Veyradier A., Meyer D. Multi‐therapeutic approach to manage delivery in an alloimmunized patient with type 3 von Willebrand disease. J Thromb Haemost 2003; 1: 190–2
- Berntorp E., Petrini P. Long‐term prophylaxis in von Willebrand disease. Blood Coag Fibrin 2005; 16: S23–6
- Federici A. B., Gianniello F., Canciani M. T., Mannucci P. M. Secondary long‐term prophylaxis in severe patients with von Willebrand disease: an Italian cohort study. Blood 2005; 106: 507a, abstract 1782