Abstract
A key role for ontologies in bioinformatics is their use as a standardized, structured terminology, particularly to annotate the genes in a genome with functional and other properties. Since the output of many genome-scale experiments results in gene sets it is natural to ask if they share a common function. A standard approach is to apply a statistical test for overrepresentation of functional annotation, often within the gene ontology. In this article we propose an alternative to the standard approach that avoids problems in overrepresentation analysis due to statistical dependencies between ontology categories. We apply methods of feature construction and selection to preprocess gene ontology terms used for the annotation of gene sets and incorporate these features as input to a standard supervised machine-learning algorithm. Our approach is shown to allow the straightforward use of an ontology in the context of data sourced from multiple experiments to learn classifiers predicting gene function as part of a cellular response to environmental stress.
INTRODUCTION
Ontologies are of growing importance in biomedical informatics and their uptake in this area may constitute one of the most successful applications to date of ontological engineering. Resources from open-source projects such as the gene ontology (GO)Footnote 1 are now nearly ubiquitous tools in bioinformatics (Bard and Rhee, Citation2004). A recent checkFootnote 2 showed the original GO article by Ashburner and The Gene Ontology Consortium (Citation2000) had over 1700 citations in PubMed Central. A number of reasons can be identified for this success from the standpoints of biology and computer science—for reviews see Bada et al. (Citation2004) and Bodenreider and Stevens (Citation2006). In this article, we are concerned with two such aspects of the gene ontology that led us to study its applicability for machine-learning problems in bioinformatics.
First, the use of ontologies such as GO provides a standard terminology for functional genomics—the objective of which is to describe the function of all genes in the genome of an organism—for example, in the analysis of gene expression data (Baldi and Hatfield, Citation2002). Thus it is important for machine-learning tools to work with such data. Second, the category definitions and hierarchical structure of GO represent a “pre-semantic web” view of ontology, where the goal of the representation was as a tool for human inspection rather than as a formal knowledge structure suitable for automated inference. The question is then how the structure of such a resource can be handled by automated systems.
The baker's yeast Saccharomyces cerevisiae is one of the key model organisms in biology and its study has led to many publicly available datasets for functional genomics research. We are in the process of adding machine-learning to a yeast dataset analysis tool currently in development, for which the domain name www.yeastinformatics.org has been reserved. Therefore a key task is to allow the use of GO annotation of yeast genes in the preparation of training sets for standard machine-learning tools.
In previous work (Bain, Citation2002) we developed a feature construction approach using formal concept analysis that demonstrated bias shift, leading to improved predictive accuracy with standard machine-learning algorithms. Feature construction as a preprocessing step is a promising approach for handling nonvector-based data as input to attribute vector-based, machine-learning methods. We investigated the applicability of this approach to learning from GO annotation data in Akand, Bain, and Temple (Citation2007). In this article we expand these results and assess the strengths and limitations of the approach.
The process of feature construction can be computationally expensive, and so in this article we extend our approach to investigate methods of feature selection as an alternative. In this approach, the GO categories are preprocessed using standard methods to select those likely to lead to good predictive accuracy (Guyon and Elisseeff, Citation2003).
The results we obtain suggest that an integrative analysis of heterogeneous genome-wide data on the behavior of cellular systems (Kanehisa, Citation2000) is possible based on standard machine-learning tools.
GENE ONTOLOGY IN FUNCTIONAL GENOMICS
The gene ontology is structured as an acyclic directed graph or DAG. Vertices or nodes are given a unique ID—a string of the form “GO:N” where N is a natural number—and a textual description intended to characterize some biological properties. There are three sub-ontologies in the gene ontology, referred to as molecular function (MF), cellular component (CC), and biological process (BP). See www.geneontology.org for more details.
In the gene ontology edges have two types, “is_a” or “part_of.” X is_a Y represents a hyponymy relation, i.e., if X is true of an object then Y is also true for it (or alternatively, Y is a hypernym of X). X part_of Y denotes meronymy (or alternatively, Y is a holonym of X). The meronomy relation is clearly intended to be different from hyponomy. However, current practice in statistical analysis of GO annotation data ignores the difference and assumes that the presence of any GO ID implies the presence of its hypernyms and holonyms, and so on recursively.
For most biologists, the key role of the gene ontology is the use of the textual descriptions as a standardized vocabulary to annotate genes in terms of their function (MF), location in the cell (CC) and involvement in biochemical pathways (BP). Gene Ontology annotations are associations made between gene products and the GO terms that describe them. A gene product is an RNA or protein product encoded by a gene. Many genes are annotated with multiple descriptions. These descriptions, each with its unique ID and place in the GO DAG, are usually referred to as GO terms or categories (used synonymously) or simply nodes.
For example, the gene known as CDC28 in the yeast Saccharomyces cerevisiae is annotatedFootnote 3 to one MF category GO:0004693 “cyclin-dependent protein kinase activity,” two CC categories GO:0005634 “nucleus” and GO:0005737 “cytoplasm” and 13 BP categories including GO:0006468 “protein amino acid phosphorylation” and GO:051446 “positive regulation of meiotic cell cycle.” Shown in Figure is part of the path to root of the MF ontology from GO:0004693.
FIGURE 1 A fragment of the annotation path for CDC28 in GO molecular function. Indentation on a line denotes refinement of a parent term on the previous line by the child term; in this example, every child has an “is_a” edge to its parent. The numbers in brackets indicate the number of gene products annotated to the GO term. Note the multiple inheritance from the parents on lines 5 and 9 to the term GO:0004672 on lines 6 and 10 and its descendants. CDC28 is annotated to the term GO:0004693 on lines 6 and 12.

Since the initiation of the gene ontology project many groups have developed specialized GO software tools to handle both the ontology itself and data (typically sets of genes) annotated with GO terms. In the former category are tools such as ontology editors, while the second contains mainly tools designed for overrepresentation or “enrichment” analysis, i.e., estimating the statistical significance of finding a set of genes annotated to a GO term.
Overrepresentation Analysis
A common setting for overrepresentation analysis is computing P-values using a particular statistical test of gene sets resulting from a genome-wide high-throughput assay, typically a gene expression microarray experiment (Baldi and Hatfield, Citation2002). In this context, most tools adopt a standard probabilistic model for the number of genes that would be found by chance to be annotated to the particular GO category of interest. Typical choices for such models, and hence significance tests, include the hypergeometric or binomial distributions, or the χ2 or Fisher's exact tests (Khatri and Drăghici, Citation2005).
A typical application of the hypergeometric distribution is the following, where we have a set of genes annotated to a particular GO term and we are interested in knowing the probability of finding that number of genes thus annotated simply by chance.Footnote 4 We assume a “background distribution” of genes, typically the total number of genes in the genome with GO annotations, or the total number of genes in the experiment. This number is n. Of this set of genes, the size of the subset annotated to the GO term of interest is m ≤ n. In the results of the experiment, the size of our set of genes is s and the number of genes in that set annotated to our term of interest is r. The probability of finding by chance r genes from s thus annotated is given by the hypergeometric distribution:
Optimistic Bias in Probability Estimates
A well-known problem in the analysis of GO annotation using a probabilistic model to estimate statistical significance is that of multiple testing, in this case, where each GO term is tested separately using Equation (Equation1). As the number of applications of a statistical test on a dataset increases so the probability of obtaining an apparently significant result (P-value below the selected threshold) increases. This is usually allowed for by essentially lowering the effective significance threshold based on the number of tests by using the Bonferroni correction or alternatives (Boyle et al., Citation2004).
In the case of overrepresentation analysis of GO annotation the issue turns out to be more complicated. The Bonferroni correction is usually thought of as being a conservative correction, i.e., the effective significance threshold is lowered more than necessary. However, Boyle et al. (Citation2004) report that in their experiments, the Bonferroni correction is not conservative enough, leading to an optimistic bias in estimating GO categories as statistically significant annotation for gene sets.
A simple qualitative argument can be developed to suggest why this is the case, and it has important consequences for the use of ontological annotation in overrepresentation analysis. The gene ontology, like many ontologies, is a generalization hierarchy. This means that any object annotated to a GO term is also implicitly annotated to all of its ancestor (i.e., more general) terms. Just by considering the relation of a node to its parents, as in Table , the effect of this in terms of multiple testing is obvious. Using the notation from Equation (Equation1) we see from Table that the hierarchical structure of the gene ontology implies a dependency between the total number m out of n genes annotated to a GO term and the number (≥m) annotated to any of its parents and hence all of its ancestors. The number of genes r in any sample of size s annotated to a GO term and its parents (≥r) shows a similar dependency relation.
TABLE 1 Tests on GO Categories Where One is More General Than the Other are Not Independent—See Text for Details
However, the Bonferroni correction assumes that each statistical test applied to the outcomes of an experiment (here, each set of genes annotated to a GO term) is independent. This can be expressed as an assumption that the values for r and m occurring in one statistical test have no relation to the values in any other test (s and n are fixed for any particular experiment). In the case of GO terms, as seen in the table, this is clearly incorrect. Once we have a GO term T annotating r genes, the probability of having other terms (i.e., the ancestors of T) annotating ≥r genes is increased. A similar argument applies in the case of values of m.
In order to deal with this bias, Boyle et al. (Citation2004) implemented an alternative correction factor based on randomly sampling s genes from the background set of n and applying Equation (Equation1) to each GO term annotating any of this set. Repeating this procedure 1000 times gives for each GO term the proportion of apparently significant gene sets under the null hypothesis of random selection, which may then be used as an adjusted P-value. Although this way of computing a correction for P-values avoids independence assumptions, it took three orders of magnitude longer to compute.
COVERAGE MATRIX
An approach developed by Carey (Citation2004) aims to account for GO structure to obtain an information measure for GO annotation. In this approach, GO edge types are ignored and edges are regarded as instances of the single relation refines(C, P) where C and P are child and parent nodes.
Terms to which a gene is annotated are referred to as the “associated terms” or simply associations for the gene. The set of associated terms for a gene is then its “ontological annotation.” Figure shows the separation between the ontology and the associations. In this diagram, the ontology is a DAG, with the edge direction going from child to parent, i.e., an edge from term T i to term T j denotes an instance of the relation refines(T i , T j ). In this case, the “terms” are the letters {a, b, c, d, e, f, g, h}, and the ontology has a single edge type, shown by the solid arrows.
FIGURE 2 Example of a DAG-structured ontology (such as GO, but with a single edge type) and associated objects (such as genes) annotated to its terms.
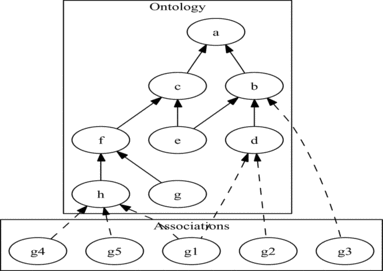
The associations in Figure are shown in a separate box below the ontology. Here the set of “genes” {g1, g2, g3, g4, g5} are linked with their associated ontology terms by the dotted arrows. Note the same gene can be annotated with multiple terms (e.g., g1), which we refer to as multiple category annotation, and that genes can be annotated to a term at any level in the ontology hierarchy (e.g., d as well as its parent b), referred to as a multiple depth of annotation.
Carey introduces the idea of an object-ontology complex in which the refinement relation from the GO DAG is represented as a binary (0, 1) matrix Γ. Γ is a square matrix V × V where V is the number of terms in the ontology. For two terms T i , T j in the ontology, Γ ij = 1 if refines(T i , T j ) for i > j, otherwise Γ ij = 0. Matrix powers Γ k represent k-step refinements of ontology terms.
A second matrix M maps P objects (here genes) to V ontology terms. In accordance with GO annotation policy, it is assumed that genes are annotated to the most specific term. All 1-step refinements of the object annotations can then be computed as C 1 = MΓ. This is generalized using the idea of coverage, where a term covers an object if that term or any refinement of it is associated with the object via the matrix M. The binary coverage matrix C (P × V) contains all such covers.
Table has the coverage matrix for the example in Figure . It is interesting to note that the coverage matrix corresponds to the interrelation between the terminological and assertion components of an ontology represented in a description logic, where they are known as the TBox and ABox, respectively (Baader et al., Citation2003). In this setting, specific genes from experiments can be thought of as ABox individuals, while the the gene ontology itself forms a TBox.
TABLE 2 Coverage Matrix for the Example in Figure
The coverage matrix can be used to calculate the probability of a term in the context of a specific object-ontology complex, i.e., a specific set of genes and their annotation in the gene ontology. The sum of the column i for a term T
i
is n
i
, i.e., the number of genes annotated to T
i
or one of its refinements. The probability of that term appearing in the annotation of the gene set is , where n is the number of occurrences of the most frequent term in the annotation, typically the root node of the ontology.
Carey proposes an information-based similarity measure between terms. The information (in bits) of term T i is −log 2 P(T i ). However, to the best of our knowledge no statistical tests for overrepresentation analysis based on the coverage matrix approach have been developed.
ONTOLOGICAL ANNOTATION FOR MACHINE-LEARNING
Overrepresentation analysis is a commonly used approach that fits well the paradigm of exploratory data analysis. This is appropriate when the purpose of a biological experiment is “data-driven” rather than “hypothesis-driven,” e.g., to group together similarly behaving genes in a microarray experiment by clustering expression profiles (Baldi and Hatfield, Citation2002). However, other experimental designs can be used that lead instead to results in which the data is divided into two or more groups. Then the task is to find a model or “hypothesis” that is the “best fit” to the data according to some criterion. In statistics, this is known as discriminant analysis and in machine-learning as supervised or classifier learning (Alpaydin, Citation2004).
If the experimental setting is appropriate then discriminant analysis can have advantages compared to overrepresentation analysis. For example, in the context of functional genomics experiments, there are problems with the approach of developing a probabilistic model then estimating statistical significance. First, simple probabilistic models may fail to take into account dependencies due to structure in a dataset. Second, the difficulty of constructing correct probabilistic models increases rapidly with the number and diversity of datasets to be used for integrative analysis, where the goal is to combine multiple sources of data from different experiments to obtain a more complete picture of the operation of biological systems.
However, there are also problems in the use of this kind of heterogeneous data with discriminant analysis or classifier learning. Algorithms of this type require example data in the form of fixed-width vectors of attribute values. Much of the data used, for example, graph data, in the yeast dataset analysis tool currently in development, is not in this format. In this article we focus on gene ontology annotation data.
In order to handle this type of annotation as data for classifier learning there are two problems to be dealt with, namely, multiple category annotation and multiple depth of annotation (see Figure ). The problem of multiple category annotation is that a given gene may have several different functions in a cell, and it may be found in several cellular processes and in different locations. Therefore, any gene is annotated with all of the categories with which it has been associated in the published scientific literature. In any particular experimental setting, however, only a subset of the known annotations of a gene will be relevant. This is known as a multi-instance problem. For propositional machine-learning algorithms this is not an easy problem to solve.
The problem of multiple depth of annotation arises from the hierarchical arrangement of ontology categories and the way in which these are used to annotate genes. For example, two genes may have a related function, but are annotated at different levels of generality. Unfortunately, to a learning algorithm this relationship is not apparent; the categories are different. The learning algorithm could be modified to deal with the concept hierarchy. However, this is not straightforward and would have to be done over again for each learning algorithm to be used, which is impractical. This problem is compounded since all genes have not been annotated to the same extent through biological experimentation, leading to variations in multiple depth of annotation.
A solution to both problems may be provided by adapting the coverage matrix previously mentioned and use the ontological annotation for input to a supervised machine-learning algorithm. Viewed in terms of graph theory, the coverage matrix represents the induced graph for a set of genes and their associations with respect to the DAG structure of the gene ontology. Non-zero entries on each row denote all terms in the set of paths from the associated terms for that gene to the root node of the ontology. The columns can be seen as Boolean attributes or features.
Given the large number of GO termsFootnote 5 it is necessary to apply either machine-learning techniques specialized to handle very high-dimensional data, or methods to select or construct “good” features. In this work we are concerned with learning human-comprehensible models, so we adopt the latter approach. The idea is to preprocess the data containing multiple category and multiple depth annotation and use properties of the probability distribution on the annotation categories to select subsets of “good” features, or construct new “intermediate” features based on such subsets.
Feature Selection
Feature selection (see Guyon and Elisseeff, Citation2003; for a recent review) is typically a preprocessing step carried out on a dataset prior to the application of machine-learning. It can have a number of objectives, such as increasing the efficiency (e.g., faster runtimes) or the effectiveness (e.g., predictive accuracy) of learning. It can also contribute towards a better understanding of the domain due to simplification of datasets and learned models. In this work we focus on supervised learning, and we have used two types of features selection: feature ranking and feature subset selection.
Feature Ranking
In feature ranking for classifier learning the goal is to evaluate each feature separately in terms of its ability to predict the class. This approach uses an information theory-based heuristic to evaluate the usefulness of individual GO terms for predicting the class label. Specifically, the method looks to maximize information gain (similar to the method used in decision-tree learning Quinlan, Citation1993). This is important, as a good feature should partition the search space into separable classes. For a feature (or attribute) with values A = {a 1, a 2,…} and a class with values C = {c 1, c 2,…} Equations (Equation2) and (Equation3) define the entropy of the class C before and after observing feature A (Hall and Holmes, Citation2003):
Here p(c i ) is the prior probability for all values for class C, and p(c j | a i ) is the conditional probability of class value c j given feature value a j . The value of entropy H ranges from 0 to 1. If all the values of a feature belong to the same class then entropy becomes zero. Otherwise if all values of a feature are random with respect to the class, this results in entropy of one.
The mutual information I(C, A) is computed as:
In Equation (Equation4), the amount the entropy of C decreases after observing A is called information gain (or mutual information). As this is a symmetrical function, the amount of information gained for C after observing A is equal to the information gain for A after observing C. High information gain indicates strong interdependence between the feature values and the class values and that the feature may be useful in a classifier. Once the information gain is computed for all features, they can be ranked and features with low ranks can be eliminated.
The aim of feature selection is to include only a subset of the original features that are more relevant to the target concept or contain more information about the class label. For each feature in the coverage matrix (i.e., induced ontology graph) for a gene set, the algorithm computes the information gain. The set of features above a minimum number of genes M is ranked and the top K among them are selected to use in the dataset. The parameters M and K filter out features with too few genes or with too little information gain. For example, in the experiments reported in a subsequent section we used M = 2 and K was set to include the top 75% of the ranked features. The algorithm for feature selection by information gain ranking—called “IG Ranker”—is in Figure .
FIGURE 3 IG Ranker-feature selection by information gain ranking.

Feature Subset Selection
The goal here is to select a “good” subset of features for a dataset. Clearly, a search for the “best” subset is not generally practical. Therefore many heuristic approaches have been proposed. In this work, we selected the correlation-based feature selection (CFS) method of Hall (Citation2000).
Details of the method are beyond the scope of this article. However, the intuitive basis for the heuristic measure used is that good subsets of features should be highly correlated with the class (i.e., likely to be relevant) while inter-correlation between features should be low (i.e., unlikely to be redundant).
The CFS algorithm implements forward selection, a greedy search (hill-climbing) method, which starts with an empty set of features and at each iteration adds the feature that has the highest value for the heuristic measure. Search terminates when no increase in this value can be obtained by adding further features to the subset.
Feature Construction
The coverage matrix is a bipartite graph since it denotes a set of edges between genes and GO terms. It can therefore be represented as a “cross table” or formal context in the framework of formal concept analysis (Ganter and Wille, Citation1999). A method of feature construction can then be derived, based on our previous results (Bain, Citation2002). The example genes in the dataset can then be reexpressed in terms of the new intermediate features which define combinations of GO categories. Constructed features can then be selected by the learning algorithm based on their utility in forming accurate models to predict the class of the genes.
Formal Concept Analysis
Detailed coverage of formal concept analysis (FCA) is in Ganter and Wille (Citation1999). In this section we follow the treatments of Godin and Missaoui (Citation1994) and Carpineto and Romano (Citation1993) since they are more oriented towards machine-learning. However, some naming and other conventions have been changed.
Definition 1 (Formal Context)
A formal context is a triple ⟨𝒟, 𝒪, ℛ ⟩. 𝒟 is a set of descriptors (attributes), 𝒪 is a set of objects, and ℛ is a binary relation such that ℛ ⊆ 𝒟 × 𝒪.
The notation ⟨x, y ⟩ ∈ ℛ or alternatively xℛy is used to express the fact that a descriptor x ∈ 𝒟 is a property of an object y ∈ 𝒪.
Definition 2 (Formal Concept)
A formal concept is an ordered pair of sets, written ⟨X, Y ⟩, where X ⊆ 𝒟 and Y ⊆ 𝒪. Each pair must be complete with respect to ℛ, which means that X′ = Y and Y′ = X, where X′ = {y ∈ 𝒪 | ∀ x ∈ X, xℛy} and Y′ = {x ∈ 𝒟 | ∀ y ∈ Y, xℛy}.
The set of descriptors of a formal concept is called its intent, while the set of objects of a formal concept is called its extent. For a set of descriptors X ⊆ 𝒟, X is the intent of a formal concept if and only if X″ = X, by composition of the ′ operator from Definition 2. A dual condition holds for the extent of a formal concept. This means that any formal concept can be uniquely identified by either its intent or its extent alone. Intuitively, the intent corresponds to a kind of maximally specific description of all the objects in the extent.
The correspondence between intent and extent of complete concepts is a Galois connection between the power set 𝒫(𝒟) of the set of descriptors and the power set 𝒫(𝒪) of the set of objects. The Galois lattice ℒ for the binary relation is the set of all complete pairs of intents and extents, with the following partial order. Given two concepts N 1 = ⟨X 1, Y 1 ⟩ and N 2 = ⟨X 2, Y 2 ⟩, N 1 ≤ N 2↔X 1 ⊇ X 2. The dual nature of the Galois connection means we have the equivalent relationship N 1 ≤ N 2↔Y 1 ⊆ Y 2.
The formal context ⟨𝒟, 𝒪, ℛ ⟩ together with ≤define an ordered set which gives rise to a complete lattice. The following version of a theorem from Godin and Missaoui (Citation1994) characterizes concept lattices.
Theorem 3 (Fundamental Theorem on Concept Lattices (Godin and Missaoui (Citation1994))
Let ⟨𝒟, 𝒪, ℛ⟩ be a formal context. Then ⟨ℒ; ≤ ⟩ is a complete latticeFootnote 6 for which the least upper bound (Sup) and greatest lower bound (Inf) are given by
Since we are concerned with concepts formed from sets of descriptors, the partial order as well as Sup and Inf definitions are given so as to relate to lattices in machine-learning rather than that which is typical in formal concept analysis. That is, the supremum Sup of all nodes in the lattice is the “most general” or top (⊤) node and the infimum Inf is the “most specific” or bottom (⊥).
Feature Construction from Concept Lattices
Treating the coverage matrix as a formal context (Definition 1), where genes are objects and GO terms are descriptors, enables the construction of concept lattices in which the formal concepts (Definition 2) contain sets of GO terms that group together sets of genes. The terms shared by such groups of genes indicate the biological properties that they have in common. An example of such a concept lattice is shown in Figure .
FIGURE 4 Concept lattice for the coverage matrix of Table 2.
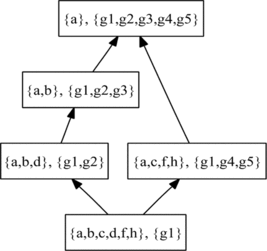
In our previous work (Bain, Citation2002) we investigated the use of concept lattices for both unsupervised and supervised learning. Both cases required the use of an information-based measure on formal concepts, similar to that of Carey (Citation2004). While both approaches are based on probability, our measure was motivated by the compressibility or algorithmic complexity (Chaitin, Citation1987) of a concept. Compressibility of structured data objects, such as strings in a formal language, or as in this case, formal concepts in a lattice, is inversely related to the probability of finding such objects by chance.
This approach was later used by us for ontology learning, essentially by extracting concepts from a concept lattice and combining them in a structured (propositional) logic program. However, while this is suitable for unsupervised learning it ignores the distribution of classes within the set of objects. Therefore, to use concepts in supervised learning we used a pseudo-MDL, a measure inspired by the Minimum Description Length (MDL criterion), in which compressibility was combined with the entropy of the class distribution of the examples in the intent of the concept. The intuition behind this is that concepts that will be useful in supervised learning will tend to have high compressibility and low class entropy, i.e., they will potentially lead to high accuracy classifiers.
Selecting Predictive Constructed Features
For the current work, where we are focused on the specific problem of handling gene ontology annotation in supervised learning, we implemented a simpler approach to feature construction. This feature construction method focuses on selecting discriminative concepts; in this case, those that discriminate well between different class values are said to be predictive.
The procedure to construct features from a concept lattice for use in supervised learning was as follows. The method shares two parameters with information gain ranking: M, the minimum number of examples to be covered by any selected concept, and K, the maximum number of concepts to be selected.
-
for each of the genes in the training set, generate the gene's GO coverage. For the example of Figure , this gives a set of “objects,” i.e., genes:
-
construct a concept lattice ℒ from the objects shown as clauses in step 1.
-
for each formal concept in ℒ with extent containing ≥M objects, evaluate the class distribution of the objects in the concept.
-
sort the concepts identified at step 3 in decreasing order of predictive accuracy for the majority class of objects in the concept.
-
select the top K concepts in the order identified at step 4, or simply all of those with accuracy above that of the frequency of the majority class (e.g., for a two-class problem, >0.5).
-
construct a table containing for each gene, a row noting whether, for each of the concepts identified at step 5 the gene is in the concept (i.e., feature is true) or not (feature is false).
-
join the table constructed at step 6 with the class values (further attributes can also be joined in the same way) to form the training set for supervised machine-learning.
This procedure is actually much more efficient than that of Bain (Citation2002) since it only involves constructing the concept lattice once. When this is done, concepts can be evaluated quickly, and there is no expensive lattice revision as in the earlier method.
Web Tool for Ontological Analysis by Machine-Learning
High-throughput, genome-wide analysis of the model organism baker's yeast Saccharomyces cerevisiae has enabled many biological attributes to be assigned to each yeast gene. These data attributes include protein-protein interactions (PPI), transcription factor-binding location analysis and protein location data. In addition, for each gene or encoded protein there are many curated sources of information that describe an associated metabolic pathway or ontology (Ashburner and The Gene Ontology Consortium, Citation2000). These data represent an invaluable resource to identify the nature of a particular gene or to define the relationship between genes. Typically, this is achieved through overrepresentation analysis of a gene set to identify significant clusters of genes belonging to each biological attribute, such as a gene ontology or metabolic pathway. This type of analysis is carried out in an interactive and exploratory manner by biologists, with the end result being one or more gene sets—these are “genes of interest” that can be used in publications or to generate new experimental hypotheses.
The problem of data analysis has become more complex and time-consuming since typically research laboratories now perform in-house, genome-wide experimentation to address niche research interests. Consequently, overrepresentation analysis needs to be performed for many interdependent gene sets. Apart from the issues of overrepresentation analysis previously discussed, this is a problem since many of the popular analysis tools process each gene set in isolation and multiple analyses must be manually collated. Typically, multiple gene sets are generated from one or more microarray experiments or deletion library screens.
Yeastinformatics
The Yeastinformatics web tools allow one to simultaneously determine which categories are overrepresented in multiple gene sets and to determine their relative distribution to determine whether these are common or unique. In addition, the Yeastinformatics web pages contain modules to facilitate: (i) the annotation of overrepresented sets; (ii) listing of all database attributes for a single gene; (iii) graphical display of all PPI for a single gene and its interactions between all connected partners; (iv) graphical display of PPI within one gene set or between two gene sets; and (v) listing of all genes bound by pairwise combinations of transcription factors.
Approximately 200,000 gene attributes, such as an interacting protein, bound transcription factors, or a metabolic pathway are stored in a MySQL database, and these data are used for overrepresentation analysis, gene annotation or, as in this article, generation of datasets for machine-learning. P-values for the overrepresentation analysis are calculated using the hypergeometric distribution with optional Bonferroni correction for multiple testing (Boyle et al., Citation2004) and only those intersections that pass the filter are tabulated. Additionally, the tables may be filtered by the numbers of intersections or not filtered at all. Calculation of hypergeometric distribution requires that the number of genes in the genome be known; however, not all genes have been verified and many are hypothetical or dubious. To allow for this, users can select which genome features to include for the calculation.
Yeastinformatics is implemented as a group of open access dynamic web pages.
Processing Gene Ontology Annotation
To complement the analysis and visualization tools in Yeastinformatics we have implemented a web interface to the database to support the generation of datasets for input to machine-learning tools.
The machine-learning approach adopted has two stages. First, the user can interactively upload datasets, browse datasets, and combine multiple datasets to form a new integrated dataset for a set of genes. Then the resulting data can be preprocessed into a suitable format and downloaded as a file for input into machine-learning tools such as the widely used Weka open-source data mining toolkit (Witten and Frank, Citation2005) or other tools such as Microsoft Excel. If the user has selected gene ontology annotation to be included, it is at the preprocessing stage that feature selection or feature construction is applied.
The feature selection method is based on information gain ranking described; we have also implemented the method of feature construction. Further processing is available via tools such as Weka, which implements feature subset selection using the CFS algorithm.
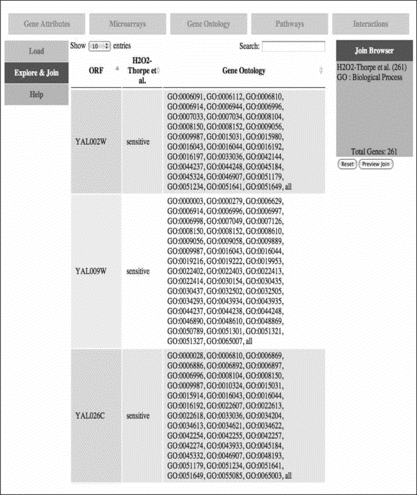
We use the MySQL database version of the gene ontology which is downloaded to update our database every month. This allows the coverage matrix to be generated efficiently via SQL queries and combined with other data for a gene set. A screen shot of the web interface is in Figure . Shown is a page from where the user has selected the biological process ontology to annotate a gene set based on one of the gene deletant screens in the database. Following this, the GO preprocessing can be run as previoulsy described and training sets generated in Weka file format for machine-learning.
FIGURE 5 Web page to combine gene ontology data with other datasets. Along top of the page are tabs to select a dataset type, such as microarray data or interactions. On the left-hand side, the user can select the mode of activity, such as uploading new data, browsing a dataset, or constructing an integrated set of multirelational data. On the right-hand side are shown the current components of the dataset being constructed. In this example, the GO biological process ontology is being integrated with oxidative stress data.
EXPERIMENTAL RESULTS
Biological Background: Cellular Network Response to Stress
The number of organisms for which the complete genome (DNA sequence) is available continues to grow at an increasing rate. Meanwhile, there have also been major advances in laboratory techniques to analyze complex cellular processes. This has ushered in a new era of cell biology termed systems biology, in which responsive phenotypes, i.e., the measurable characteristics of the organism in response to environmental or genetic perturbations, can be investigated genome-wide, i.e., by collecting data on the activity of all the organism genes simultaneously. In this way we can investigate the cellular network response of the genes that give rise to an observed phenotype as the downstream effect of an external stimulus through signal transduction.
For example, when cells adapt to sudden changes in the environment, cellular network responses include the action of sets of transcription factors (proteins) to activate sets of genes involved in biochemical pathways. Such a responsive subnetwork of the cell is referred to as the genetic regulatory network (GRN). The protein products of co-expressed genes in a GRN combine to form interacting molecular machines that produce a responsive cellular phenotype. This responsive subnetwork is partly described by the PPI network. In turn, proteins act to regulate cellular metabolism in pathways of biochemical reactions, and by subtle feedback mechanisms, their own GRNs and PPI networks.
The bakers and brewers yeast Saccharomyces cerevisiae is a key model organism for systems biology, due to the ease with which genetic manipulation can be carried out. Virtually all areas of cell biology have benefited from the use of yeast as a model organism to study processes and pathways relevant to higher eukaryotes. Importantly, many fundamental processes in yeast are conserved through to humans. Data describing yeast cellular network responses are derived from high-throughput, genome-wide experimental techniques, the development of which continues unabated. However, although a decade has passed since the sequencing of the complete yeast genome, there is still a sizeable subset of yeast genes with no known molecular function. Even for those with a designated function this often denotes only part of their likely cellular role. Yet yeast is one of the most intensively studied organisms with a relatively small genome. A key reason that knowledge on gene function is not greater is that the computational techniques and tools which will provide biologists with systematic ways to integrate the data resources and generate hypotheses about the function of cellular networks are not yet in place. This study aims to contribute towards that goal.
Overview of Experiments
We evaluated our approach to learning from ontological annotation with a comparative study of different methods on two biological datasets. We selected two problems on yeast systems biology and formulated them as classification problems. In both cases, the task was to learn a classifier to predict a selected behavior or phenotype of interest for genes (or their products), given a profile of other measures for those genes, i.e., a set of attributes or features.
We chose real problems, in the sense that any progress with them may be of interest to biologists. This enabled us both to obtain quantitative results on the performance of different methods and validate the learned classifiers in biological terms. The first case study was on the prediction of protein expression in response to the addition of hydrogen peroxide (H2O2) to yeast cells to produce “oxidative stress.” The second task was more difficult—predicting the behavior of genes when yeast cells were challenged by a range of multiple stress agents.
Machine-learning is typically focused on the generation of predictive models, e.g., classifiers with high predictive accuracy on datasets. However, such models can sometimes lack in explanatory power. In this work we are concerned with learning predictive models but we also require them to be comprehensible; hence our approach uses decision tree learning (Quinlan, Citation1993; Witten and Frank, Citation2005).
Since this was a preliminary study, and we wanted to focus on the effects of representation, i.e., attributes used rather than the choice of learning algorithms, we limited our attention to a single algorithm. We also had a requirement that the classifiers should be comprehensible to biologists. This indicated an approach such as decision-tree learning. We selected the Weka implementation (Witten and Frank, Citation2005) of the C4.5 decision-tree induction system (Quinlan, Citation1993) called J48. For these experiments we used the default parameters for J48 and ran 10 replicates of 10-fold cross-validation to obtain a mean predictive accuracy.
Case Study 1: Predicting Protein Expression
A preliminary “proof of principle” experiment was carried out to test the main aims of this work. First, we aimed to investigate whether a supervised learning approach using a standard machine-learning algorithm was suitable to perform an integrative analysis of high-throughput results from multiple molecular biology experiments. A second aim was to compare feature selection and construction approaches as described above to investigate the use of gene ontology annotation in such integrated datasets.
Experiment 1: Protein Expression Given Gene Expression
As an example application of integrating datasets on yeast genes, we uploaded two sets of data on the same 92 genes to our database. These two datasets give different “snapshots” of the yeast cellular network response to the addition of hydrogen peroxide to the cells' growth environment. This environmental oxidant places the yeast cells under oxidative stress, causing the suppression of some normal functioning and the activation of cellular defence mechanisms. This provides a useful experimental tool for studying cellular responses to environmental stress. As an additional test, we also included data on other stresses to see if any effect of a common stress response system would be observed.
-
Classification task. In the article by Godon et al. (Citation1998), the authors identified 56 proteins whose synthesis was stimulated and 36 that were repressed under oxidative stress caused by exposure of yeast cells to hydrogen peroxide. This was a proteomics study, i.e., the results obtained reflected changes in the total composition of proteins in the cell using comparative two-dimensional gel electrophoresis.
-
Attributes. In the article by Causton et al. (Citation2001) microarray data was collected on the cellular network response by yeast to six different environmental stresses, including hydrogen peroxide. In contrast to the proteomics study previously mentioned, this data was on the transcriptional response in terms of mRNA levels in the cell observed at 8–10 time points over a period of around 2 hours. This reflects genes that are “turned on” or “turned off” in response to the addition of the stress-inducing agent to the cellular environment. Data for each stress condition comprises a time-series, with mRNA levels recorded at irregular intervals following initial exposure to the stressor.
-
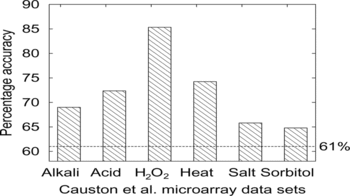
Results. The results are summarized in the histogram of Figure . These show that gene expression under H2O2 is a good predictor of protein expression (mean accuracy 85.3%). Of the remaining five conditions, three (heat, acid, and alkali) show some predictivity (mean accuracy 69.0–74.2%). The other two conditions—salt and sorbitol—have mean accuracy 65.8% and 64.8%, respectively. However, this is only slightly above the baseline accuracy of simply predicting the majority class for all examples (60.9%), shown as the dotted line across the histogram in Figure .
FIGURE 6 Accuracy of predicting protein expression given six microarray datasets (see text for details). The dotted horizontal line shows the baseline accuracy of 61% obtained by simply predicting the majority class for all genes.
Experiment 2: Protein Expression Given GO Features
-
Classification task The classification task was the same as that in the previous section.
-
Attributes.The set of attributes was limited to those gene ontology categories in the coverage matrix of the 92 genes to be classified. Each of the three GO sub-ontologies BP, CC, and MF was used to generate separate coverage matrices. Each set of GO categories was then used as the basis for four methods of feature selection or construction as follows:
No selection – no feature selection or construction; used as a baseline for comparison.
IG ranker – the information gain ranking method, with M = 2 and K set to select the top-ranked 75% of features.
CFS – correlation-based feature selection; the Weka implementation was used with default parameter settings.
Conduce – uses formal concept analysis to construct new features ranked by their predictive ability with M = 2 and K set to select all features.
-
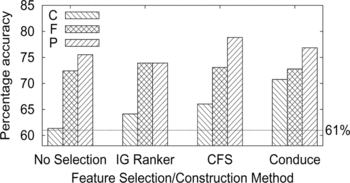
Result.The results are summarized in the histogram of Figure . These show that gene ontology categories can provide predictive power, although less so than gene expression under H2O2. The highest performance was obtained with the CFS method of feature subset selection on BP (mean accuracy 78.8%). However, the performance range between subontologies for each selection or construction method is relatively large. Conduce has the smallest range (mean accuracy 70.8–76.8%) and No Selection the largest (mean accuracy 61.3–75.5%). For each selection method, the ordering in terms of decreasing accuracy was BP, MF, and CC.
FIGURE 7 Accuracy of predicting protein expression given cellular component (C), molecular function (F), and biological process (P) subontologies of the gene ontology. Four feature selections and construction methods are compared for each subontology (see text for details). The dotted horizontal line shows the baseline accuracy of 61% obtained by simply predicting the majority class for all genes.
Experiment 3: Protein Expression Given Microarray Data and GO Features
This approach was discussed in Akand et al. (Citation2007). With the addition of features from the MF ontology in this experiment the predictive accuracy of decision tree learning showed a slight increase to 87% correct, with a slightly smaller tree size (five leaves, nine nodes), compared to the use of the Causton et al. (Citation2001) microarray data alone. The tree learned is shown in Figure . In this tree, the feature “node_25” stands for the following set of molecular function GO terms:
-
/%molecular/_function; GO:0003674
-
/%catalytic activity ; GO:0003824
-
/%transferase activity ; GO:0016740
-
/%transferase activity, transferring alkyl or aryl (other than methyl) groups; GO:0016765
-
/%methionine adenosyltransferase activity; GO:0004478
FIGURE 8 A decision tree for protein induction repression learned with gene ontology features. Ovals are attribute tests (“Peroxide t” means microarray data at time t), classifications are at leaves. See text for details.
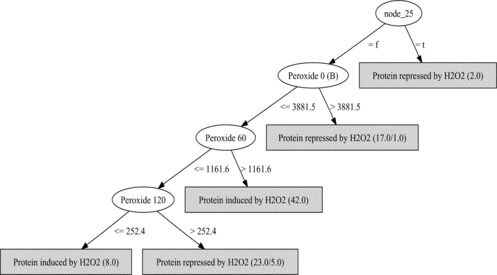
Although this is biologically plausible, the question of whether it has significance in the context of this dataset is left to future work.
We also investigated adding features from the other subontologies, BP and CC separately, in all pairwise combinations, and all together using Conduce. However, at around 79–80%, predictive accuracy was not as high for these combinations.
Case Study 2: Predicting General vs. Specific Stress Response
The Saccharomyces Genome Deletion Project (Winzeler et al., Citation1999) is a set of yeast strains in each of which exactly one gene from the genome has been systematically removed. About 4800 of these deletant strains are viable (in the remainder, the deleted gene is essential for life). Strains from this set can then be studied to see the effects of genes under different conditions. Roughly, the methodology enables the inference of a functional role for a gene in response to some stress if its deletant shows some growth defect under that stress.
Biologists have carried out many “screens” of the deletant set—selecting a subset of genes and subjecting each of the corresponding deletants to that stress. Unlike our first case study, where a fairly clear relationship between the target class and the microarray data would be expected, the screen data presents a more interesting yet more difficult problem, since many mechanisms could be hypothesized to be the cause of the observed effect of a gene deletion. In a nutshell, case study 1 had the aim of learning to predict an intracellular response given measurements of intracellular concentration of mRNA plus GO annotation, whereas in this case study we aim to learn to predict an extracellular response, given the same data. This is in fact more representative of a typical laboratory analysis.
We have collected a set of 26 screens on a total of 1094 deletants. Each screen represents deletant sensitivity to a chemical or environmental stress—the “phenotype” is that cells are sensitive (to a given stress) when the gene is deleted. These included multiple screens from Thorpe et al. (Citation2004) and Tucker and Fields (Citation2004). Initial results on individual screens from both these studies indicated two issues with the approach we developed in case study 1.
First, for each individual screen we have, in most cases, simply a set of genes deemed “sensitive.” For supervised learning, it is not clear what the set of “not-sensitive” genes should be. Since only a subset of the genes are tested in a screen, one possibility is to take those deletants tested but not found to be sensitive. But in most cases, this is a much smaller proportion than those that are sensitive, leading to a very skewed class distribution. It also ignores the fact that other genes not tested in the screen could be “sensitive” or “not-sensitive.”
Second, Thorpe et al. (Citation2004) noted that for many of the screens in their study there was little correlation between microarray data and the deletant sensitivity phenotype. This was borne out in our attempt to use the microarray data under various stress conditions to predict the response to H2O2 stress in screens from both Thorpe et al. (Citation2004) and Tucker and Fields (Citation2004), where we found no improvement in predictive accuracy over predicting the majority class.
-
Classification task. Since many of the 26 screens contain genes sensitive only to that stress, we defined instead a “meta-level” classification problem, in terms of the number of screens under which a deletant was found to be sensitive. We found 607 deletants sensitive to exactly one or two screens. While no gene was sensitive to more than eleven screens, we found 409 out of 1016 were sensitive to three or more screens. These could be “key” deletants—part of a general cellular stress response system—whereas the other 607 could be involved in responses more specific to the particular stress-inducing agent. The training set therefore contained 409 genes labelled “general-responders” and 607 labelled “specific-responders.” We will refer to this task as predicting the “response-class.”
-
Attributes. We envisaged three experiments for this classification task. In the first experiment, the task was to predict the response class given the microarray datasets. However, as we found with the H2O2 screens, and as observed by Thorpe et al. (Citation2004), there was no correlation between the microarray data and the response class. This resulted in no decision tree being found that exceeded the baseline accuracy of 60% obtained by simply predicting the majority class for all genes. This led us to abandon the experiment.
The second experiment involved predicting the response class given GO features. We used the same approach as before, with the exception of Conduce, which was unable to run to completion on the enlarged training set. However, we also investigated a method of feature selection based on ranking all GO terms by their P-value computed by the hypergeometric distribution of Equation (Equation1), as follows. For all genes in the general responders (resp. specific-responders) class, we collected all GO terms with P-value less than 0.1. The union of these sets of GO terms was then used as a set of Boolean features—a feature has the value true if a gene is annotated to the term; otherwise it is false.
Thirdly, we aimed to investigate the combined representation of microarray data plus GO features. However, as with the first experiment, we found the microarray data gave no increase in predictive accuracy, and the results were not significantly different from using GO features alone.
-
Result. The results are summarized in the histogram of Figure and Table . We also obtained some initial results on the method of feature ranking using the hypergeometric distribution P-values. We selected the biological process subontology, as that was the best performing, and achieved a 10-fold cross-validation accuracy of 82.5% in a tree of only 19 leaves. Since these were not obtained using the 10 replicates of 10-fold cross-validation methodology, we have omitted them from Table .
FIGURE 9 Accuracy of predicting general vs. specific deletant sensitivity to multiple stresses given cellular component (C), molecular function (F), and biological process (P) subontologies of the gene ontology. Three feature selection and construction methods are compared for each subontology (see text for details). The dotted horizontal line shows the baseline accuracy of 60% obtained by simply predicting the majority class for all genes.
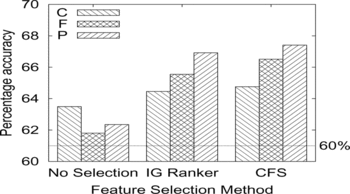
TABLE 3 Accuracies of Predicting General vs. Specific Deletant Sensitivity to Multiple Stresses with GO Biological Process Features. Note that°, • Denote Statistically Significant Improvement or Degradation, Respectively, with Respect to “No Selection”
DISCUSSION
We have investigated two separate but empirically related problems to test our approach to incorporating GO terms into machine-learning. In both, we compared the use of gene expression data alone, GO terms alone, and both together. The first problem was one of predicting an “intra-cellular” phenotype, the second predicting an “extracellular” phenotype. In the first case, we found that both types of information in isolation were comparable in terms of predictive accuracy, and their combination yielded no significant extra performance. The second case was a considerably more difficult problem, and here the use of GO terms enabled some gains in predictive accuracy, although gene expression data, separately or in combination, gave no improvement.
We are able to conclude that our results support the hypothesis that GO annotation can be useful in the context of supervised machine-learning.
Case Study 1: Predicting Protein Expression
There is a relationship between protein synthesis and gene expression. For example, Godon et al. (Citation1998) noted that the alterations they observed in the expression of proteins in response to hydrogen peroxide would be likely to involve a transcriptional component. In particular, the observed genomic response to oxidative stress strongly suggested an element of transcriptional control. Accordingly, we designed a classification task to predict the protein response observed in the Godon et al. (Citation1998) data in terms of attributes involving transcriptional response to hydrogen peroxide, or any of the other stressors.
Two effects are suggested by the results in Figure . One is a specific response to H2O2 by transcription of genes leading to expression of corresponding proteins. The other is that some nonspecific response, varying over the non-H2O2 stresses, may be occurring in the transcriptional data and is correlated with a subset of the proteins expressed under H2O2 stress.
The effect of using gene ontology categories alone for predicting protein expression as seen in Figure is interesting. First, using the highest accuracy obtained using microarray data alone is better than the highest accuracy obtained using any of the feature selection or construction methods on GO categories alone. Second, none of the feature selections or construction methods are clearly superior to any other. Although CFS with BP has the highest accuracy, Conduce on BP has next highest accuracy, and has the least variability over all three subontologies.
Third, it is noticeable that accuracy for each of the subontologies with all four different methods follows the ordering BP, MF, and CC. Interestingly, this is the same ordering as the number of terms in each—in fact, BP had around twice the number of terms as MF (approximately 16,000 vs. 8000) at the time of the experiments (with approximately 2000 terms for CC).
One interpretation of this is that GO categories are predictive of a reasonably large subset of the genes in the training set, as long as the ontology has sufficient “resolution”—in other words, that there is sufficient detail and coverage within the ontology to provide accurate definitions of the target class. Based on the number of terms alone, the BP would a priori be the most likely to have the highest resolution.
For BP and MF, there is relatively little difference between the accuracy obtained by using all GO terms in the training set and the complicated methods of feature subset selection and construction, CFS, and Conduce. Using the simpler feature ranking by information gain actually reduced accuracy relative to using all the features, presumably since this leaves out some useful features. However, on CC it is interesting that the best accuracy was obtained with Conduce. This suggests that there may be some advantage in feature construction when the ontology may have lower resolution, since the “formal concepts plus decision tree” representation is the most powerful of those used in our approach.
Case Study 2: Predicting General vs. Specific Stress Response
There are two principal messages from the results. First, as expected, microarray data alone or in combination with GO features makes no significant contribution to being able to predict which response class a gene is in. Second, GO features do enable a significant improvement in accuracy over predicting the default class, although only when some method of feature subset selection is used. Taken together with the results for predicting protein expression, this supports the central hypothesis of the article.
Although further work is needed to confirm the results when using a ranking based on the hypergeometric test, it is instructive to examine the tree learned from the biological process ontology. There are 53 GO terms with a P-value below 0.01 for the general responder class, and only 8 for the specific responders. Only one of these terms is in common between the two subsets. This suggests that using these GO terms as features will lead to good predictive accuracy and that is the case.
Examining the tree, the top 6 out of 19 leaves each contain ≥10 genes. The corresponding GO terms are:
-
transcription [66]
-
response to DNA damage stimulus [40]
-
protein transport [38]
-
regulation of translation [31]
-
endocytosis [13]
-
ion transport [10]
It might seem that the importance of the “transcription” category contradicts the finding that microarray data is not predictive for the response class problem. However, initial inspection suggests that this category denotes genes involved in the control of transcription, rather than being themselves changed in expression. An example of this would be the transcription factor, Yap1p, (the protein corresponding to the gene YAP1). Yap1p is the best characterized component of cellular response to reactive oxygen species (Temple, Perrone, and Dawes, Citation2005).
Related Work
Graph-Based Similarity and Overrepresentation Analysis
The problem of bias in overrepresentation analysis of GO annotation is still under active research. For example, several proposals for dealing with this bias in estimates by taking into consideration the graph structure and resulting dependencies in the ontology have been recently proposed (Alexa, Rahnenfuhrer, and Lengauer, Citation2006; Grossmann, Bauer, Robinson, and Vingron, Citation2007). However, the more general problem of dealing with dependencies in the complex data types, such as interaction networks in an integrative setting, remains.
In the context of machine-learning, an alternative to the preprocessing approach we have described in this article is to build ontology-handling directly into the machine-learning algorithm. This is a long-standing idea in machine-learning; a recent approach was implemented by Zhang, Caragea, and Honavar (Citation2005). They incorporated “attribute-value trees” into a decision tree learning system. However, for ontologies of the size and complexity of the gene ontology it is not clear how well this approach will scale. Additionally, building in ontology-handling requires modifying each machine-learning algorithm one wishes to use, whereas preprocessing the training data into a standard format can allow the use of any standard algorithm.
A potential problem with our approach lies in the use of formal concept analysis as the basis for our feature construction approach. Since each concept in the lattice has a set of descriptors that is “closed” with respect to the objects in its extent, the features that can be constructed are, in a sense, maximally specific. However, this is a form of inductive bias (Mitchell, Citation1997) that may not be appropriate. In particular, it is not clear that this is an appropriate bias for the often noisy data that gene sets constitute. Although, since more general concepts may also be included as features, this may not be a critical problem, it should be investigated as part of future work. There are also known issues with the scalability of formal concept analysis, and this will also need to be investigated.
Feature-Extraction for Machine-Learning Using Closed Concepts
In general, the reason GO annotation causes problems for vector-based machine-learning is that it is a graph-based data representation. For example, the coverage matrix approach could be used directly as a set of attribute vectors for such algorithms. This would lead, though, to very high-dimensional datasets (there are approaching 30,000 terms in the current gene ontology, leading to a coverage matrix with up to 30,000 columns). However, it is possible that kernel methods could be used, since they are often appropriate for high-dimensional data. We have chosen not to take this approach at this stage since we seek comprehensible models rather than “black-boxes” for use in the Yeastinformatics tools. Furthermore, with the addition of multiple graph-based data sources, such as protein-protein interactions, the data dimensionality could quickly rise to the order of 106 or even higher. Nonetheless, this could be investigated as part of further work.
A general property of graph-based data representations for machine-learning is their sparseness when converted to vector format. However, other data sources in our Yeastinformatics database have this property, such as annotation of genes by the biological pathways in which they are involved, such as pathways in the KEGG database (Kanehisa and Goto, Citation2002). A preliminary experiment, using the feature construction method with KEGG pathway annotation replacing GO annotation, resulted in successful incorporation of pathway features in the learned decision tree. We plan to investigate this further.
Description Logics
Our approach may be seen as deduction followed by induction: first, the transitive closure of the GO refines relation is computed relative to the set of genes in the training set; then a machine-learning algorithm searches for a hypothesis with good estimated predictive accuracy. In principle, the same approach should be possible using a more powerful ontology language. In this setting, however, it is likely to be necessary to control more strictly both the deductive and inductive phases of the process, via devices such as language restrictions, search constraints, and heuristics. Restrictions to subsets of first-order logic are well-studied in description logics (Baader et al., Citation2003), and these are widely proposed as a basis for representation of and reasoning in ontologies.
Wroe, Stevens, Goble, and Ashburner (Citation2003) was an early proposal to move the gene ontology to a description logic framework. This was motivated by the need for semantic analysis of the gene ontology by the use of description logic to enable validation, extension, and classification tasks. Currently, multiple formats of GO are available for download, including OWL, MySQL, and Prolog; we are using the latter two in our work.
Computing the coverage matrix can be done by bottoms-up breadth-first traversal of the gene ontology from the gene associations. Then constructing a concept lattice by treating the coverage matrix as a formal context amounts to finding the minimal common graph paths for gene subsets. This appears to be related to the classification problem in description logics, and we plan to investigate possible connections as part of further work. Since GO is available in OWL format it makes sense to pursue this approach. Although the simple formalism of GO itself probably does not make this worthwhile; the possibility of applying this approach to ontologies in richer representations is one of our research goals.
The Gene Ontology Next Generation Project (GONG) is developing a staged methodology to evolve the current representation of the gene ontology into DAML+ OIL in order to take advantage of the richer formal expressiveness and the reasoning capabilities of the underlying description logic. Each stage provides a step level increase in formal explicit semantic content with a view to supporting validation, extension, and multiple classification of the gene ontology.
However, it is not clear that description logics are always the best choice for ontology construction tasks; Stevens et al. (Citation2007) found that using OWL for modelling complex biological knowledge was only partially successful due to limitations of the formalism.
Bioinformatics Approaches
Antonov and Mewes (Citation2006) have proposed a method of finding combinations of ontology terms that is related to our method of feature construction and selection. Their method searches for algebraic combinations of basic categories, such as GO terms. Since they use set intersection, union, and difference their representation is quite powerful. The exact relation to our approach is not clear, but given the general nature of the hypothesis representation language of decision trees it should be possible to accomplish the same or more powerful combinations in our approach using feature construction.
Guo et al. (Citation2005) integrate gene expression profiles from microarray data with GO categories by reducing the vector of measurements to a point score. They can then learn a decision tree to classify samples based on combinations of the profile-based GO-derived attributes. This is clearly related to our approach, except their aim is to classify samples (e.g., microarrays taken from patients), whereas we are aiming to classify genes.
CONCLUSIONS
Overrepresentation analysis applied to gene ontology annotation of gene sets obtained from high-throughput experiments was reviewed and the problem with bias resulting from dependencies due to the structure of the ontology was described.
We proposed an alternative approach that avoids the need for development of a probabilistic model by which significance can be assessed. By adopting a discriminant or supervised learning methodology, we enable both the integration of heterogeneous data sources in a common “systems biology” framework and the use of gene ontology annotation without directly relying on statistical tests to compute P-values.
Building on previous work we implemented a method for feature construction based on concept lattices to compute the common GO annotations for subsets of genes. Features selected from the concept lattice by a simple discriminative measure were then supplied to a decision tree learning algorithm. We further examined a number of feature selection algorithms. In two case studies, we found that GO annotation can be incorporated into a learned tree to enable prediction significantly above default accuracy. This was found both with and without the integration of microarray data.
As part of future work it is planned to extend the use of the approach to other datasets to enable a more detailed evaluation, both in application to GO annotation and other nonvector-based attributes for machine-learning.
One of the reasons for using decision tree learning in this work is that they are designed to produce comprehensible classifiers. This is an important advantage over “black-box” methods that are opaque to human inspection—even though they may be highly accurate, biologists will have less confidence in their outputs.
However, decision trees have an inbuilt tendency to return the smallest tree giving good classification performance on the data. When interpreting the output of a decision tree, there may be a question as to which other attributes of the data not appearing in the relevant part of the tree may be associated with an output classification. We intend to investigate this problem as part of further work.
Thanks to Rohan Williams for useful discussions, and to Eneko Fernandes-Brèis and Jane Sivieng for work on the database and website. Thanks also to the anonymous reviewers for their helpful comments. This research was partly supported by a grant from the Faculty of Engineering, University of New South Wales.
Notes
www.geneontology.org.
As of October 2009, www.pubmedcentral.nih.gov.
www.yeastgenome.org, accessed February 2009.
This treatment follows that of Boyle et al. (Citation2004), although we have corrected an error in the formula they present.
Over 26,000 as of February 2009.
Given a nonempty ordered set P, if for all S ⊂ P there exists a least upper bound and a greatest lower bound then P is a complete lattice.
REFERENCES
- Akand , E. , M. Bain , and M. Temple . 2007 . Learning from ontological annotation: an application of formal concept analysis to feature construction in the gene ontology . In: Proc. Third Australasian Ontology Workshop (AOW-2007) , eds. T. Meyer and A. Nayak , Vol. 85 , 15 – 23 . Conferences in Research and Practice in Information Technology, Australian Computer Society, Inc.
- Alexa , A. , J. Rahnenfuhrer , and T. Lengauer . 2006 . Improved scoring of functional groups from gene expression data by decorrelating GO graph structure . Bioinformatics 22 ( 13 ): 1600 .
- Alpaydin , E. 2004 . Introduction to Machine Learning. Cambridge , MA : MIT Press .
- Antonov , A. , and H. Mewes . 2006 . Complex functionality of gene groups identified from from high-throughput data . Journal of Molecular Biology 363 : 289 – 296 .
- Ashburner , M. , and The Gene Ontology Consortium . 2000 . Gene ontology: tool for the unification of biology . Nature Genetics 25 ( 1 ): 25 – 29 .
- Baader , F. , D. Calvanese , D. McGuinness , D. Nardi , and P. Patel-Schneider . 2003 . The Description Logic Handbook: Theory, Implementation, and Applications. Cambridge , UK : Cambridge University Press .
- Bada , M. , R. Stevens , C. Goble , Y. Gil , M. Ashburner , J. A. Blake , J. M. Cherry , M. Harris , and S. Lewis . 2004 . A short study on the success of the gene ontology . Web Semantics: Science, Services and Agents on the World Wide Web 1 ( 2 ): 235 – 240 .
- Bain , M. 2002 . Structured features from concept lattices for unsupervised learning and classification . In: AI 2002: Proc. of the 15th Australian Joint Conference on Artificial Intelligence , eds. B. McKay and J. Slaney , 557 – 568 . LNAI 2557 , Berlin : Springer .
- Baldi , P. , and W. Hatfield . 2002 . DNA Microarrays and Gene Expression. Cambridge , UK : Cambridge University Press .
- Bard , J. , and S. Rhee . 2004 . Ontologies in biology: design, applications and future challenges . Nature Reviews Genetics 5 : 213 – 222 .
- Bodenreider , O. , and R. Stevens 2006 . Bio-ontologies: current trends and future directions . Briefings in Bioinformatics 7 ( 3 ): 256 – 274 .
- Boyle , E. , S. Weng , J. Gollub , H. Jin , D. Botstein , J. M. Cherry , and G. Sherlock . 2004 . GO::TermFinder – open source software for accessing gene ontology information and finding signficantly enriched Gene Ontology terms associated with a list of genes . Bioinformatics 20 ( 18 ): 3710 – 3715 .
- Carey , V. J. 2004 . Ontology concepts and tools for statistical genomics . Journal of Multivariate Analysis 90 : 213 – 228 .
- Carpineto , C. , and G. Romano . 1993 . GALOIS: An order-theoretic approach to conceptual clustering . In: Proc. 10th Intl. Conf. on Machine Learning. Los Altos , CA : Morgan Kaufmann, pp . 33 – 40 .
- Causton , H. , B. Ren , S. Koh , C. T. Harbison , E. Kanin , E. G. Jennings , T. I. Lee , H. L. True , E. S. Lander , and R. A. Young . 2001. Remodeling of yeast genome expression in response to environmental changes. Molecular Biology of the Cell 12:323–337.
- Chaitin , G. 1987 . Information, Randomness and Incompleteness – Papers on Algorithmic Information Theory. Singapore : World Scientific Press .
- Ganter , B. , and R. Wille . 1999 . Formal Concept Analysis: Mathematical Foundations. Berlin : Springer .
- Godin , R. , and R. Missaoui . 1994 . An incremental concept formation approach for learning from databases . Theoretical Computer Science 133 : 387 – 419 .
- Godon , C. , G. Lagniel , J. Lee , J. M. Buhler , S. Kieffer , M. Perrot , H. Boucherie , M. B. Toledano , and J. Labarre . 1998 . The H202 Stimulon in Saccharomyces cerevisiae . Journal of Biological Chemistry 273 ( 34 ): 22480 – 22489 .
- Grossmann , S. , S. Bauer , P. Robinson , and M. Vingron . 2007 . Improved detection of overrepresentation of gene ontology annotations with parent–child analysis . Bioinformatics 23 ( 22 ): 3024 .
- Guo , Z. , T. Zhang , X. Li , Q. Wang , J. Xu , H. Yu , J. Zhu , H. Wang , C. Wang , E. J. Topol , and S. Rao . 2005 . Towards precise classification of cancers based on robust gene functional expression profiles . BMC Bioinformatics 6 ( 1 ): 58 .
- Guyon , I. , and A. Elisseeff . 2003 . An introduction to variable and feature selection . Journal of Machine Learning Research 3 : 1157 – 1182 .
- Hall , M. 2000 . Correlation-based feature selection for discrete and numeric class machine learning . In: Proc. Seventeenth Intl. Conference of Machine Learning. San Mateo , CA : Morgan Kaufmann , pp. 359 – 366 .
- Hall , M. , and G. Holmes . 2003 . Benchmarking attribute selection techniques for discrete class data mining . IEEE Transactions on Knowledge and Data Engineering 15 ( 6 ): 1437 – 1447 .
- Kanehisa , M. 2000 . Post-Genome Informatics. Oxford : Oxford University Press .
- Kanehisa , M. , and S. Goto . 2002 . KEGG for computational genomics . In: Current Topics in Computational Molecular Biology , eds. T. Jiang , Y. Xu , and M. Zhang , 301 – 315 . Boston : MIT Press .
- Khatri , P. , and S. Drăghici . 2005 . Ontological analysis of gene expression data: current tools, limitations, and open problems . Bioinformatics 21 ( 18 ): 3587 – 3595 .
- Mitchell , T. 1997 . Machine Learning. New York : McGraw-Hill .
- Quinlan , J. R. 1993 . C4.5: Programs for Machine Learning. San Mateo , CA : Morgan Kaufmann .
- Stevens , R. , M. Aranguren , K. Wolstencroft , U. Sattler , N. Drummond , M. Horridge , and A. Rector . 2007 . Using OWL to model biological knowledge . International Journal of Human-Computer Studies 65 : 583 – 594 .
- Temple , M. , G. Perrone , and I. Dawes . 2005 . Complex cellular responses to reactive oxygen species . Trends in Cell Biology 15 ( 6 ): 319 – 326 .
- Thorpe , G. , C. Fong , N. Alic , V. Higgins , and I. Dawes . 2004 . Cells have distinct mechanisms to maintain protection against different reactive oxygen species: oxidative-stress-response genes . Proc. Natl. Acad. Sci. USA 101 ( 17 ): 6564 – 6569 .
- Tucker , C. , and S. Fields . 2004 . Quantitative genome-wide analysis of yeast deletion strain sensitivities to oxidative and chemical stress . Comparative and Functional Genomics 5 : 216 – 224 .
- Winzeler , E. , D. Shoemaker , A. Astromoff , H. Liang , K. Anderson , B. Andre , R. Bangham , R. Benito , J. D. Boeke , et al. . 1999 . Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis . Science 285 : 901 – 906 .
- Witten , I. , and E. Frank . 2005 . Data Mining, () , 2nd ed. . San Francisco , CA : Morgan Kaufmann .
- Wroe , C. , R. Stevens , C. Goble , and M. Ashburner , 2003 . A methodology to migrate the gene ontology to a description logic environment using DAML +OIL. In: Proc. of the Pacific Symposium on Biocomputing , pp. 624 – 635 .
- Zhang , J. , D. Caragea , and V. Honavar . 2005 . Learning ontology-aware classifiers . In: DS-2005: Proc. Discovery Science Conference , pp. 308 – 321 .