Abstract
Immunoglobulin A (IgA) nephropathy is the most common form of primary glomerulonephritis worldwide, and approximately 20% to 30% of adult patients with the disorder develop chronic renal failure within 20 years. This type of nephropathy is also an important risk factor for chronic renal failure in children. The pathogenesis of IgA nephropathy is still unknown, and treatment remains controversial. Microscopic hematuria and recurrent episodes of macroscopic hematuria are the most common clinical manifestations of this condition in children. This article describes the case of a young girl who presented with steroid-resistant nephrotic syndrome unaccompanied by hematuria. Renal biopsy findings were consistent with IgA nephropathy. The patient's condition was a rare clinical manifestation of IgA nephropathy.
Introduction
Immunoglobulin A nephropathy (IgAN) is the most common form of primary glomerulonephritis. This condition is characterized by prominent and diffuse immunoglobulin A (IgA) deposits in the glomerular mesangium, which are detectable on immunofluorescence microscopy.Citation[1] There are three morphologic forms of IgAN: primary IgAN, secondary IgAN, and Henoch-Schönlein purpura. Primary IgAN, also known as Berger's disease, is an immune complex-mediated glomerulonephritis.Citation[2] Henoch-Schönlein purpura is a vasculitic and systemic variant of primary IgAN. Secondary IgAN can occur with hepatobiliary or autoimmune diseases, bacterial and viral infections, or neoplasia.
IgAN was initially considered a benign disease with a favorable outcome; however, data from long-term follow-up studies have shown that 20% to 30% of adult patients progress to chronic renal failure within 20 years of disease onset. More recent reports have also suggested that this condition may not take a benign course in children.Citation[3-5] Yoshikawa and colleagues reported that 5% of 258 children with IgAN developed chronic renal failure by 5 years after onset of the condition, 6% were in chronic renal failure by 10 years, and 11% were in chronic renal failure by 15 years.Citation[6]
Microscopic hematuria and recurrent episodes of macroscopic hematuria are the most common clinical manifestations of IgAN in children. We report the case of a young girl with a rare clinical presentation of IgAN: steroid-resistant nephrotic syndrome unaccompanied by hematuria.
Case Report
A 5-year-old girl presented to our hospital with periorbital edema and cushingoid appearance. Two months earlier, she had visited another center with complaints of periorbital and pretibial swelling, and that workup had revealed hypoalbuminemia (1.6 g/dL), hypercholesterolemia (300 mg/dL), and proteinuria (300 mg/dL). She had been diagnosed with minimal-change nephrotic syndrome. Six weeks of treatment with prednisolone 60 mg/m2/day had failed to achieve remission, and she was thus referred to our pediatric nephrology unit.
The patient's family history was significant for renal disease. Her 1.5-year-old sister had bilateral vesicoureteral reflux (first degree on the right, third degree on the left), and one of her grandmothers had died of chronic renal failure. Physical examination revealed height and weight in the 3rd and 10th percentiles for age, respectively, and blood pressure 100/60 mmHg. Apart from cushingoid appearance, the exam findings were unremarkable. Laboratory investigations revealed hypoalbuminemia (2.8 g/dL), hypercholesterolemia (299 mg/dL), serum C-reactive protein (1.8 mg/L), and erythrocyte sedimentation rate (17 mm/h). Serum levels of electrolytes, calcium, phosphorus, alkaline phosphatase, blood urea nitrogen, and creatinine were all within normal limits. Microscopic examination of the urine showed no hematuria or casts. The patient's daily urinary protein excretion was 110 mg/m2/h. Serum levels of complement C3 (1.5 g/L), C4 (1.4 g/L), and IgA (1.4 g/L) were all in the normal range. Serologic tests for hepatitis B surface antigen, hepatitis C antibody, antinuclear antibodies, double-stranded DNA antibodies, and antineutrophil cytoplasmic antibodies were all negative. Renal ultrasonography showed nothing abnormal. Because steroid-resistant nephrotic syndrome was suspected, renal biopsy was performed.
Renal Biopsy Findings
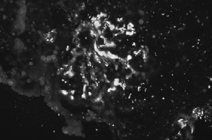
A total of 56 glomeruli were examined under the light microscope. In one glomerulus, a crescent formation occupied one-third of the glomerulus structure. Several other glomeruli showed a minimal lobulation and synechia. All 56 glomeruli exhibited prominent, diffuse mesangial cell proliferation and less prominent increased matrix ( and ) There was no fibrosis in the interstitium, and all vascular structures appeared normal. Staining for amyloid fibrils was negative. Electron microscopy revealed electron-dense deposits in the mesangial matrix and along the subendothelial margin of the glomerular basement membrane. Immunofluorescence microscopy revealed IgA deposits (strong positive) and complement C3 deposits (granular positive) in the mesangium (). There was no staining for C1q, IgM, or IgG. The findings were consistent with IgAN.
Figure 1.
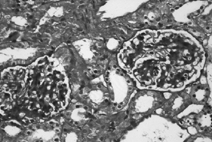
Figure 2.
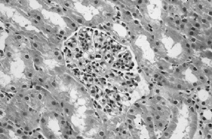
Figure 3.
Clinical Course
As noted, prior to arrival at our center, the patient had been diagnosed with nephrotic syndrome but had shown no response to a 6-week course of prednisolone 60 mg/m2/day. Based on the biopsy findings, the decision was made to combine another immunosuppressive agent with the steroid. Thus, she was started on cyclophosphamide (2 mg/kg/day) and prednisolone (40 mg/m2 on alternate days). She was also prescribed fish oil (1.5 g/day) and captopril (0.3 mg/kg) to address the proteinuria. After 2 months on this regimen, the patient still had proteinuria (46 mg/m2/h), so the previous immunosuppressive protocol was discontinued and she was started on a combination of prednisolone (0.5 mg/kg/day), azathioprine (2 mg/kg/day), and dipyridamole (2 mg/kg/day). By the third month on this regimen, the patient's daily protein excretion had normalized. Azathioprine was discontinued at 6 months. At the time of writing, the patient was still taking fish oil and captopril, and had experienced no relapse. Her renal function has been normal throughout 2 years of follow-up.
Discussion
The etiology and pathogenesis of IgAN are still not fully understood, but substantial evidence suggests this is an immune-mediated disease in which circulating IgA immune complexes are deposited in the glomeruli. Onset of IgAN may be associated with upper respiratory or gastrointestinal infections. More recent findings suggest patients with IgAN have decreased mucosal immunity, a function that is largely driven by IgA.Citation[7] Some affected individuals exhibit increased production of IgA1 in the bone marrow, and this may be responsible for the elevated serum IgA levels.Citation[1], Citation[8&9] However, reports indicate that only 30% to 70% of adult patients with IgAN and 8% to 16% of children with this condition have high levels of circulating IgA immune complexes.Citation[6], Citation[10]
IgAN can occur at all ages, but is most often diagnosed in the second or third decade of life. Males are affected more frequently than females, and the reported male:female ratios range from 2:1 to 6:1.Citation[11] The clinical presentation of IgAN varies, ranging from asymptomatic urine abnormalities to acute renal failure. Five different clinical syndromes have been identified: macroscopic hematuria, asymptomatic microscopic hematuria and/or proteinuria, acute nephritic syndrome characterized by hematuria associated with hypertension and/or renal failure, nephrotic syndrome, and mixed nephritic-nephrotic syndrome.
Several studies of IgAN in children have shown that more than 80% of all patients have microscopic hematuria. Indeed, recurrent macroscopic hematuria has traditionally been considered the hallmark of childhood IgAN.Citation[12] Nephrotic edema is reported to occur in approximately 10% of patients with this condition.Citation[13] The unusual association of IgAN and minimal-change nephrosis has been reported previously,Citation[14-16] and these patients responded to steroid therapy. To our knowledge, no previous report has documented onset of IgAN in childhood that is associated with steroid-resistant nephrotic syndrome but unaccompanied by microscopic or macroscopic hematuria. In our unusual pediatric case, steroid-resistant nephrotic syndrome was a clinical manifestation of IgAN.
Owing to the diversity of clinical presentations of IgAN and the variable rate of progression to renal failure, the optimal therapy for patients with this form of nephropathy remains controversial.Citation[13], Citation[17] The treatment options include prophylactic antibiotics or tonsillectomy to prevent potential entry of microbial antigens, glucocorticoids, and other immunosuppressive drugs and immunomodulators, and plasma exchange to remove circulating immune complexes. A more recent controlled study in adults with IgAN and proteinuria showed that 2 years of treatment with fish oil slowed the rate at which renal function deteriorated.Citation[18] In addition, other more recent studies have indicated that angiotensin-converting enzyme inhibitors can reduce protein excretion and preserve renal function in affected adults.Citation[19]
As mentioned, there is continued controversy about the most appropriate treatment for patients with IgAN. Several studies in children with this condition have shown that degree of proteinuria is correlated with morphologic severity of the glomerular lesions and that severe proteinuria at the time of biopsy predicts poor outcome.Citation[20&21] Thus, children with IgAN who have severe proteinuria are candidates for specific therapeutic interventions. Glucocorticoids are only beneficial for IgAN patients who have nephrotic syndrome, minimal histopathologic changes, and relatively well-preserved renal function.
Saint-Andre and colleaguesCitation[14] were the first to note the unusual association between mesangial IgA deposition and minimal-lesion nephrosis in adults. Subsequently, similar cases were reported in children.Citation[15&16] It is now established that there is a subset of patients with steroid-responsive nephrotic syndrome who have this distinct clinical syndrome (minimal-change disease plus IgA deposits). Patients with this combination of disorders are at low risk for progression to end-stage renal disease, possibly because they respond favorably to steroid treatment. Relapses of nephrosis and steroid dependence have also been described in adults with steroid-responsive IgAN.Citation[22] Our pediatric case was different from steroid-responsive cases in that the patient was resistant to steroids but did not have other factors associated with poor prognosis, such as hypertension, renal failure at the time of disease, tubulointerstitial changes, and prominent crescent formation.
Research on adults with IgAN has shown that combined treatment with prednisolone for 2 years, and cyclophosphamide for 3 months followed by azathioprine, effectively preserves renal function at 3 years.Citation[23] In another study, a regimen of cyclophosphamide, dipyridamole, and warfarin initially reduced proteinuria and stabilized renal function in this patient group; however, a 5-year posttrial assessment revealed no difference in renal function between treatment and control groups.Citation[24] Our pediatric patient did not respond to cyclophosphamide therapy. Andreoli and coworkersCitation[20] studied the efficacy of 1 year of treatment with prednisolone (60 mg/m2/day for 8 weeks followed by 60 mg/m2 on alternate days for the remainder of the year) and azathioprine in 10 children with severe IgAN. They observed a decrease in proteinuria and an improvement in histologic activity score. Yoshikawa and colleagues investigated 78 children with newly diagnosed severe IgAN,Citation[25] and found that treatment with prednisolone, azathioprine, heparin-warfarin, and dipyridamole for 2 years effectively reduced proteinuria and decreased the intensity of mesangial IgA deposits. Our 5-year-old patient responded well to a regimen of azathioprine, prednisolone, and dipyridamole. More recently, tacrolimus and mycophenolate mofetil have been suggested as new treatments for IgAN, and responses to these agents have been favorable.Citation[26-28]
The case described in this article is unusual in that the child had IgAN associated with steroid-resistant nephrotic syndrome. This rare combination of disorders was successfully treated with low-dose steroid, azathioprine, and dipyridamole.
References
- Galla J H. IgA nephropathy. Kidney Int. 1995;47:377-387. [PUBMED], [INFOTRIEVE], [CSA]
- Berger J, Hinglais N. Intercapillary deposits of IgA-IgG. J Urol Nephrol. 1968;74:694-695. [CSA]
- Berg U B. Long term-follow up of renal function in IgA nephropathy. Arch Dis Child. 1991;66:588-592. [PUBMED], [INFOTRIEVE], [CSA]
- Hogg R J, Silva F G, Wyatt R J, Reisch J S, Argyle J C, Savino D. Prognostic indicators in children with IgA nephropathy—report of the Southwest Pediatric Nephrology Study Group. Pediatr Nephrol. 1994;8:15-20. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Wyatt R J, Kritchevsky S B, Woodford S Y, , et al. IgA nephropathy: long-term prognosis for pediatric patients. J Pediatr. 1995;127:913-919. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Yoshikawa N, Ito H, Nakamura H. IgA nephropathy in children from Japan. Clinical and pathological features. Child Nephrol Urol. 1989;9:191-199. [CSA]
- Feehally J, Allen A C. Pathogenesis of IgA nephropathy. Ann Med Interne. 1999;150:91-98. [CSA]
- Harper S J, Pringle J H, Wicks A C, , et al. Expression of J chain mRNA in duodenal IgA plasma cells in IgA nephropathy. Kidney Int. 1994;45:836-844. [PUBMED], [INFOTRIEVE], [CSA]
- Layward L, Allen A C, Hattersley J M, Harper S J, Feehally J. Elevation of IgA in IgA nephropathy is localized in the serum and not saliva and is restricted to the IgA1 subclass. Nephrol Dial Transplant. 1993;8:25-28. [PUBMED], [INFOTRIEVE], [CSA]
- Kher K K, Makker S P, Moorthy B. IgA nephropathy (Berger's disease). A clinicopathologic study in children. Int J Pediatr Nephrol. 1983;4:11-18. [PUBMED], [INFOTRIEVE], [CSA]
- D'Amico G. The commonest glomerulonephritis in the world: IgA nephropathy. Q J Med. 1987;64:709-727. [PUBMED], [INFOTRIEVE], [CSA]
- Linne T, Aperia A, Broberger O, Bergstrand A, Bohman S O, Rekola S. Course of renal function in IgA glomerulonephritis in children and adolescents. Acta Pediatr Scand. 71:735-743. [CSA]
- Yoshikawa N, Tanaka R, Iijima K. Pathophysiology and treatment of IgA nephropathy in children. Pediatr Nephrol. 2001;16:446-457. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Saint-Andre J P, Simard C L, Spiesser R, Houssin A. Nephrotic syndrome in a child, with minimal glomerular lesions and mesangial IgA deposits. Nouv Presse Med. 1980;9:531-532. [PUBMED], [INFOTRIEVE], [CSA]
- Mustonen J, Pasternek A, Rantala I. The nephritic syndrome in IgA glomerulonephritis: response to corticosteroid therapy. Clin Nephrol. 1983;20:172-176. [PUBMED], [INFOTRIEVE], [CSA]
- Habib R, Girardin E, Gagnadoux M F, Hinglais N, Levy M, Broyer M. Immunopathological findings in idiopathic nephrosis: clinical significance of glomerular immune deposits. Pediatr Nephrol. 1988;2:402-408. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Wyatt R J, Hogg R J. Evidence-based assessment of treatment options for children with IgA nephropathies. Pediatr Nephrol. 2001;16:156-167. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Donadio J J, Jr., Bergstralh E J, Offord K P, Spencer D C, Holley K E. A controlled trial of fish oil in IgA nephropathy. Mayo Nephrology Collaborative Group. N Engl J Med. 1994;331:1194-1199. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Yoshida H, Mitarai T, Kawamura T, , et al. Role of the deletion polymorphism of the angiotensin converting enzyme gene in the progression and therapeutic responsiveness of IgA nephropathy. J Clin Invest. 1995;96:2162-2169. [PUBMED], [INFOTRIEVE], [CSA]
- Andreoli S P, Yum M N, Bergstein J M. IgA nephropathy in children: significance of glomerular basement membrane deposition of IgA. Am J Nephrol. 1986;6:28-33. [PUBMED], [INFOTRIEVE], [CSA]
- Yoshikawa N, Ito H, Nakamura H. Prognostic factors in childhood IgA nephropathy. Nephron. 1992;60:60-67. [PUBMED], [INFOTRIEVE], [CSA]
- Lai K N, Lai F M, Ho C P, Chan K W. Corticosteroid therapy in IgA nephropathy with nephrotic syndrome: a long-term controlled trial. Clin Nephrol. 1986;26:174-180. [PUBMED], [INFOTRIEVE], [CSA]
- Balladie F W, Roberts I S. Controlled prospective trial of prednisolone and cytotoxics in progressive IgA nephropathy. Nephrol Dial Transplant. 1996;11:1684. [CSA]
- Woo K T, Lee G S, Lau Y K, Chiang G S, Lim C H. Effects of triple therapy in IgA nephritis: a follow-up study 5 years later. Clin Nephrol. 1991;36:60-66. [PUBMED], [INFOTRIEVE], [CSA]
- Yoshikawa N, Ito H, Sakai T, , et al. A controlled trial of combined therapy for newly diagnosed severe childhood IgA nephropathy. The Japanese Pediatric IgA Nephropathy Treatment Study Group. J Am Soc Nephrol. 1999;10:23-33. [CSA]
- Loeffer K, Gowrishankar M, Yiu V. Tacrolimus therapy in pediatric patients with treatment-resistant nephrotic syndrome. Pediatr Nephrol. 2004;19(3):281-287. [CSA], [CROSSREF]
- Chen X, Chen P, Cai G, , et al. A randomized control trial of mycophenolate mofetil treatment in severe IgA nephropathy. A randomized control trial of mycophenolate mofetil treatment in severe IgA nephropathy [Abstract]. 2002. [CSA]
- Julian B A. Treatment of IgA nephropathy. Semin Nephrol. 2000;20(3):277-285. [PUBMED], [INFOTRIEVE], [CSA]