Abstract
In a previous study we found that an episode of acute subclinical nephrotoxicity with gentamicin (G) (but not that induced by another proximal tubular cell nephrotoxin: ferric nitrilotriacetate, FeNTA), paradoxically reduced the progression of renal function and injury in uninephrectomized rats with nephrotic glomerular disease due to Adriamycin nephropathy (AN). Here, we hypothesized that subclinical exposure to G reduces early renal cortical tubulointerstitial inflammation and NF-κB activation in AN. To test this hypothesis, male Wistar rats with established AN received either G (10, 40, or 80 mg/kg by daily s.c.i. for 3 days), FeNTA (1.25, 5, or 10 mg/kg by a single i.p.i.), or vehicle (n = 8 per group), 13 to 15 days after disease induction. Although G and FeNTA caused acute tubular necrosis in a dose-dependant manner (day 17), only the highest doses (10 mg/kg and 80 mg/kg) produced an acute elevation in the serum creatinine. On day 33, chronic tubulointerstitial inflammation (tubular atrophy, interstitial ED-1 + /CD8 + cell accumulation) and NF-κB activation were exacerbated only in the groups that caused functional nephrotoxicity. These data suggest that: 1) the protective effect of subclinical G nephrotoxicity in chronic AN does not involve early changes in interstitial inflammation or NF-κB activation; and 2) a single episode of G exposure must be accompanied by clinically apparent nephrotoxicity in order to accelerate progression in a nonuremic model of chronic glomerular disease.
Introduction
Nephrotoxins such as aminoglycosides, radiocontrast, amphotericin, and anaesthetic compounds cause acute renal failure, in part, by direct injury to proximal tubular epithelial cells (PTECs).Citation[1] It widely accepted that exposure to nephrotoxins during the course of chronic glomerular disease (CGD) could hasten progression to end-stage kidney failure by exacerbating tubulointerstitial injury.Citation[2&3] Paradoxically, in a previous longterm study, we found that an episode of subclinical nephrotoxicity (defined as pathological renal injury without a rise in the serum creatinine) induced by gentamicin [but not ferric nitrilotriacete (FeNTA); another proximal tubular cell nephrotoxin] reduced the decline in renal function, glomerulosclerosis, and tubulointerstitial injury in a rat model of CGD (Adriamycin nephropathy, AN).Citation[4] The mechanisms involved in this novel type of acquired resistance were not known.
Nuclear factor (NF)-κβ is a highly conserved family of transcription factors that has a critical role in mediating inflammation, apoptosis, and growth in chronic disease.Citation[5] In early AN, the renal cortical activation of NF-κB is increased and correlated with the severity of tubulointerstitial injury.Citation[6] Moreover, suppression of NF-κB activation in established AN, with the antioxidant, pyrrolidine dithiocarbamate (PDTC), attenuated tubulointerstitial inflammation.Citation[6] These data suggest that NF-κB has an important role in mediating disease progression in this model.
In other experimental systems (such as the repeated exposure of monocytes to lipopolysaccharide) attenuation of NF-κB has been postulated to be one of the mechanisms involved in mediating acquired resistance to further tissue injury.Citation[7] These observations led us to hypothesize that an episode of subclinical gentamicin nephrotoxicity reduces renal cortical NF-κB activation in AN. To investigate this hypothesis, the dose-dependant effects of a single episode of gentamicin- and FeNTA-induced nephrotoxicity on renal cortical tubulointerstitial injury and NF-κB activation in early AN were examined in this study.
Materials and Methods
Male Wistar rats (6–8 weeks old) (n = 56) were supplied by the Animal Care Facility, Westmead Hospital and allowed free access to food and water. The AN was induced on day 0, by a single intravenous injection of doxorubicin hydrochloride (7.5 mg/kg).Citation[6] On day 10, baseline 24-hour urinary protein and creatinine were measured. On day 13, rats were stratified into seven groups (n = 8 per group) according to body weight, baseline proteinuria, and endogenous creatinine clearance (CrCl). The groups received: 1) vehicle (saline, day 14 to day 16, by s.c. or i.p.i.); 2) gentamicin (G) (10, 40, or 80 mg/kg, day 14 to day 16, by s.c.i.) or FeNTA (1.25, 5, or 10 mg/kg, by a single i.p.i. on day 16). Sterile FeNTA was prepared as previously describedCitation[4] and gentamicin was obtained from David Bull Laboratories (Melbourne, Australia). The doses of nephrotoxins were determined from preliminary experiments and a previous study,Citation[4] and designed to induce mild, moderate, or severe acute tubular necrosis in AN. Higher doses of the nephrotoxins (> 80 mg/kg gentamicin or > 10 mg/kg FeNTA) caused diffuse tubular necrosis and early mortality due to severe acute renal failure in AN, whereas doses less than 1.25 mg/kg FeNTA or 10 mg/kg gentamicin caused no tubular necrosis.
Animals were pair fed, and body weight and food intake were measured daily. Proteinuria and CrCl were determined on days 17, 21, and 33. Groups of animals were sacrificed either on day 17 (n = 3 per group) or day 33 (n = 5 per group) for analysis of acute and chronic effects on tubulointerstitial injury, respectively. On the day of sacrifice, animals were anesthetized with ketamine:xylazine, a mid-line laparotomy was performed and both kidneys were removed, as previously described. The Animal Ethics Committee, University of Sydney at Westmead Hospital, approved all experimental protocols.
Formalin-fixed paraffin sections (3 µm), were stained with periodic-acid Schiff (PAS). Cortical tubulointerstitial injury was assessed by both a semiquantitative scoring system and quantitative morphometric analysis. For semiquantitative analysis, 20 random cortical fields were viewed at × 200 magnification and graded according to the percentage area occupied by tubular injury (tubular atrophy, dilatation) and interstitial inflammation (0 = normal; 1 = less than 25%; 2 = 26%–50%; 3 = 51%–75%; 4 = 76%–100%). For quantitative morphometry, 10 random cortical fields were viewed on a computer screen using a video camera ( × 400). The cross-sectional cell height of cortical tubules and interstitial volume, was measured by line and area morphometric measurements respectively, using image analysis software (Optimas version 5.2, Optimas Corporation, Seattle, WA).Citation[6] The number of interstitial monocyte (ED-1) and cytotoxic T-lymphocytes (OX-8) were assessed by immunohistochemistry on acetone-fixed frozen sections. Nonspecific staining was blocked with normal rabbit serum (1:5) followed by either mouse anti-rat ED-1 (1:400, Serotec, Oxford, England) or mouse anti-rat OX8 (1:100, Serotec, Oxford, England) for 1 h at room temperature. Anti-mouse rabbit antibody (1:50 in 1% rat serum, Dako Australia, Sydney, Australia) and mouse peroxidase-antiperoxidase complex (PAP, Z0259, 1:100; Dako Australia, Sydney, Australia) were applied sequentially for 25 minutes each at room temperature. The mean numbers of 3,3-diaminobenzidine tetrahydrochloride positive ED-1 or OX-8 positive cells were determined from 10 nonoverlapping cortical fields (× 400, measuring 0.075 mm2 each).
Extraction of nuclear protein and electrophoretic mobility shift assay for NF-κB and activator protein (AP)-1 were performed as previously described.Citation[6], Citation[8] Competition and supershift assay were performed to confirm the specificity of the retarded bands.
All data were expressed as mean ± SEM. Comparisons between experimental groups were performed using the independent t-test and Mann-Whitney U test for parametric and nonparametric data respectively. A P value less than 0.05 indicated a significant difference between groups.
Results
No animals died during the study. Groups administered the highest doses of FeNTA (10 mg/kg) and G (80 mg/kg), gained less body weight than vehicle (Day 33: AN + vehicle: 295 ± 18; AN + G: 244 ± 13; AN + FeNTA: 272 ± 15 g; P = 0.05). Body weight in the other groups was similar to vehicle-treated animals. Serum creatinine was elevated only in groups that received the highest doses of FeNTA and G (). The recovery of the serum creatinine occurred earlier in the FeNTA group (day 22), whereas it was only partial by day 33 in the G group (). The serum creatinine (or CrCl, data not shown), did not change significantly in the other groups receiving nephrotoxins (compared to vehicle) (), even though acute tubular necrosis did occur at these lower doses in AN (see the following information). There were no differences in urinary protein excretion in the experimental groups at any timepoint (urinary protein:creatinine ratio on day 33: AN + vehicle: 72.2 ± 6.5; AN + G: 76.4 ± 8.1; AN + FeNTA: 77.8 ± 5.1 mg/µmol; P = not significant).
Table 1. Serum creatinine (µmol/L) in the experimental groups
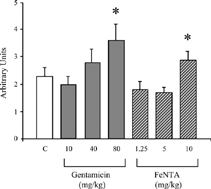
Histological examination on day 17 confirmed that animals treated with either nephrotoxin developed dose-dependant degrees of acute tubular necrosis (). On day 33 the histological changes of AN consisted of cortical tubular atrophy, interstitial cell accumulation, and volume expansion (). By semiquantitative analysis, cortical tubulointerstitial injury was exacerbated only in the groups that received the highest doses of either FeNTA or G ( and ). By quantitative morphometric analysis, interstitial volume and cortical tubular atrophy (as assessed by a reduction in cross-sectional tubule cell height) were increased in these groups, compared to vehicle (). Similarly, interstitial ED-1 and CD8 cell accumulation in AN was worsened by high-dose FeNTA and G ().
Table 2. Quantitative morphometric analysis of the tubulointerstitial inflammation in the experimental groups on day 33
Figure 1 The acute effect of gentamicin (G, 10–80 mg/kg) and ferric nitrilotriacetate (Fe, 1.25–10 mg/kg) on cortical tubulointerstitial injury in Adriamycin nephropathy (AN) on day 17. Areas of focal acute tubular necrosis are present, in a dose-dependant manner, in the groups treated with nephrotoxins (Magnification, × 400, PAS stain).
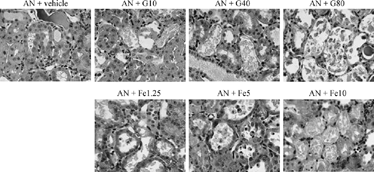
Figure 2 The subacute effect of gentamicin (G) and ferric nitrilotriacetate (Fe) on tubulointerstitial inflammation in Adriamycin nephropathy (AN) on day 33. Chronic tubulointerstitial inflammation in AN is exacerbated by high-dose G and Fe treatment (PAS stain).
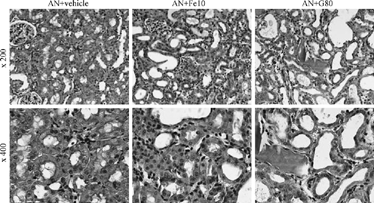
Figure 3 Semiquantitative cortical tubulointerstitial injury score in vehicle (C)-, gentamicin- and ferric nitrilotriacetate (FeNTA)-treated rats with Adriamycin nephropathy on day 33. *P < 0.05 vs. C.
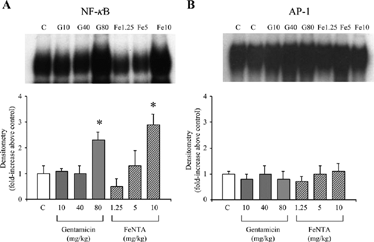
The DNA-binding activity of both AP-1 and NF-κB were increased in renal cortical nuclear extracts from rats with AN. On day 33, NF-κB DNA-binding activity (but not AP-1) was exacerbated only in the groups that received the highest doses of either FeNTA or G, compared to vehicle ().
Figure 4 The DNA-binding activity of nuclear factor (NF)-κB (A) and activator protein (AP)-1 (B) in renal cortical nuclear extracts from vehicle (C)-, gentamicin-, and ferric nitrilotriacetate (FeNTA)-treated rats with Adriamycin nephropathy on day 33 of the study. Top and lower panels show representative autoradiographs and mean densitometry (n = 5 per group) respectively. *P < 0.05 vs. C.
Discussion
The results of this study demonstrate that the acute administration of nephrotoxic doses of two site-specific proximal tubular cell nephrotoxins (gentamicin and FeNTA) to rats with established AN exacerbated underlying tubulointerstitial inflammation and renal cortical NF-κB activation. In contrast, lower doses, that caused mild acute tubular necrosis and subclinical nephrotoxicity, were not sufficient to alter early tubulointerstitial inflammation or renal cortical NF-κB activation.
Previously, in a uninephrectomized model of Adriamycin nephropathy, we reported that an episode of subclinical nephrotoxicity induced by gentamicin (but not FeNTA) at week 6, completely prevented the decline in creatinine clearance and partially reduced glomerulosclerosis and tubulointerstitial fibrosis at week 14.Citation[4] The present study was performed to investigate the potential role of NF-κB in mediating the protective effect of gentamicin in AN. However, we were unable to demonstrate that an early downregulation in NF-κB activation (as described in some models of acquired resistance)Citation[7] and tubulointerstitial inflammation were mechanisms to explain the protective effect of subclinical gentamicin nephrotoxicity on the longterm progression of chronic AN. Other possibilities that could be involved include the upregulation of antioxidant proteins (such as heme-oxygenase-1)Citation[9] and/or specific effects of gentamicin on the lysosomal processing of filtered plasma proteins.Citation[4]
In previous studies, inhibition of renal cortical NF-κB activation with PDTC reduced cortical tubulointerstitial injury in AN.Citation[6] In the present study, exacerbation of interstitial inflammation and tubular atrophy with nephrotoxins was associated with augmentation of renal cortical NF-κB (but not AP-1). These correlative data provide further evidence that NF-κB activation is involved in the pathogenesis of chronic tubulointerstitial injury in progressive renal diseases.
Avoiding exposure to nephrotoxins is one of the main therapeutic cornerstones in the clinical management of preventing disease progression in chronic kidney disease. Our experimental data suggests that an episode of subclinical nephrotoxicity, at least following a short exposure to gentamicin, does not have any adverse long-term effects in a nonuremic nephrotic model of glomerular disease. However, the clinical scenario in humans is more complex than that that can be replicated in the laboratory. In the clinical setting, patients have impaired renal function and are exposed to multiple nephrotoxic insults, sometimes on repeated occasions. This may be an explanation for the divergence of our experimental data with retrospective clinical studies. For example, Aubia and colleagues showed that subclinical nephrotoxin exposure (including aminoglycosides and possibly anesthesia) accelerated the decline in renal function by more than 2-fold in humans with impaired renal function due to diabetic nephropathy.Citation[10] Similarly, aminoglycoside exposure accelerated the decline in loss of residual renal function in peritoneal dialysis patients with end-stage renal failure.Citation[11&12]
In summary, the cytoprotective effect of subclinical gentamicin nephrotoxicity in AN is not due to an early downregulation in renal cortical NF-κB activation, and involves other mechanisms that require further investigation. In addition, the acute exposure to clinically nephrotoxic doses of gentamicin and FeNTA worsened renal cortical tubulointerstitial injury in AN through mechanisms that may involve the upregulation of NF-κB.
Acknowledgments
This study was supported by the Australian Kidney Foundation (GKR) and project grant 970721 from the National Health Medical Research Council of Australia (DCH).
References
- Bennett, W M. Drug nephrotoxicity: an overview.Ren. Fail. 1997, 19 (2), 221–224. [PUBMED], [INFOTRIEVE], [CSA]
- Caring for Australians with Renal Impairment (Draft guidelines); Prevention of Progression of Renal Disease (avoiding superimposed renal insults). Available at: http://www.kidney.org.au/cari/drafts/new/prevention_avoidinsults.html. Accessed October 18, 2004.
- Kidney Disease Outcomes Quality Initiative (K/DOQI). K/DOQI clinical practice guidelines on hypertension and antihypertensive agents in chronic kidney disease.Am. J. Kidney Dis. 2004, 43 (5 Suppl 1), S1–S290. [CSA]
- Rangan, G K.; Wang, Y.; Tay, Y.-C.; Chen, L.; Harris, D C.H. Mild gentamicin nephrotoxicity reduces the progression of chronic adriamycin nephrosis.Nephrology 1998, 4, 57–64. [CSA], [CROSSREF]
- Wardle, E N. Nuclear factor-κB for the nephrologist.Nephrol. Dial. Transplant. 2001, 16, 1764–1768. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Rangan, G K.; Wang, Y.; Tay, Y C.; Harris, D C.H. Inhibition of NF-κB activation reduces cortical tubulointerstitial injury in proteinuric rats.Kidney Int. 1999, 56, 118–134. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Ziegler-Heitbrock, H W. Molecular mechanism in tolerance to lipopolysaccharide.J. Inflam. 1995, 45, 13–26. [CSA]
- Rangan, G K.; Wang, Y.; Tay, Y C.; Harris, D C.H. Cytokine gene expression in adriamycin nephropathy: effects of antioxidant NF-κB inhibitors in established disease.Nephron 2000, 86, 482–490. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Aubia, J.; Hojman, L.; Chine, M.; , et al. Hypertension and nephrotoxicity in the rate of decline in kidney function in diabetic nephropathy.Clin. Nephrol. 1987, 27, 15–20. [PUBMED], [INFOTRIEVE], [CSA]
- Nath, K A.; Vercellotti, G M.; Grande, J P.; , et al. Heme protein-induced chronic renal inflammation: suppressive effect of heme oxygenase-1.Kidney Int. 2001, 59, 106–117. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Shemin, D.; Maaz, D.; St Pierre, D.; Kahn, S I.; Chazan, J A. Effect of aminoglycoside use on residual renal function in peritoneal dialysis patients.Am. J. Kidney Dis. 1999, 43, 14–20. [CSA]
- Singhal, M K.; Bhaskaran, S.; Vidgen, E.; Bargman, J M.; Vas, S I.; Oreopoulos, D G. Rate of decline of residual renal function in patients on continuous peritoneal dialysis and factors affecting it.Perit. Dial. Int. 2000, 20, 429–438. [PUBMED], [INFOTRIEVE], [CSA]