
Abstract
Purpose
Familial hyperaldosteronism type 1 (FH-1) is an autosomal dominant form of primary aldosteronism (PA), featuring a marked phenotypic heterogeneity, ranging from mild forms of PA and arterial hypertension (HT) to severe forms complicated by stroke at a young age. Affected patients usually reach the fertile age; hence, transmission of the disease to offspring is common. Notwithstanding this, only anecdotal reports of FH-1 in pregnancy exist and recommendations for treatment remain vague.
Materials and Methods and Results
We herein report on a novel FH-1 pedigree featuring very severe HT, fatal aortic dissection, and high rate of early stroke, where a young FH-1 woman was successfully managed throughout pregnancy with low-dose dexamethasone.
Conclusions
Based on this experience and on available information on pathophysiology of FH-1 in pregnancy, the pros and cons of dexamethasone administration in the treatment of FH-1 in pregnancy are also discussed.
Introduction
Familial hyperaldosteronism type 1 (FH-1), also known as glucocorticoid-remediable-aldosteronism (GRA) (OMIM: 103,900), is an autosomal dominant form of primary aldosteronism (PA) featuring a marked phenotypic heterogeneity, ranging from mild to severe forms of arterial hypertension (HT) and aldosteronism complicated by stroke, either ischaemic or haemorrhagic, at a young age.
The discovery of a chimeric gene highlighted the molecular basis of FH-1 in 1992 as this gene derives from an unequal crossing-over of two genes that are 95% homologous and map close on chromosome 8q24 [Citation1]: the CYP11B1 gene encoding for 11-β hydroxylase, the enzyme forming cortisol, and the CYP11B2 gene that encodes for aldosterone synthase. The adrenocorticotropic hormone (ACTH) responsive elements in the chimeric gene accounted for the ectopic expression of aldosterone synthase, the CYP11B2 product, in the adrenocortical zona fasciculata and for FH-1 remittance in response to glucocorticoid treatment. Genetic testing now permits a conclusive diagnosis and institution of a glucocorticoid treatment to suppress the ACTH-drive, which corrects the hyperaldosteronism and normalises the high blood pressure (BP) values.
FH-1 affects < 1% of PA patients, but its prevalence could be markedly underestimated since PA is common and often overlooked in hypertensive patients. Although fatal strokes occurring at a young age in FH-1 have been reported, most patients show a mild clinical PA phenotype, and therefore reach the fertile age, which allows transmission of the disease to offspring. Nonetheless, only scant anedoctal cases of FH-1 during pregnancy have been reported [Citation2–5].
We herein report on a novel FH-1 pedigree featuring severe hypertension and a high prevalence of early onset stroke, entailing a young affected woman who was successfully managed throughout pregnancy with low-dose dexamethasone. Based on this experience and on available information on pathophysiology of FH-1 in pregnancy, some recommendations for treatment of this condition are given.
Case presentation and pedigree
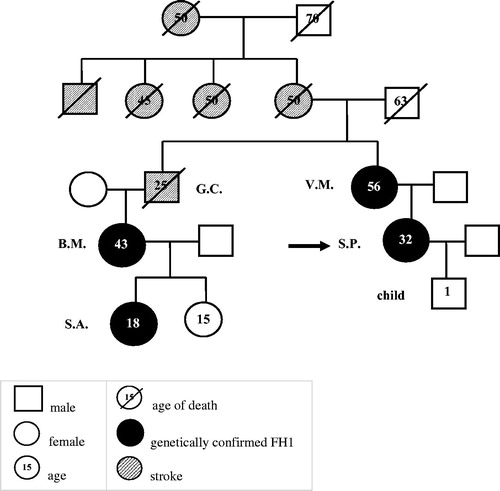
The proband (S.P.) was a 27 years-old Caucasian professional volley player with a 7 years history of hypertension. She was diagnosed with PA in another hospital, but no subtyping was undertaken. At the time of the diagnosis, her plasma aldosterone concentration (PAC) was 70.9 ng/dl (n.v. < 15 ng/dl), plasma renin activity (PRA) was 0.30 ng/ml/hour (n.v. 0.65–2.61), corresponding to a DRC value of 2.5 mIU/L. The ARR, as calculated with the ARR-App [Citation6], was 290 ng/mIU (n.v. < 20.6). In 2009, since low-dose spironolactone (25 mg o.d.) did not control her high BP values, she was referred to our ESH Centre of Excellence, where she was presented with her 51-years old mother (V.M.). A strong family history of arterial hypertension and cerebrovascular disease emerged. V.M. also reported a history of HT first detected at age 20 years, for which she was on treatment with spironolactone 25 mg o.d. and amiloride/hydrochlorothiazide 2.5/25 mg o.d. with poor control of her BP values. The family pedigree () was characterised by a high prevalence of stroke: V.M.’s mother and her three uncles had a stroke before age 50 years. V.M.’s brother (G.C.) also suffered from repeated strokes and died of aortic rupture at age of 25 years. G.C.’s 38 years-old daughter (B.M.) was reported to have received a diagnosis of an undefined form of PA at another hospital. B.M.’s daughter (S.A.) was discovered to have HT during childhood and, after being tested positive for FH-1, was successfully treated with dexamethasone 0.25 mg o.d. and potassium canrenoate 100 mg o.d.
Figure 1. Family tree of the proband.
Neither the proband (S.P.) nor the mother (V.M.) reported any history of pre-eclampsia, abortion, cardio- or cerebro-vascular disease and their physical examination was unremarkable except for stage 1 HT and a mid-systolic click, indicating the presence of a mitral valve prolapse, which was confirmed at transthoracic echocardiography in both.
Both S.P. and V.M. showed a florid form of PA () and genetic testing using long-PCR confirmed the diagnosis of FH-1 (). They were started on low dose dexamethasone at 0.50 mg o.d. in S.P. and 0.375-0.75 mg o.d. in V.M., in the evening, which normalised BP and PAC values () and lowered cortisol levels in both patients. Considering the occurrence of stroke in their pedigree (), both S.P. and MV were submitted to cerebral angio-MR that allowed to rule out any vascular abnormalities.
Figure 2. Genetic confirmation of the proband (S.P.) and her mother (V.M.). Agarose gel electrophoresis showing the results of long PCR for FH1 diagnosis of the daughter SP (lane 2 and lane 3) and of the mother VM (lane 4 and lane 5). In lane 2 and 4 are showed the PCR products of the wildtype CYP11B2, while in lane 3 and 5 are showed the chimeric gene amplicons.
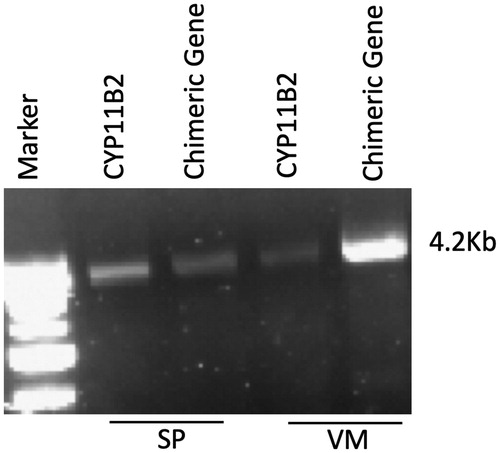
Table 1. Clinical and biochemical features of the proband (S.P.) and her mother (V.M.).
In June 2013, S.P. became pregnant and, given her normal BP values and serum K+ levels and the expected fall of BP during the first trimester, she was advised to stop dexamethasone treatment and to close monitor BP and serum K+ values during gestation. At the end of the second trimester, because of increasing BP, she was started again on low-dose dexamethasone (0.25 mg o.d. in the evening), which immediately normalised her BP values. Ultrasonography showed a regular foetal development during gestation. A natural delivery of a healthy normal weight male baby occurred at term with no complications. The postnatal course was uneventful and the newborn growth was normal. After the delivery, she restarted dexamethasone dose she was taking before pregnancy (0.50 mg o.d.) with benefit ().
Notwithstanding multiple invitations and exhaustive information on the importance of an early diagnosis of FH-1 and early institution of targeted treatment for the prevention of cerebrovascular abnormalities, S.P. refused a free genetic test for FH-1 on baby’s saliva DNA. A FH-1 test was offered again in 2019, when she informed us that her 5-years old child was found to have severe HT resistant to treatment, but unfortunately she refused.
Discussion and conclusion
In this FH-1 woman from a pedigree with a very high rate of premature severe cardiovascular events, low-dose dexamethasone treatment warranted control of arterial hypertension and hyperaldosteronism before and during the last part of gestation. Upon discovery of pregnancy, treatment was stopped; notwithstanding this, BP and hyperaldosteronism remained well controlled during the first trimester and the first part of the second trimester. Overall, available data [Citation2] and the present case suggest the safety of discontinuing such treatment for the foetus and the mother during such period.
During the second and third trimester, immediate control of the high BP values was accomplished by the reintroduction of low-dose dexamethasone treatment, which provided a regular course of the pregnancy, and the natural delivery of an apparently healthy baby at term. The occurrence of a severe form of drug-resistant hypertension in the child at age 5 years suggested inheritance of the FH-1 gene. Unfortunately, this could not be verified at genetic testing notwithstanding exhaustive information on the importance of an early diagnosis of FH-1. Concerns about labelling the child as affected by a genetic disease were possible reasons for this weird decision.
Only anecdotal cases of FH-1 management during pregnancy exist [Citation2–5], but some pathophysiological considerations can be made. During normal pregnancy, aldosterone secretion remains tightly regulated, allowing the mother to cope with changes of salt intake, and to develop body volumes expansion. The parallel increase of progesterone and aldosterone is key for normal development of the placenta. Following the placental production of corticotropin releasing hormone (CRH), ACTH and cortisol levels also increase and, considering that aldosterone production is ACTH-dependent in FH-1, it was hypothesised that this could exacerbate hypertension during pregnancy [Citation7].
However, the increase of progesterone, which acts as aldosterone-antagonist on the mineralocorticoid receptor (MR), might counterbalance this exacerbation, and can account for the normalisation of blood pressure and PAC during the first trimester of pregnancy and possibly throughout pregnancy as noticed in two reported FH-1 cases [Citation2,Citation3]. Moreover, it was also shown that progesterone, but not oestradiol, inhibited the chimeric isoforms of aldosterone synthase in HEK-293 cells transiently transfected with vectors containing the full chimeric CYP11B2 cDNAs [Citation8]. At variance with the reported cases [Citation2,Citation3], our patient showed a BP normalisation only during the first part of pregnancy, after which reinstitution of dexamethasone treatment became necessary owing to high BP values.
Compared to other glucocorticoids, dexamethasone is known to be more potent in suppressing ACTH. Nonetheless, it was contended that prednisolone and hydrocortisone would be preferable to dexamethasone in pregnancy, because they are inactivated more effectively by placental 11-β-hydroxysteroid dehydrogenase type 2 (11-β-HSD-2). Considering the higher potency of dexamethasone in suppressing ACTH and the fact that FH-1 treatment is for the mother and not for the foetus, upon recurrence of severe hypertension during late pregnancy, it seemed logical to us to administer to our patient the lowest dose of dexamethasone that had warranted normotension and control of hypokalaemia. The happy end of this pregnancy testified the safety and effectiveness of this choice.
Acknowledgements
Prof. Rossi managed the case and drafted the manuscript. Dr. Sanga participated in the acquisition of the data and collected data from literature. Dr. Lenzini performed the genetic testing. Prof. Seccia revised the manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability statement
Data sharing is not applicable to this article as no datasets were generated or analysed during the current study.
Additional information
Funding
References
- Lifton RP, Dluhy RG, Powers M, et al. A chimaeric llβ-hydroxylase/aldosterone synthase gene causes glucocorticoid-remediable aldosteronism and human hypertension. Nature. 1992;355(6357):262–265.
- Campino C, Trejo P, Carvajal CA, et al. Pregnancy normalized familial hyperaldosteronism type I: A novel role for progesterone. J Hum Hypertens. 2015;29(2):138–139.
- Hamilton E, O’Callaghan C, O’Brien RM, et al. Familial hyperaldosteronism type 1 in pregnancy. Intern Med J. 2009;29(2):138–139.
- Wyckoff JA, Seely EW, Hurwitz S, et al. Glucocorticoid-remediable aldosteronism and pregnancy. Hypertension. 2000. 35(2):668–672.
- Mulatero P, Cella SM, Di Williams TA, et al. Glucocorticoid remediable aldosteronism: Low morbidity and mortality in a four-generation Italian pedigree. J Clin Endocrinol Metab. 2002;87(7):3187–3191.
- Rossi GP, Bisogni V. A useful tool to improve the case detection rate of primary aldosteronism: the aldosterone -renin ratio (ARR)-App. J Hypertens. 2016. 34(5):1019–1021.
- Escher G. Hyperaldosteronism in pregnancy. Ther Adv Cardiovasc Dis. 2009;3(2):123–132.
- Vecchiola A, Lagos CF, Fuentes CA, et al. Different effects of progesterone and estradiol on chimeric and wild type aldosterone synthase in vitro. Reprod Biol Endocrinol. 2013;11:76.