Abstract
The multiligand receptor RAGE (receptor for advanced glycation end-products) is emerging as a central mediator in the immune/inflammatory response. Epidemiological evidence accruing in the human suggests upregulation of RAGE's ligands (AGEs, S100/calgranulins, high mobility group box-1 (HMGB1), and amyloid β-peptide and β-sheet fibrils) and the receptor itself at sites of inflammation and in chronic diseases such as diabetes and neurodegeneration. The consequences of ligand-RAGE interaction include upregulation of molecules implicated in inflammatory responses and tissue damage, such as cytokines, adhesion molecules, and matrix metalloproteinases. In this review, we discuss the localization of RAGE and its ligand families and the biological impact of this axis in multiple cell types implicated in chronic diseases. Lastly, we consider findings from animal model studies suggesting that although tissue-damaging effects ensue from recruitment of the ligand-RAGE interaction, in distinct settings, adaptive and repair/regeneration outcomes appear to override detrimental effects of RAGE. As RAGE blockade moves further into clinical development, clarifying the biology of RAGE garners ever-increasing importance.
The biology of RAGE—interactions with multiple ligands
The receptor for advanced glycation end-products (RAGE) is a multiligand receptor of the immunoglobulin superfamily of cell surface molecules. RAGE was first identified as a cell surface receptor for the products of non-enzymatic glycation and oxidation of proteins and lipids which form in such settings as diabetes, renal failure, aging, and inflammation, the advanced glycation end-products (or AGEs) Citation[1]. The observation that RAGE bound non-AGE ligands, including certain members of the S100/calgranulin family of polypeptides (such as S100A12 and S100B), high mobility group box-1 (HMGB1), amyloid-β peptide and β-sheet fibrils, and Mac-1 Citation[2–5], provided pivotal insights into integral roles for RAGE in multiple stages of the inflammatory response. Further, RAGE is expressed on multiple cell types implicated in both the earliest to the later and progressive phases of inflammation, including neutrophils, monocytes/macrophages, T and B lymphocytes, dendritic cells (DCs) and endothelial cells (ECs). In this review, we shall outline the identified roles for RAGE and its ligands in these cell types and discuss selected inflammatory-type diseases in which RAGE has been demonstrated to contribute. Collectively, studies to date suggest a compelling role for RAGE as a therapeutic target in chronic diseases in which inflammation is a central factor.
Key messages
Epidemiological evidence accruing in the human suggests upregulation of RAGE's ligands (AGEs, S100/calgranulins, high mobility group box-1 (HMGB1), and amyloid β-peptide and β-sheet fibrils) and the receptor itself at sites of inflammation and in chronic diseases such as diabetes and neurodegeneration.
The consequences of ligand-RAGE interaction include upregulation of molecules implicated in inflammatory responses and tissue damage, such as cytokines, adhesion molecules, and matrix metalloproteinases.
Insights from studies in animal models suggest key roles for RAGE in vascular and inflammatory stresses that contribute to the pathogenesis and complications of chronic diseases, such as diabetes, autoimmunity and inflammation, and neurodegeneration.
RAGE and roles in neutrophil biology
RAGE expression has been detected on neutrophils, both at the mRNA and protein levels. AGEs bind to RAGE-expressing neutrophils, with Kd ≈3.7 nM in a manner blocked by ligand-RAGE decoys and antagonists, including soluble RAGE (sRAGE, the extracellular ligand-binding RAGE domain), antibodies to RAGE or antibodies to a specific AGE (carboxymethyl lysine or CML albumin) Citation[6]. Although AGEs are a heterogeneous group of structures, CML-AGEs have been demonstrated to be specific AGEs that bind RAGE and activate signal transduction pathways Citation[7].
The first studies probing the role of RAGE specifically in neutrophils focused on diabetes and the particular observation that diabetes may be associated with increased susceptibility to recurrent infections. Compared to control albumin, AGE-albumin inhibited transendothelial migration of neutrophils and the production of reactive oxygen species (ROS) induced by the pathogen Staphylococcus aureusCitation[6]. Although AGE-albumin enhanced phagocytosis of this pathogen by neutrophils, bacterial killing was suppressed by AGE in a manner dependent on RAGE Citation[6].
Abbreviations
As RAGE is expressed not only on neutrophils, but on molecules with which neutrophils interact in the inflammatory response, the relationships between RAGE on inflammatory cells versus endothelial or epithelial cells have been addressed. It is well established that RAGE is highly expressed on EC Citation[1]. RAGE-dependent leukocyte adhesion to EC is mediated, at least in part, by the β2-integrin Mac-1 Citation[5]. In an animal model of thioglycollate-induced peritonitis, leukocyte recruitment was significantly impaired in mice devoid of RAGE. Further, although enhanced leukocyte recruitment to the inflamed peritoneum was observed in diabetic mice, it was markedly reduced in the presence of the ligand decoy sRAGE, or in diabetic RAGE null mice Citation[5].
Neutrophil responses in diabetes may also be affected by modification of the extracellular matrix. Toure and colleagues recently employed neutrophils retrieved from healthy human volunteers; they found that glycoxidation of collagen caused increased adhesion of neutrophils compared to their behavior on non-glycoxidated collagen Citation[8]. The increased adhesion of neutrophils to AGE-collagen was suppressed by antibodies to RAGE and by inhibitors of phosphatidylinositol-3 (PI3) kinase, but not by inhibitors of mitogen-activated protein (MAP) kinases Citation[8]. These considerations provided further mechanistic support for the suppression of host defenses that occur in AGE-enriched settings, such as diabetes.
The role of RAGE in mediating neutrophil migration in distinct settings, such as across the intestinal epithelium, has also been addressed. RAGE expression was established in intestinal epithelial cells, primarily at the lateral membranes close to the apical cell junction complexes Citation[9]. In vitro, the expression of RAGE was low in homeostatic conditions but was increased in the presence of inflammatory cytokines, such as interferon-γ and/or tumor necrosis factor-α. Further, intestinal epithelium from subjects with inflammatory bowel disease (a RAGE ligand-enriched environment) revealed higher levels of RAGE than tissue retrieved from control individuals without inflammatory bowel disease Citation[9]. In an in vitro transmigration assay, RAGE mediated neutrophil adhesion to and migration across intestinal epithelial monolayers in a manner dependent on the binding of RAGE to β2 integrin (CD11b/CD18) Citation[9].
Recent studies suggest that HMGB1 may promote Mac-1-dependent neutrophil recruitment. HMGB1 induced rapid recruitment of neutrophils in an in vivo model; this was prevented in mice deficient in Mac-1, but not those deficient in a distinct integrin, leukocyte function-associated antigen-1 (LFA-1) Citation[10]. The effects of HMGB1 were dependent on the expression of RAGE on neutrophils, but not on EC, as demonstrated by experiments using bone marrow chimeras Citation[10]. These studies reiterated earlier evidence that a central transcription factor activated by RAGE relevant to stress responses was nuclear factor-kappa B (NF-kB) Citation[10], Citation[11].
Interestingly, one of the first-identified S100/calgranulin ligands of RAGE, S100A12, is a factor produced by inflammatory cells, especially neutrophils. Studies examining levels of S100A12 in the human (and particularly in neutrophils) suggest that its expression is increased in plasma and synovial membranes in rheumatoid and psoriatic arthritis Citation[12], in synovial fluid and serum in juvenile rheumatoid arthritis Citation[13], in pulmonary tissue and bronchoalveolar lavage fluid in respiratory distress syndrome and lung inflammation Citation[14], in serum and sputum in infection in cystic fibrosis Citation[15], and in colonic tissue and serum in chronic active inflammatory bowel disease Citation[16].
Taken together, these findings link RAGE and its ligands to neutrophil-endothelial/epithelial biology, thereby implicating RAGE in initiating and sustaining events in infection and autoimmunity.
RAGE and roles from monocytes/macrophages to microglia: expression and function
RAGE is expressed on monocytes/macrophages, and some of the first insights into mechanisms by which the ligands of RAGE mediated their cellular actions were elucidated from studies performed on monocytes/macrophages. The first demonstration of RAGE's role in macrophages was made using human peripheral blood-derived monocytes. Direct binding of AGE to monocytes was revealed using radiolabeled AGE-albumin with Kd ≈80 nM in a manner prevented by antibodies to RAGE Citation[17]. Chemotaxis and haptotaxis of monocytes in response to AGE-albumin were dependent on RAGE, and in vivo, when polytetrafluoroethylene mesh was impregnated with AGE-albumin, a florid mononuclear cell infiltrate resulted in the graft mesh 4 days after implantation, compared with much sparser infiltration when the mesh was incubated with control albumin Citation[17]. These studies suggested that in AGE-enriched environments, monocytes/macrophage activation, in part via RAGE, might contribute to exaggerated vascular inflammation and stress.
Although these studies were performed with heterogeneous AGEs, later studies, using specific RAGE ligand CML-AGE species revealed that CML-ovalbumin stimulated monocyte/macrophage migration and activated NF-kB via RAGE Citation[7]. In addition, a form of β2 microglobulin modified with AGE (and highly relevant to amyloid fibrils which form in dialysis-related amyloidosis), AGE-β2 microglobulin, bound immobilized RAGE and human mononuclear phagocytes (Kd ≈ 50–80 nM) Citation[18]. Induction of mononuclear phagocyte RAGE-dependent chemotaxis and induction of tumor necrosis factor (TNF)-α by AGE-β2 microglobulin, but not by native β2 microglobulin, were shown in these studies Citation[18]. These studies suggested potential links of RAGE to inflammatory conditions characterized by bone and joint destruction.
In addition to AGEs, the RAGE ligand S100A12 mediated chemotaxis of human peripheral blood-derived mononuclear phagocytes as well. In RAGE-expressing Bv2 microglia-type cells, S100A12 stimulated production of interleukin (IL)-1β and TNF-α protein and activated NF-kB, in a manner suppressed in the presence of insertion of a dominant negative (DN) form of RAGE Citation[3]. The construct encoding DN RAGE consists of the ligand-binding extracellular domain and the membrane-spanning domain; solely the cytoplasmic domain is deleted. Thus, ligands may bind to this form of RAGE but are unable to stimulate signal transduction. In addition to S100A12, HMGB1 stimulates monocyte migration and adhesion in a manner suppressed by DN RAGE Citation[19].
In addition to induction of cellular migration and generation of cytokines, RAGE-dependent effects on monocytes may also include delay in apoptosis and increased production of ROS Citation[20]. RAGE mediates generation of ROS in monocytes/macrophages largely via activation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, as illustrated by studies using mice deficient in gp91phox Citation[21]. Compared to wild-type macrophages, macrophages retrieved from gp91phox-deficient mice showed blunted responses to AGEs Citation[21].
RAGE ligands likely contribute to amplification of inflammatory mechanisms by multiple mechanisms, including those stimulated by HMGB1. HMGB1 is secreted both by activated monocytes and macrophages, and by necrotic or damaged cells Citation[22]. HMGB1 null necrotic cells fail to promote inflammatory responses, thus illustrating the central role of this molecule in these processes. It is likely that release of HMGB1 stimulates inflammation on a range of neighboring cells (inflammatory, vascular, and other cells, depending on the tissue), at least in part via RAGE. It is possible, however, that such effects are mediated in part by toll receptors (toll-like receptor (TLR) 2, 4, and 9 as examples), as these molecules have been shown to bind to HMGB1 as well Citation[23], Citation[24].
A recent in vivo study provided support for the role of HMGB1 and RAGE in inflammation in a rat model of necrotizing enterocolitis induced by hypoxia and formula-feeding in neonatal rats. After this injury, examination of the distal ileum revealed intestinal inflammation with upregulation of HMGB1 and RAGE, in parallel with increased expression of apoptosis-related proteins, inducible nitric oxide synthase (iNOS), and cyclo-oxygenase-2 (COX-2) Citation[25]. However, treatment of the animals with semapimod, a macrophage deactivator, blocked upregulation of all of these inflammatory mediators and protected the animals against the development of necrotizing enterocolitis Citation[25].
RAGE is also expressed in microglia, monocyte/macrophage-like cells of the central nervous system Citation[4]. The brains of human subjects with Alzheimer's disease (AD) revealed increased expression of RAGE in neuronal, microglial, and endothelial cells when compared to age-matched subjects without AD Citation[4]. The RAGE ligand amyloid-β peptide (Aβ) bound RAGE on cultured Bv2 microglial cells in a dose-dependent saturable manner. Aβ-RAGE interaction on cultured microglial cells induced their migration, production of TNF-α, and activation of NF-kB Citation[4].
Relevance to the human in microglial biology was suggested by the work of Lue and colleagues. These investigators cultured microglia from AD versus control brain and showed increased macrophage-colony-stimulating factor (M-CSF) production in the presence of Aβ1-42 in AD-derived microglia, a process that was dependent on RAGE, as evidenced by blockade in the presence of anti-RAGE F(ab’)2 fragments Citation[26]. Further, Walker and colleagues performed gene array studies on microglia retrieved from AD brains and treated with Aβ and found upregulation of multiple proinflammatory cytokines and matrix metalloproteinases (MMPs) Citation[27]. Further, an S100/calgranulin, S100A8, was also identified as one of the chronically activated genes in this study.
In studies distinct from AD, monocytes retrieved from diabetic subjects revealed higher levels of tissue factor compared to cells retrieved from non-diabetic individuals, and, in vitro, incubation of monocyte-like U937 cells with AGEs resulted in increased expression of tissue factor Citation[28]. These findings underscore the role of circulating/monocyte inflammatory factors in diabetes. Other studies in human diabetic mononuclear phagocytes link RAGE to phosphorylation of pleckstrin and mechanisms associated with production of inflammatory mediators Citation[29].
RAGE and lymphocytes: priming the pump in inflammatory mechanisms
In addition to monocytes/macrophages, central roles for RAGE in lymphocyte properties of migration and priming have recently been identified. One of the first studies to test the role of ligand-RAGE interaction on lymphocytes was performed using the S100/calgranulin S100A12. Exposure of S100A12 to peripheral blood mononuclear cells resulted in increased expression of IL-2, in a manner suppressed by excess sRAGE or anti-RAGE immunoglobulin G (IgG).
Murine models of diseases with prominent roles for T lymphocytes were studied in order to probe the mechanisms by which T lymphocyte RAGE might contribute to inflammatory responses. Induction of experimental autoimmune encephalomyelitis (EAE) in mouse models of multiple sclerosis was found to depend importantly on RAGE, as RAGE blockade suppressed EAE induced either by myelin basic protein (MBP) peptide or encephalitogenic T lymphocytes, or when EAE occurred spontaneously in T cell receptor (TCR)-transgenic mice devoid of endogenous TCR-α or TCR-β chains Citation[30]. A prominent role for RAGE-dependent T lymphocyte migration into the central nervous system (CNS) was revealed in these studies. RAGE signaling in T lymphocytes (CD4) was essential in migration responses, as mice expressing DN RAGE in T lymphocytes (CD4) displayed resistance to MBP-induced EAE Citation[30].
Chen and colleagues showed that both human T and B lymphocytes expressed RAGE, and they tested the role of this receptor in mechanisms linked to the development of autoimmunity and type 1 diabetes (T1D) in non-obese diabetic (NOD)/scid mice subjected to adoptive transfer of diabetogenic splenocytes from NOD mice Citation[31]. In those studies, compared to vehicle administration, treatment with sRAGE resulted in increased time to the development of T1D and, in parallel, suppression of expression of proinflammatory cytokines in the islets (including TNF-α and IL-1β) Citation[31]. RAGE blockade was associated with increased expression of anti-inflammatory cytokines IL-10 and TGF-β in the islets, but had no effect when preactivated CD4+ T cell clones were administered, suggesting roles for RAGE in the differentiation of T lymphocytes to more mature and pathogenic phenotypes during the late stages of T1D Citation[31].
The direct effects of RAGE on T lymphocyte proliferation were studied in murine and human donor-reactive T lymphocyte priming studies Citation[32]. Incubation of murine peripheral blood mononuclear cells with sRAGE resulted in a significant decrease in lymphocyte proliferation versus IgG control-treated cultures, and the effects were dose-dependent Citation[32]. Further, blocking antibodies to RAGE, but not non-immune Ig resulted in a significant decrease in lymphocyte proliferation. These findings were extended to human lymphocyte proliferation triggered by alloresponses. Treatment with sRAGE, or pretreatment with antibodies to RAGE, significantly reduced lymphocyte proliferation Citation[32]. In vivo, in a murine model of orthotopic allogeneic heterotopic heart transplantation, treatment with sRAGE increased survival of the graft and greatly suppressed infiltration of lymphocytes and macrophages into the allograft Citation[32].
To further probe the role of RAGE in T lymphocyte priming, experiments were performed using OT II T cells as a model of TCR specifically recognizing ovalbumin (OVA) and OVA-immune complexes (IC) Citation[33]. Transfer of RAGE-deficient OT II cells into OVA-immunized hosts resulted in reduced proliferative responses that were even further reduced when these cells were infused into RAGE null hosts Citation[33]. In vitro, RAGE null T lymphocytes displayed markedly impaired proliferative responses to nominal and alloantigens, in parallel with reduced expression of interferon (IFN)-γ and IL-2 Citation[33].
Specific roles for HMGB1 in T cell activation were illustrated by Dumitriu and colleagues. Human DCs were shown to actively release HMGB1 and, via RAGE, mediate clonal expansion, survival, and functional polarization of naive T lymphocytes Citation[34]. Signaling mechanisms linked to these observations included activation of mitogen-activated protein kinases (MAPs) and NF-kB Citation[34].
RAGE and dendritic cell biology
In addition to release of HMGB1 by DC, thereby potentially leading to RAGE-dependent responses, DC themselves express RAGE. However, studies to date are not conclusive on roles for RAGE in DC properties. Moser and colleagues employed multiple strategies to test these concepts. RAGE null DC (retrieved from bone marrow) did not display abnormalities in antigen processing or presentation to T lymphocytes Citation[33]. Further, these authors reported that RAGE was not required for DC migration in vivo or for DC:T cell conjugate stability in vitroCitation[33].
In contrast to these findings, Dumitriu and colleagues showed that plasmacytoid DC express RAGE and that treatment of these cells with anti-RAGE IgG suppressed HMGB1-mediated maturation and production of IFN-γ Citation[35]. Others showed that HMGB1 mediated migration of monocyte-derived, immature DCs (but not mature DCs) in a manner suppressed by anti-RAGE IgG Citation[36]. In vitro, blockade of RAGE blocked upregulation of CCL19 and CCL12 in maturing DC Citation[37]. However in vivo, Manfredi and colleagues used both magnetic resonance imaging (MRI) and immunohistochemistry to demonstrate that RAGE-deficient DC injected into mouse foot-pads displayed reduced migration to popliteal lymph nodes compared to RAGE-expressing DC Citation[38]. These studies, in contrast to those of Moser and colleagues Citation[33], suggested that RAGE is necessary for DC maturation and homing. Future studies are required to resolve these apparent differences—clearly with meticulous attention to the details of DC source and treatment conditions.
In this context, fascinating insights into functional roles for RAGE in liver-derived DC are emerging. In murine models of severe liver injury, such as that induced by massive (85%) hepatectomy, immunohistochemistry studies revealed that a principal RAGE-expressing cell type in the liver remnant is the CD11c-expressing DC Citation[39]. When wild-type mice were treated with sRAGE and subjected to massive hepatectomy, improved survival and markers of regeneration resulted Citation[39]. In transgenic mice expressing DN RAGE under control of the macrophage scavenger receptor type A promoter (MSR), which drives expression of DN RAGE in cells of mononuclear phagocytic lineage and DC, survival and regeneration of the remnant were enhanced compared to wild-type litter-mates Citation[39]. Although roles for RAGE in non-DC might have been the proximate cause underlying these findings, it remains possible that DC RAGE in the liver critically contributes to failure of regeneration.
Taken together, recent studies probing roles for RAGE in T lymphocytes and DC suggest seminal roles for this pathway in adaptive immunity. In the sections to follow, we present evidence that RAGE importantly contributes to immune/inflammatory diseases based on experiments in animal models and association studies in human beings.
RAGE and models of disease
RAGE and delayed type hypersensitivity (DTH)
A first test of the role of RAGE and its ligand families in non-diabetic mice was explored in murine models of foot-pad inflammation. In the first studies, when RAGE ligand S100A12 was directly injected into murine foot-pad, an influx of inflammatory cells ensued in a manner dependent on RAGE Citation[3]. Compared to treatment with non-immune F(ab’)2 fragments of IgG or treatment with protein vehicle, murine serum albumin (MSA), CF-1 mice treated with sRAGE or with F(ab’)2 fragments of either anti-RAGE or anti-S100A12 IgG revealed significantly decreased foot-pad inflammation after S100A12 injection, in parallel with decreased influx of inflammatory cells Citation[3].
These studies were extended to a model of delayed type hypersensitivity in CF-1 mice. Mice were sensitized by injections of methylated bovine serum albumin (BSA) (mBSA, not a RAGE ligand); 3 weeks later, mBSA was injected into the foot-pad and, in a typical DTH response, elicited influx of inflammatory cells, development of foot-pad swelling and erythema, and in many sites formation of granulomata. Administration of sRAGE or F(ab’)2 fragments of anti-RAGE or anti-S100A12 IgGs greatly suppressed foot-pad inflammation in terms of erythema, edema, and influx of inflammatory cells compared to respective vehicle treatments Citation[3].
Examination of the underlying mechanisms shed first light on key roles for RAGE in inflammatory signaling. In those studies, we focused on NF-kB and the inflammatory cytokine, TNF-α in vivo. Compared with the contralateral foot-pad (the latter subjected to sensitization with mBSA, but without local challenge), nuclear extracts from the mBSA-injected foot-pad revealed a 6-fold increase in activation of NF-kB, as detected by electrophoretic mobility shift assays (EMSA). In the presence of sRAGE, the intensity of the gel shift band was substantially reduced as contrasted with results using nuclear extracts from foot-pads appropriately prepared/challenged with mBSA but treated only with the vehicle. Consistent with the protective effect of interrupting RAGE interaction with S100A12, administration of anti-RAGE/anti-S100A12 F(ab')2 to mBSA-sensitized/challenged mice caused a 70% decrease in activation of NF-kB versus non-immune F(ab')2. An important consequence of RAGE ligation by S100A12 was increased expression of inflammatory mediators, likely due, at least in part, to NF-kB. Compared with DTH mice receiving vehicle (MSA), animals treated with sRAGE displayed a 3.1-fold decrease in levels of TNF-α protein in foot-pad tissue. Similar beneficial effects were observed upon treatment with anti-S100A12 or anti-RAGE F(ab')2Citation[3].
RAGE and colitis
The ligand families of RAGE have been shown to accumulate in human colitis and indeed represent markers of the degree of inflammation Citation[16]. Interestingly, more than one class of RAGE ligands have been demonstrated in colitis tissue. Gut tissue retrieved from subjects with inflammatory bowel disease revealed extensive accumulation of S100/calgranulins and CML-AGE-modified proteins Citation[40]. In parallel with increased expression of RAGE in these colitis tissues, activation of NF-kB was noted. In vivo, CML-modified proteins, including CML-modified S100s, retrieved from human colitis tissue activated NF-kB and increased inflammation when injected, by rectum, into wild-type but not RAGE null mice Citation[40]. In vitro, incubation of endothelial cells with these RAGE ligands activated NF-kB in a manner suppressed in the presence of sRAGE or inhibitors of p44/42 or p38 MAP kinase Citation[40]. Other studies in human and murine colitis-associated carcinogenesis suggested roles for S100A8/S100A9, via RAGE, in the development of tumors in a mouse model Citation[41]. In addition to AGEs and S100/calgranulins, distinct studies illustrated roles for HMGB1 in colitis. A marked increase in serum levels of HMGB1 was identified in mice subjected to chemically induced colitis Citation[42]. Although these studies did not specifically address the role of RAGE, they illustrated that blocking antibodies to HMGB1 reduced inflammation and, in parallel, the number of tumors Citation[42]. These studies linked HMGB1-dependent colonic inflammation to the development of tumors.
Roles for RAGE ligands in colitis in murine models were further supported by earlier experiments in mice deficient in IL-10. These animals, susceptible to intestinal inflammation, revealed marked decreases in activated NF-kB in colonic tissue and inflammatory markers when treated chronically with sRAGE compared to vehicle Citation[3].
RAGE and arthritis
In addition to epidemiological evidence identifying RAGE ligands such as S100/calgranulins and HMGB1 in human arthritis tissue and/or joint fluid Citation[12], Citation[13], Citation[43], murine models of arthritis have been studied using bovine type II collagen Citation[44]. Mice immunized and challenged with bovine type II collagen develop clinical and histological evidence of arthritis. When treated with sRAGE compared with vehicle, joint swelling/erythema scores were reduced, in parallel with reduced joint levels of cytokines and MMPs Citation[44]. Of interest, a specific polymorphism of human RAGE, G82S, is in linkage disequilibrium with HLA-DR4, thereby linking RAGE and this variant to rheumatoid arthritis in the human Citation[44].
RAGE and sepsis
Key questions regarding the feasibility of RAGE-directed therapy in chronic diseases in humans revolve around the potential impact of RAGE antagonism on innate responses to overwhelming infections (such as sepsis) and specific infections. Multiple studies in mice have established that chronic blockade of RAGE (using sRAGE or rat anti-murine RAGE monoclonal antibody) or genetic deletion of RAGE prolongs survival compared to respective vehicle or litter-mate control animals upon cecal ligation and puncture-induced sepsis Citation[45], Citation[46]. These studies suggest that RAGE is involved in later stages of hyperinflammation upon stimulation with infectious pathogens or overwhelming polymicrobial gut inflammation. Importantly, tissue colony counts of Gram-positive and Gram-negative bacteria in this model did not differ between vehicle versus RAGE antibody-treated animals Citation[45], Citation[46].
Consistent with this concept, mice infected with Listeria monocytogenes displayed decreased lethality when treated with antibodies to RAGE versus vehicle Citation[46]. Similarly, RAGE null and RAGE heterozygous null (+/ − ) mice demonstrated increased survival compared to RAGE-expressing animals subjected to infection with this organism Citation[46].
These findings in distinct models of massive infection/inflammation do not rule out primal roles for RAGE in mediating clearance of specific pathogens, but, nevertheless, do add to the growing body of evidence that RAGE mounts exaggerated tissue-damaging responses to pathogens and not the initial phase of immune attack or clearance.
Heart failure—linking RAGE to cardiac damage via inflammatory signaling
In sepsis, the release of RAGE ligands may target multiple cell types and tissues. Among these is the heart and, in particular, cardiomyocytes. In vivo (injection of massive doses of lipopolysaccharide into mice) and in vitro, using cultured cardiomyocytes, roles for S100A8/9, via RAGE, in cardiac failure linked to septic type conditions were illustrated Citation[47]. This ligand-receptor interaction was associated with suppression of cardiac contractility by modulation of intracellular calcium fluxes and the association of S100A8/9 with sarco/endoplasmic reticulum ca2+ -ATPase 2a (SERCA2a) and ryanodine receptor 2 complex Citation[47]. In those studies, S100A8/9 were shown to be products of inflammation-challenged cardiac cells, thus suggesting that release of RAGE ligand from injured cells perpetuates organ damage.
In the heart, RAGE plays critical roles in the response to inflammatory signals, such as that induced by ischemia/reperfusion (I/R) in the absence or presence of diabetes. I/R in the heart generates AGEs, S100/calgranulins, and HMGB1 and, in a RAGE-dependent manner, stimulates upregulation of oxidative and inflammatory stresses, coupled with proapoptotic forces Citation[48], Citation[51]. In the isolated perfused heart and/or model of transient occlusion and reperfusion of the left anterior descending coronary artery, administration of sRAGE, inhibitors of HMGB1, or genetic deletion of RAGE results in significantly reduced infarct size, together with preservation of cardiac function and metabolic integrity (levels of adenosine triphosphate (ATP)). Key roles for MAP kinase, particularly c-Jun NHCitation[2]-terminal kinase (JNK) MAP kinase, and signal transducers and activators of transcription (STAT) signaling, via RAGE, in cardiac injury in I/R were demonstrated in these studies Citation[48–51].
Additional noteworthy experiments in these reports implicated RAGE itself in the production of AGE ligands (particularly CML-AGE) in cardiac I/R. Thus, at least in part by oxidative stress or yet-to-be identified mechanisms, RAGE-dependent cellular stresses provide a milieu conducive for on-going generation of ligand in I/R. Specifically, in the isolated perfused heart in diabetic mice, levels of AGEs, particularly CML AGEs, were higher in wild-type mouse hearts after I/R versus RAGE-deficient hearts Citation[49].
Taken together, these studies suggest strongly that I/R generates RAGE ligands that signal to initiate damage in the heart. Further general support for RAGE-dependent mechanisms in I/R injury was evident from studies in the liver, in which RAGE blockade greatly attenuated I/R injury in C57BL/6 mice Citation[52]. Although RAGE null mice were not assessed in that study, it was shown that in liver I/R, administration of sRAGE significantly improved survival, in parallel with diminished tissue-damaging inflammation and apoptosis Citation[52].
RAGE and inflammation—front row seats in chronic disease
RAGE-dependent signaling appears to significantly contribute to chronic diseases in which inflammation plays a key role. A specific example of such a chronic disease setting is diabetes—both types 1 and 2. Multiple reports are accruing to suggest that hyperglycemia exerts both direct and indirect damage. Among hyperglycemia's many actions is the production of AGEs. The actions of AGEs may result from multiple underlying etiologies; included among these are formation of cross-links and damage to basement membrane and cellular structures, as well as activation of RAGE. Earliest studies suggested that AGE-RAGE interaction on EC provoked rapid production of ROS and activation of inflammatory signaling cascades, together with upregulation of cytokines, adhesion molecules, and permeability factors in these cells Citation[1], Citation[7], Citation[11], Citation[53].
Recent experiments in coronary arterioles reaffirmed the strong link between diabetes and vascular inflammation. Abnormal dilation of vessels in db/db type 2 diabetic mice in response to acetylcholine was reversed in the presence of sRAGE Citation[54]. Coronary arterioles from diabetic mice revealed increased expression of TNF-α, and increased mRNA and protein expression of NADPH oxidase subunits NOX-2, p22(phox) and p40(phox) Citation[54]. Administration of sRAGE reduced expression of this cytokine and the key components of the NADPH oxidase enzyme Citation[54].
Thus, it was not surprising that diabetes-accelerated progression of atherosclerosis in mice devoid of apolipoprotein E was attenuated by administration of sRAGE or by genetic deletion of the receptor in models of type 1 and/or type 2 diabetes Citation[55–59]. Particularly noteworthy in these studies was the observation that inflammation, oxidative stress, and activation of NF-kB were significantly increased in diabetic vasculature compared with non-diabetic vasculature in the apolipoprotein E null background. However, in the presence of blockade/deletion of RAGE, the reduction in atherosclerosis was accompanied by marked reduction in vascular inflammation Citation[55–59].
It is important to note that RAGE ligands, AGEs, S100/calgranulins, and HMGB1, accumulate even in non-diabetic aortas of apolipoprotein E-deficient mice and that sRAGE and/or genetic deletion of RAGE exerts benefit in reducing both vascular inflammation, endothelial dysfunction (aortic rings), and atherosclerosis Citation[57], Citation[60]. In primary murine EC retrieved from the aortas of wild-type, but not EC retrieved from EC DN RAGE (driven by the preproendothelin-1 promoter) or RAGE-deficient mice, incubation with either S100B or AGE-containing oxidized low-density lipoprotein (LDL) upregulated vascular cell adhesion molecule (VCAM)-1 in a manner dependent on JNK MAP kinase signaling Citation[60]. Similar key roles for RAGE in inflammation in human aortic EC were demonstrated as well by the effects of siRNA designed to reduce RAGE expression Citation[60].
Beyond frank atherosclerosis, vascular inflammation most certainly contributes to other complications of diabetes as well. For example, in the isolated perfused heart, introduction of DN RAGE in EC (preproendothelin (PPET)-1 promoter) in diabetic and non-diabetic mice exerted protection against the adverse consequences of I/R Citation[49].
RAGE and signaling to key transcription factors—targeting NF-kB and Egr-1 to the inflammatory response
The diverse effects of RAGE in distinct cell types may be explained, at least in part, by the multiple signaling pathways activated by RAGE ligand-RAGE interaction. Multiple members of the MAP kinase family may be activated by RAGE; ligand-RAGE interaction activates p44/p42 (ERK, extracellular-regulated kinase) MAP kinase, p38 MAP kinase, and JNK MAP kinases Citation[60], Citation[61]. Further, AGE-mediated activation of ras and src kinase via RAGE in smooth muscle cells (SMC) is a key step in activation of NF-kB Citation[62], Citation[63]. The specific signaling pathways triggered by RAGE are influenced by the context of stimulatory signals; in the setting of arterial injury, for example, RAGE-mediated activation of JAK/STAT pathways critically impacts on SMC proliferation and migration Citation[64]. In monocytes/macrophages, NF-kB is a central target of ligand-RAGE. Recent studies suggested that in cultured microglial cells, RAGE-mediated upregulation of cyclo-oxygenase-2 required recruitment of cdc42/rac and JNK MAP kinase signal transduction Citation[65]. Cdc42/rac also plays key roles in neurite outgrowth in neuroblastoma cells in a manner dependent on RAGE Citation[66].
In addition to NF-kB, which plays central roles in proinflammatory signaling stimulated by RAGE ligands, recent studies have uncovered the novel finding that regulation of early growth response-1 (Egr-1) in hypoxic mouse hearts and primary EC is dependent on RAGE signaling Citation[67]. Mice deficient in RAGE failed to upregulate Egr-1 mRNA and protein in response to hypoxia in these organs/cells, and EMSA revealed that RAGE-deficient hearts or EC did not display increased Egr-1 activity in hypoxia Citation[67]. In these studies, RAGE-dependent upregulation of Egr-1 in hypoxia was dependent on rapid activation of protein kinase C, particularly the βII isoform, via stimulation of JNK MAP kinase signaling Citation[67]. Of note, although neither S100/calgranulin nor HMGB1 could be identified in the supernatants of primary EC in hypoxia, rapid (within 10 min) increase in AGE epitopes was observed Citation[67].
These findings suggest that multiple small biochemical species produced rapidly in acute cellular stress (hypoxia) or chronically (atherosclerosis) likely serve to trigger signaling, at least in part via RAGE, to stimulate mechanisms that enhance vascular inflammation and mediate tissue damage.
Summary and hypotheses
What might be deduced from these data is the conclusion that RAGE action is purely damaging. Emerging evidence suggests that this is an oversimplification. Analogous to beneficial and key roles for NF-kB in survival of hepatocytes Citation[68], blockade of RAGE and in specific phases and contexts of immune responses may be deleterious. Thus, although in models of severe sepsis, infection with Listeria monocytogenes, and massive hepatectomy, blocking or deleting RAGE is advantageous, distinct RAGE contexts reveal contrary results.
For example, key roles for RAGE in T lymphocyte priming may have implications for natural and adaptive responses to vaccination—as well as to exaggerated immune or autoimmune responses. This remains to be tested. Further, inflammatory responses are central to certain forms of tissue repair. Intriguingly, although blockade of RAGE did not negatively impact cutaneous wound healing in diabetic or non-diabetic animals Citation[69], or liver regeneration after massive hepatectomy Citation[39], in non-diabetic mice subjected to unilateral crush of the sciatic nerve, blockade of RAGE or its ligands caused delays in regeneration, as measured by motor and sensory conduction velocities and myelinated fiber density regeneration Citation[70]. Direct and beneficial roles for RAGE signaling in cells of mononuclear phagocyte lineage or in peripheral neurons (using DN RAGE transgenic mice) were demonstrated, as regeneration was impaired in these animals compared with wild-type litter-mate mice Citation[71].
What might explain these findings? One possibility is the pattern and/or chronicity of ligand upregulation may directly impact the RAGE-dependent outcome as, in chronic diabetic peripheral nerve damage (neuropathy), blockade or deletion of RAGE suppresses functional and pathological indices of damage Citation[72], Citation[73]. Further, the generalized or localized distribution of RAGE ligand in distinct settings may be a central factor. For example, Limana and colleagues administered HMGB1 locally to the injured myocardium in a rodent model beginning 4 hours after the infarction, and in this setting augmented repair to the infarcted heart resulted, in part via accelerated recruitment of stem cells Citation[74]. Although these authors did not demonstrate that these actions were via RAGE, many investigators have illustrated key roles for RAGE in mediating the impact of HMGB1. More recently, transgenic mice expressing HMGB1 on the α-myosin heavy chain promoter subjected to myocardial infarction demonstrated reduced necrosis and smaller infarct size Citation[75]. In this specific setting, the expression of transgenic HMGB1 was specifically limited to the heart and, indeed, the cardiomyocyte. Thus, as opposed to wild-type mice in which HMGB1 action in macrophages was deemed to be the infarct-provoking culprit Citation[51], via RAGE, in these distinct studies, local, cardiac-specific benefit of HMGB1 in healing the heart was observed. Importantly, however, it remains to be seen if these adaptive effects of HMGB1 in the heart occurred due to RAGE action.
The situation in the adaptive immune response is clearly more complex, involving multiple cell types both recruited to and in situ in vulnerable tissue as the immune response is initiated, sustained, and, usually, terminated. In this context, much evidence suggests that in autoimmune or chronic inflammatory diseases in which RAGE blockade has shown benefit in animal models (such as colitis, arthritis, EAE), striking and sustained upregulation of RAGE ligands is a prominent feature.
It is important to note that in the overall context of RAGE biology, it was not possible in this review to cover every cell type expressing RAGE and the biological impact of ligand-RAGE interaction. For example, the specific actions of RAGE on endothelial cells play critical roles in immune modulation. Thus, illustrates a partial summary of the cell types expressing RAGE and the consequences of ligand-stimulated RAGE action.
Table I. Cell types expressing RAGE (partial list): examples of the impact of RAGE ligands and selected references.
Collectively, the wide-ranging and sometimes paradoxical effects of RAGE signaling highlight the need to gather definitive evidence on the pros and cons of RAGE (). What must emerge from such investigations is a picture of the biological ‘safety’ profile of blocking RAGE, particularly in chronic and unrelenting disorders such as diabetes, Alzheimer's disease, and autoimmunity. Such data will provide essential information for the design of RAGE-directed therapy in the clinic.
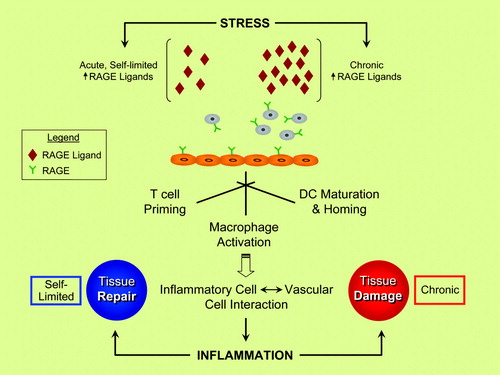
Figure 1. RAGE and inflammatory responses—repair versus injury? Evidence suggests that the ligand families of RAGE (receptor for advanced glycation end-products) are generated by both acute and chronic stresses. RAGE is expressed by key cell types linked to inflammatory responses, such as neutrophils, T lymphocytes, monocytes/macrophages, and dendritic cells (DC). Activation of RAGE in these cell types stimulates inflammation mechanisms, and, via RAGE ligand-stimulated upregulation of adhesion molecules and chemokines on endothelial cells (ECs), sets the stage for vascular inflammation and perturbation—harbingers of tissue damage. In acute stress, such as in injury to the peripheral nervous system, rapid and self-limited upregulation and release of RAGE ligands stimulate inflammatory mechanisms that contribute to tissue repair. In marked contrast, in chronic disease settings such as diabetes, autoimmunity, aging, and neurodegeneration, the long-term and sustained accumulation of RAGE ligands in the tissues crosses a threshold at some level, thereby triggering mechanisms that mediate chronic stress and, ultimately, tissue damage. A key challenge in the translational biology of RAGE is the identification of strategies to block the maladaptive consequences of RAGE ligation while preserving innate adaptive repair pathways.
Acknowledgements
The authors gratefully acknowledge funding from the United States Public Health Service and the Juvenile Diabetes Research Foundation. The authors are grateful to Ms Latoya Woods for her excellent assistance in the preparation of this manuscript. Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Schmidt AM, Vianna M, Gerlach M, Brett J, Ryan J, Kao J, et al. Isolation and characterization of binding proteins for advanced glycosylation end products from lung tissue which are present on the endothelial cell surface. J Biol Chem. 1992; 267: 14987–97
- Hori O, Brett J, Slattery T, Cao R, Zhang J, Chen M, et al. The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin: mediation of neurite outgrowth and co-expression of RAGE and amphoterin in the developing nervous system. J Biol Chem. 1995; 270: 25752–61
- Hofmann MA, Drury S, Fu C, Qu W, Taguchi A, Lu Y, et al. RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell. 1999; 97: 889–901
- Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A, et al. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer's disease. Nature. 1996; 382: 685–91
- Chavakis T, Bierhaus A, Al-Fakhri N, Schneider D, Witte S, Linn T, et al. The pattern recognition receptor (RAGE) is a counterreceptor for leukocyte integrins: a novel pathway for inflammatory cell recruitment. J Exp Med. 2003; 198: 1507–15
- Collison KS, Parhar RS, Saleh SS, Meyer BF, Kwaasi AA, Hammami MM, et al. RAGE-mediated neutrophil dysfunction is evoked by advanced glycation end products (AGEs). J Leukoc Biol. 2002; 71: 433–44
- Kislinger T, Fu C, Huber B, Qu W, Taguchi A, Yan SD, et al. N-ε (carboxymethyl)lysine modifications of proteins are ligands for RAGE that activate cell signalling pathways and modulate gene expression. J Biol Chem. 1999; 274: 31740–9
- Toure F, Zahm JM, Garnotel R, Lambert E, Bonnet N, Schmidt AM, et al. Receptor for advanced glycation end-products (RAGE) modulates neutrophil adhesion and migration on glycoxidated matrix. Biochem J. 2008; 416: 255–61
- Zen K, Chen CX, Chen YT, Wilton R, Liu Y. Receptor for advanced glycation endproducts mediates neutrophil migration across intestinal epithelium. J Immunol. 2007; 178: 2483–90
- Orlova VV, Choi EY, Xie C, Chavakis E, Bierhaus A, Ihanus E, et al. A novel pathway of HMGB1-mediated inflammatory cell recruitment that requires Mac-1 integrin. EMBO J. 2007; 26: 1129–39
- Yan SD, Schmidt AM, Anderson G, Zhang J, Brett J, Zou YS, et al. Enhanced cellular oxidant stress by the interaction of advanced glycation end products with their receptors/binding proteins. J Biol Chem. 1994; 269: 9889–97
- Foell D, Kane D, Bresnihan B, Vogl T, Nacken W, Sorg C, et al. Expression of pro-inflammatory protein S100A12 (EN-RAGE) in rheumatoid and psoriatic arthritis. Rheumatology (Oxford) 2003; 42: 1383–9
- Foell D, Wittkowski H, Hammerschmidt I, Wulffraat N, Schmeling H, Frosch M, et al. Monitoring neutrophil activation in juvenile rheumatoid arthritis by S100A12 serum concentrations. Arthritis Rheum. 2004; 50: 1286–95
- Wittkowski H, Sturrock A, van Zoelen MA, Viermann D, van der Poll T, Hoidal JR, et al. Neutrophil-derived S100A12 in acute lung injury and respiratory distress syndrome. Crit Care Med. 2007; 35: 1369–75
- Foell D, Seeliger S, Vogl T, Koch HG, Maschek H, Harms E, et al. Expression of S100A12 in cystic fibrosis. Thorax. 2003; 58: 613–7
- Foell D, Kucharzik T, Kraft M, Vogl T, Sorg C, Domschke W, et al. Neutrophil derived human S100A12 (EN-RAGE) is strongly expressed during chronic active inflammatory bowel disease. Gut. 2003; 52: 847–53
- Schmidt AM, Yan SD, Brett J, Mora R, Nowygrod R, Stern D. Regulation of human mononuclear phagocyte migration by cell surface-binding proteins for advanced glycation end products. J Clin Invest. 1993; 91: 2155–68
- Miyata T, Hori O, Zhang J, Yan SD, Ferran L, Iida Y, et al. The receptor for advanced glycation end products (RAGE) is a central mediator of the interaction of AGE-beta2microglobulin with human mononuclear phagocytes via an oxidant-sensitive pathway. Implications for the pathogenesis of dialysis-related amyloidosis. J Clin Invest. 1996; 98: 1088–94
- Rouhiainen A, Kuja-Panula J, Wilkman E, Pakkanen J, Stenfors J, Tuominen RK, et al. Regulation of monocyte migration by amphoterin. Blood. 2004; 104: 1174–82
- Hou FF, Miyata T, Boyce J, Yuan Q, Chertow GM, Kay J, et al. beta[2] Microglobulin modified with advanced glycation end-products delays monocyte apoptosis. Kidney Int. 2001; 59: 990–1002
- Wautier MP, Chappey O, Corda S, Stern DM, Schmidt AM, Wautier JL. Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. Am J Physiol Endocrinol Metab. 2001; 280: E685–94
- Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002; 418: 191–5
- Park JS, Gamboni-Robertson F, He Q, Svetkauskaite D, Kim JY, Strassheim D, et al. High mobility group box 1 protein interacts with multiple toll-like receptors. Am J Physiol Cell Physiol. 2006; 290: C917–24
- Tian J, Avalos AM, Mao SY, Chen B, Senthil K, Wu H, et al. Toll-like receptor-9 dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol. 2007; 8: 487–96
- Zamora R, Grishin A, Wong C, Boyle P, Wang J, Hackam D, et al. High mobility group box 1 protein is an inflammatory mediator in necrotizing enterocolitis: protective effect of the macrophage deactivator semapimod. Am J Physiol Gastrointest Liver Physiol. 2005; 289: G643–52
- Lue LF, Walker DG, Brachova L, Beach TG, Rogers J, Schmidt AM, et al. Involvement of microglial receptor for advanced glycation endproducts (RAGE) in Alzheimer's disease: identification of a cellular activation mechanism. Exp Neurol. 2001; 171: 29–45
- Walker DG, Link J, Lue LF, Dalsing-Hernandez JE, Boyes BE. Gene expression changes by amyloid beta peptide stimulated human postmortem brain microglia identify activation of multiple inflammatory processes. J Leukoc Biol. 2006; 79: 596–610
- Ichikawa K, Yoshinari M, Iwase M, Wakisaka M, Doi Y, Iino K, et al. Advanced glycosylation end products induced tissue factor expression in human monocyte-like U937 cells and increased tissue factor expression in monocytes from diabetic patients. Atherosclerosis. 1998; 136: 281–7
- Ding Y, Kantarci A, Badwey JA, Hasturk H, Malabanan A, Van Dyke TE. Phosphorylation of pleckstrin increases proinflammatory cytokine secretion by mononuclear phagocytes in diabetes mellitus. J Immunol. 2007; 179: 647–54
- Yan SSD, Wu ZY, Zhang HP, Furtado G, Chen X, Yan SF, et al. Suppression of experimental autoimmune encephalomyelitis by selective blockade of encephalitogenic T-cell infiltration of the central nervous system. Nat Med. 2003; 9: 287–93
- Chen Y, Yan SS, Colgan J, Zhang HP, Luban J, Schmidt AM, et al. Blockade of the late stages of autoimmune diabetes by inhibition of the receptor for advanced glycation end products. J Immunol. 2004; 173: 1399–405
- Moser B, Szabolcs MJ, Ankersmit HJ, Lu Y, Qu W, Weinberg A, et al. Blockade of RAGE suppresses alloimmune reactions in vitro and delays allograft rejection in murine heart transplantation. Am J Transplant. 2007; 7: 293–302
- Moser B, Desai DD, Downie MP, Chen Y, Yan SF, Herold K, et al. Receptor for advanced glycation end products expression on T cells contributes to antigen-specific cellular expansion in vivo. J Immunol. 2007; 179: 8051–8
- Dumitriu IE, Baruah P, Valentinis B, Voll RE, Herrmann M, Nawroth PP, et al. Release of high mobility group box 1 by dendritic cells controls T cell activation via the receptor for advanced glycation end products. J Immunol. 2005; 174: 7506–15
- Dumitriu IE, Baruah P, Bianchi ME, Manfredi AA, Rovere-Querini P. Requirement of HMGB1 and RAGE for the maturation of plasmacytoid dendritic cells. Eur J Immunol. 2005; 35: 2184–90
- Yang D, Chen Q, Yang H, Tracey KJ, Bustin M, Oppenheim JJ. High mobility group box-1 protein induces the migration and activation of human dendritic cells and acts as an alarmin. J Leukoc Biol. 2007; 81: 59–66
- Dumitriu IE, Bianchi ME, Bacci M, Manfredi AA, Rovere-Querini P. The secretion of HMGB1 is required for the migration of maturing dendritic cells. J Leukoc Biol. 2007; 81: 84–91
- Manfredi AA, Capobianco A, Esposito A, De Cobelli F, Canu T, Monno A, et al. Maturing dendritic cells depend on RAGE for in vivo homing to lymph nodes. J Immunol. 2008; 180: 2270–5
- Cataldegirmen G, Zeng S, Feirt N, Ippagunta N, Dun H, Qu W, et al. RAGE limits regeneration after massive liver injury by coordinated suppression of TNF-alpha and NF-kB. J Exp Med. 2005; 201: 473–84
- Andrassy M, Igwe J, Autschbach F, Volz C, Remppis A, Neurath MF, et al. Posttranslationally modified proteins as mediators of sustained intestinal inflammation. Am J Path. 2006; 169: 1223–37
- Turovskaya O, Foell D, Sinha P, Vogl T, Newlin R, Nayak J, et al. RAGE, carboxylated glycans and S100A8/A9 play essential roles in colitis-associated carcinogenesis. Carcinogenesis. 2008; 29: 2035–43
- Maeda S, Hikiba Y, Shibata W, Ohmae T, Yanai A, Ogura K, et al. Essential roles of high mobility group box-1 in the development of murine colitis and colitis-associated cancer. Biochem Biophys Res Commun. 2007; 360: 394–400
- Pisetsky DS, Erlandsson-Harris H, Andersson U. High mobility group box protein 1 (HMGB1): an alarmin mediating the pathogenesis of rheumatic disease. Arthritis Res Ther. 2008; 10: 209
- Hofmann MA, Drury S, Hudson BI, Gleason MR, Qu W, Lu Y, et al. RAGE and arthritis: the G82 polymorphism amplifies the inflammatory response. Genes Immun. 2002; 3: 123–35
- Liliensiek B, Weigand MA, Bierhaus A, Nicklas W, Kasper M, Hofer S, et al. Receptor for advanced glycation end products (RAGE) regulates sepsis but not the adaptive immune response. J Clin Invest. 2004; 113: 1641–50
- Lutterloh EC, Opal SM, Pittman DD, Keith JC, Jr, Tan XY, Clancy BM, et al. Inhibition of the RAGE products increases survival in experimental models of severe sepsis and systemic infection. Crit Care. 2007; 11: R122
- Boyd JH, Kan B, Roberts H, Wang Y, Walley KR. S100A8 and S100A9 mediate endotoxin induced cardiomyocyte dysfunction via receptor for advanced glycation end products. Circ Res. 2008; 102: 1239–46
- Bucciarelli LG, Kaneko M, Ananthakrishnan R, Harja E, Lee LK, Hwang YC, et al. Receptor for advanced-glycation end products: key modulator of myocardial ischemic injury. Circulation. 2006; 113: 1226–34
- Bucciarelli LG, Ananthakrishnan R, Hwang YC, Kaneko Song F, Sell DR, et al. RAGE and modulation of ischemic injury in the diabetic myocardium. Diabetes. 2008; 57: 1941–51
- Aleshin A, Ananthakrishnan R, Li Q, Rosario R, Lu Y, Qu W, et al. RAGE modulates myocardial injury consequent to LAD infarction via impact on JNK and STAT signaling in a murine model. Am J Physiol. 2008; 294: H1823–32
- Andrassy M, Volz HC, Igwe JC, Funke B, Eichberger SN, Kaya Z, et al. High mobility group box-1 in ischemia-reperfusion injury of the heart. Circulation. 2008; 117: 3216–26
- Zeng S, Feirt N, Goldstein M, Guarrera J, Ippagunta N, Ekong U, et al. Blockade of receptor for advanced glycation end product (RAGE) attenuates ischemia and reperfusion injury in the liver in mice. Hepatology. 2004; 39: 422–32
- Schmidt AM, Hori O, Chen JX, Li JF, Crandall J, Zhang J, et al. Advanced glycation endproducts interacting with their endothelial receptor induce expression of vascular cell adhesion molecule-1 (VCAM-1) in cultured human endothelial cells and in mice. A potential mechanism for the accelerated vasculopathy of diabetes. J Clin Invest. 1995; 96: 1395–403
- Gao X, Zhang H, Schmidt AM, Zhang C. AGE/RAGE produces endothelial dysfunction in coronary arterioles in type 2 diabetic mice. Am J Physiol Heart Circ Physiol. 2008; 295: H491–8
- Park L, Raman KG, Lee KJ, Lu Y, Ferran LJ, Jr, Chow WS, et al. Suppression of accelerated diabetic atherosclerosis by the soluble receptor for advanced glycation endproducts. Nat Med. 1998; 4: 1025–31
- Kislinger T, Tanji N, Wendt T, Qu W, Lu Y, Ferran LJ, Jr, et al. Receptor for advanced glycation end products mediates inflammation and enhanced expression of tissue factor in vasculature of diabetic apolipoprotein E null mice. Arterioscler Thromb Vasc Biol. 2001; 21: 905–10
- Bucciarelli LG, Wendt T, Qu W, Lu Y, Lalla E, Rong LL, et al. RAGE blockade stabilizes established atherosclerosis in diabetic apolipoprotein E null mice. Circulation. 2002; 106: 2827–35
- Wendt T, Harja E, Bucciarelli L, Qu W, Lu Y, Rong LL, et al. RAGE modulates vascular inflammation and atherosclerosis in a murine model of type 2 diabetes. Atherosclerosis. 2006; 185: 70–7
- Soro-Paavonen A, Watson AM, Li J, Paavonen K, Koitka A, Calkin AC, et al. Receptor for advanced glycation end products (RAGE) deficiency attenuates the development of atherosclerosis in diabetes. Diabetes. 2008; 57: 2461–9
- Harja E, Bu DX, Hudson BI, Chang JS, Shen X, Hallam K, et al. Vascular and inflammatory stresses mediate atherosclerosis via RAGE and its ligands in Apo E -/- mice. J Clin Invest. 2008; 118: 183–94
- Taguchi A, Blood DC, del Toro G, Canet A, Lee DC, Qu W, et al. Blockade of amphoterin/RAGE signaling suppresses tumor growth and metastases. Nature. 2000; 405: 354–60
- Lander HM, Tauras JM, Ogiste JS, Hori O, Moss RA, Schmidt AM. Activation of the receptor for advanced glycation end products triggers a p21(ras)-dependent mitogen-activated protein kinase pathway regulated by oxidant stress. J Biol Chem. 1997; 272: 17810–4
- Reddy MA, Li SL, Sahar S, Kim YS, Xu ZG, Lanting L, et al. Key role of Src kinase in S100b-induced activation of the receptor for advanced glycation end products in smooth muscle cells. J Biol Chem. 2006; 281: 13685–93
- Sakaguchi T, Yan SF, Yan SD, Rong LL, Sousa M, Belov D, et al. Arterial restenosis: central role of RAGE-dependent neointimal expansion. J Clin Invest. 2003; 111: 959–72
- Bianchi R, Adami C, Giamanco I, Donato R. S100b binding to RAGE in microglia stimulates cox-2 expression. J Leukocy Biol. 2007; 81: 108–18
- Huttunen H.J, Fages C, Rauvala H. Receptor for advanced glycation end products (RAGE) mediated neurite outgrowth and activation of NF-kappaB require the cytoplasmic domain of the receptor but different downstream signaling pathways. J Biol Chem. 1999; 274: 19919–24
- Chang JS, Wendt T, Qu W, Kong L, Zou YS, Schmidt AM, et al. Oxygen deprivation triggers upregulation of early growth response-1 by the receptor for advanced glycation end products. Circ Res. 2008; 102: 905–13
- Bellas R.E, FitzGerald M.J, Fausto N, Sonenshein G.E. Inhibition of NF-kB activity induces apoptosis in murine hepatocytes. Am J Pathol. 1997; 151: 891–6
- Goova MT, Li J, Kislinger T, Qu W, Lu Y, Bucciarelli LG, et al. Blockade of receptor for advanced glycation end-products restores effective wound healing in diabetic mice. Am J Pathol. 2001; 159: 513–25
- Rong LL, Trojaborg W, Qu W, Kostov K, Yan SD, Gooch C, et al. Antagonism of RAGE suppresses peripheral nerve regeneration. FASEB J. 2004; 18: 1812–7
- Rong LL, Yan SF, Wendt T, Hans D, Pachydaki S, Bucciarelli LG, et al. RAGE modulates peripheral nerve regeneration via recruitment of both inflammatory and axonal outgrowth pathways. FASEB J. 2004; 18: 1818–25
- Bierhaus A, Haslbeck KM, Humpert PM, Liliensiek B, Dehmer T, Morcos M, et al. Loss of pain perception in diabetes is dependent on a receptor of the immunoglobulin superfamily. J Clin Invest. 2004; 114: 1741–51
- Toth C, Rong LL, Yang C, Martinez J, Song F, Ramji N, et al. Receptor for advanced glycation end products (RAGE) and experimental diabetic neuropathy. Diabetes. 2008; 57: 1002–17
- Limana F, Germani A, Zacheo A, Kajstura J, Di Carlo A, Borsellino G, et al. Exogenous high mobility group box 1 protein induces myocardial regeneration after infarction via enhanced cardiac C-kit+ cell proliferation and differentiation. Circ Res. 2005; 97: e73–83
- Kitahara T, Takeishi Y, Harada M, Nizeki T, Suzuki S, Sasaki T, et al. High mobility group box 1 restores cardiac function after myocardial infarction in transgenic mice. Cardiovasc Res. 2008; 80: 40–6
- Guo XH, Huang QB, Chen B, Wang SY, Li Q, Zhu YJ, et al. Advanced glycation end products induce actin rearrangement and subsequent hyperpermeability of endothelial cells. APMIS. 2006; 114: 874–83
- Hsieh HL, Schafer BW, Weigle B, Heizmann CW. S100 protein translocation in response to extracellular S100 is mediated by receptor for advanced glycation endproducts in human endothelial cells. Biochem Biophys Res Commun. 2004; 316: 949–59
- Kaji Y, Amano S, Usui T, Oshika T, Yamashiro K, Ishida S, et al. Expression and function of receptors for advanced glycation end products in bovine corneal endothelial cells. Invest Ophthalmol Vis Sci. 2003; 44: 521–8
- Fiuza C, Bustin M, Talwar S, Tropea M, Gerstenberger E, Shelhamer JH, et al. Inflammation-promoting activity of HMGB1 on human microvascular endothelial cells. Blood. 2003; 101: 2652–60
- Yamagishi S, Fujimori H, Yonekura H, Yamamoto Y, Yamamoto H. Advanced glycation endproducts inhibit prostacyclin production and induce plasminogen activator inhibitor-1 in human microvascular endothelial cells. Diabetologia. 1998; 41: 1435–41
- Mackic JB, Stins M, McComb JG, Calero M, Ghiso J, Kim KS, et al. Human blood brain barrier receptors for Alzheimer's amyloid-beta 1-40. Asymmetrical binding, endocytosis, and transcytosis at the apical side of brain microvascular endothelial cell monolayer. J Clin Invest. 1998; 102: 734–43
- Bierhaus A, Illmer T, Kasper M, Luther T, Quehenberger P, Tritschler H, et al. Advanced glycation end product (AGE)-mediated induction of tissue factor in cultured endothelial cells is dependent on RAGE. Circulation. 1997; 96: 2262–71
- Schmidt AM, Hasu M, Popov D, Zhang JH, Chen J, Yan SD, et al. Receptor for advanced glycation end products (AGEs) has a central role in vessel wall interactions and gene activation in response to circulating AGE proteins. Proc Natl Acad Sci U S A. 1994; 91: 8807–11
- Wautier JL, Wautier MP, Schmidt AM, Anderson GM, Hori O, Zoukourian C, et al. AGEs on the surface of diabetic erythrocytes bind to the vessel wall via a specific receptor inducing oxidant stress in the vasculature: a link between surface-associated AGEs and diabetic complications. Proc Natl Acad Sci U S A. 1994; 91: 7742–6
- Warboys, CM, Toh, HB, Fraser, P. AGEs rapidly increase retinal microvascular permeability via RAGE activation of NADPH oxidase. Invest Ophthalmol Vis Sci. Nov 7, 2008, (Epub ahead of print).
- Shaw SS, Schmidt AM, Banes AK, Wang X, Stern DM, Marrero MB. S100B-RAGE-mediated augmentation of angiotensin II-induced activation of JAK2 in vascular smooth muscle cells is dependent on PLD2. Diabetes. 2003; 52: 2381–8
- Yoon YW, Kang TS, Lee BK, Chang W, Hwang KC, Rhee JH. Pathobiological role of advanced glycation endproducts via mitogen-activated protein kinase dependent pathway in the diabetic vasculopathy. Exp Mol Med. 2008; 40: 398–406
- Korenaga K, Micci MA, Taglialatela G, Pastricha PJ. Suppression of nNOS expression in rat enteric neurones by the receptor for advanced glycation end-products. Neurogastroenterol Motil. 2006; 18: 392–400
- Wang L, Li S, Jungalwala FB. Receptor for advanced glycation end products (RAGE) mediates neuronal differentiation and neurite outgrowth. J Neurosci Res. 2008; 86: 1254–66
- Origlia N, Righi M, Capsoni S, Cattaneo A, Fang F, Stern DM, et al. Receptor for advanced glycation end product-dependent activation of p38 mitogen activated protein kinase contributes to amyloid-beta-mediated cortical synaptic dysfunction. J Neurosci. 2008; 28: 3521–30
- Hassid BG, Nair MN, Ducruet AF, Otten ML, Komotar RJ, Pinsky DJ, et al. Neuronal RAGE expression modulates severity of injury following transient focal cerebral ischemia. J Clin Neurosci. 2008; 16: 302–6
- Kobayashi T, Oku H, Komori A, Okuno T, Kojima S, Obayashi H, et al. Advanced glycation end products induce death of retinal neurons via activation of nitric oxide synthase. Exp Eye Res. 2005; 81: 647–54
- Arancio O, Zhang HP, Chen X, Lin C, Trinchese F, Puzzo D, et al. RAGE potentiates Abeta-induced perturbation of neuronal function in transgenic mice. EMBO J. 2004; 23: 4096–105
- Kogel D, Peters M, Konig HG, Hashemi SM, Bui NT, Arolt V, et al. S100b potently activates p65-c-Rel transcriptional complexes in hippocampal neurons: clinical implications for the role of S100B in excitotoxic brain injury. Neuroscience. 2004; 127: 913–20
- Srikrishna G, Huttunen HJ, Johansson L, Weigle B, Yamaguchi Y, Rauvala H, et al. N-glycans on the receptor for advanced glycation end products influence amphoterin binding and neurite outgrowth. J Neurochem. 2002; 80: 998–1008
- Huttunen HJ, Kuja-Panula J, Rauvala H. RAGE signaling induces CREB-dependent chromogranin expression during neuronal differentiation. J Biol Chem. 2002; 277: 38635–46
- Du Yan S, Zhu H, Fu J, Yan SF, Roher A, Tourtellotte WW, et al. Amyloid beta peptide receptor for advanced glycation endproduct interaction elicitis neuronal expression of macrophage colony stimulating factor: a proinflammatory pathway in Alzheimer's disease. Proc Natl Acad Sci U S A. 1997; 94: 5296–301
- Sousa MM, Yan SD, Stern D, Saraiva MJ. Interaction of RAGE with transthyretin triggers nuclear transcription factor (NF-kB) activation. Lab Invest. 2000; 80: 1101–10
- Huttunen HJ, Kuja-Panula J, Sorci G, Agneletti AL, Donato R, Rauvala H. Coregulation of neurite outgrowth and cell survival by amphoterin and S100 proteins through RAGE activation. J Biol Chem. 2000; 275: 40096–105
- Sousa MM, Du Yan S, Fernandes R, Guimaraes A, Stern D, Saraiva MJ. Familial amyloid polyneuropathy: RAGE dependent triggering of neuronal inflammatory and apoptotic pathways. J Neurosci. 2001; 21: 7576–86