Abstract
Strumpell's familial spastic paraplegia is a rare hereditary disease, clinically characterized by progressive disturbance of gait. Focal Segmental Glomerulosclerosis (FSGS) is a frequent glomerulopathy, with an extremely rare familial subtype. The cases of two brothers with Strumpell's disease are reported, who also developed glomerular renal disease, most probably familial FSGS. The genetics of the two disorders, Strumpell's paraplegia and familial FSGS, are discussed, together with the possibility of a parallel transmission.
INTRODUCTION
Familial spastic paraplegia or Strumpell's hereditary spastic paraplegia is a rare neurological disease characterized by progressive spasticity and muscle weakness of mainly the lower extremitiesCitation[1] and pathology findings of degeneration of the lateral corticospinal tracts and, sometimes, the posterior columns of the spinal cord.Citation[2] Strumpell's paraplegia belongs to the family of upper motor neuron diseases and is inherited in three ways: 1) an autosomal dominant, where three genetic loci, situated on chromosomes 14q, 2p, and 15q, are considered responsible;Citation[3] 2) an autosomal recessive, where a gene located on chromosome 8q has been implicated, but its sequence has not been determined in full yet;Citation[4] and 3) a sex-linked recessive inheritance, the rarest form; two genetic loci (xq28 and xq21-q22) have been described.Citation[4] Muscle spasticity of the lower extremities and the resulting impaired gait, hyperreflexia, and familial occurrence of cases are considered to be pathognomonic of this rare neuropathy.Citation[2] Moreover, lower-extremity somatosensory evoked potentials (EPs) are disturbed. Lifespan of affected individuals is unaffected, with the exception of the recessive forms of the disease.
Focal segmental glomerulosclerosis (FSGS) is a major cause of end-stage renal disease (ESRD) and it encompasses idiopathic (sporadic), secondary, and familial FSGS, the latter being extremely rare and of particular interest, because the study of its genetic basis sheds light into the pathophysiology of the whole disorder.Citation[5] Familial FSGS has been attributed to mutations of genes encoding proteins involved in the cytoskeletal structure and function of glomerular podocytes. Its mode of inheritance is of the autosomal recessive or dominant type and histopathologic and clinical features are common with those of the idiopathic form.
Herein we report the case of two brothers suffering from hereditary spastic paraplegia and accompanying glomerulopathy, most probably FSGS. No other case of Strumpell's disease complicated with renal involvement has been, to our knowledge, reported, and, inversely, familial FSGS has never been linked to hereditary spastic paraplegia.
CASE PRESENTATION
Case 1
The older of the two brothers, 40 years of age, had first developed spasticity of the lower extremities at the age of 8 and a gradually worsening gait disturbance. Three years ago he developed ankle edema and hypertension. He was now referred to our Department because of renal insufficiency. His father had suffered from chronic renal failure (CRF) of unknown etiology, had undergone dialysis for years, and died at the age of 58. The patient's mother and sister were of good health, whereas his brother had already been diagnosed with spastic paraplegia and undergone renal transplantation (see case 2).
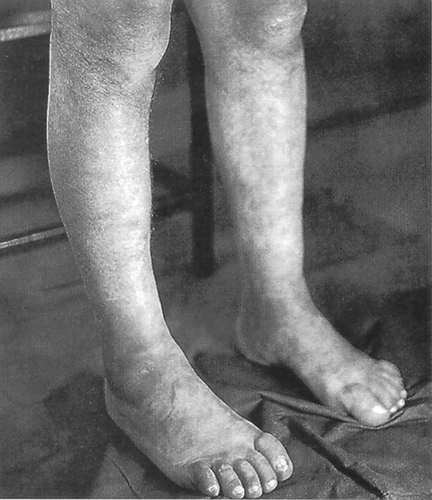
On admission, the patient had a spastic gait, ankle edemas, and arterial hypertension (180/100 mmHg); clinically, pes cavus was evident (). Funduscopy revealed grade II hypertensive retinopathy. Neurological examination revealed: normal cognitive function, sensory pathways, and cranial nerves; spasticity and muscle weakness of the lower extremities; hyperactive tendon reflexes with clonus, a positive jaw reflex, and a positive Babinski sign bilaterally. Visual and upper-extremity somatosensory EPs were normal, whereas lower-extremity potentials were unobtainable. Audiometry was unremarkable. Laboratory results were as follows: Hct 36%; urea 170 mg/dL, serum creatinine 3.5 mg/dL, uric acid 10.5 mg/dL, potassium 4.4 mEq/L, sodium 139 mEq/L, total calcium 8.7 mg/dL, phosphorus 4 mg/dL, total protein 7.1 g/dL, albumin 3.8 g/dL, total cholesterol 230 mg/dL, triglycerides 200 mg/dL; and protein 2 g/24 h-urine collection. Ultrasonography revealed symmetrical kidneys, 10 cm of length, with a smooth border. Histocompatibility antigens were as follows: A11, A30, B13, B51, DR4, DR7.
Figure 1 Typical appearance of lower extremities in the older of the two brothers (case 1) with Strumpell's disease.
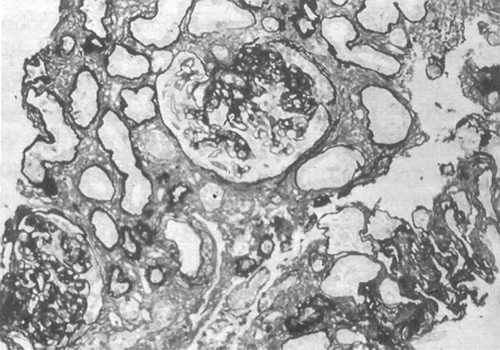
The patient underwent renal biopsy, in which 13 glomeruli were included, 6 of them being totally scarred. The rest demonstrated a marked dilatation and sclerosis of the mesangium, with formation of sclerosing bridges with the Bowman capsule, which, in some glomeruli, showed hyperplasia of the epithelial cells. The glomerular base membrane was edematous, poly-helical, and with no spikes or double border. The urinary tubules revealed at sites ischemic atrophy, while others appeared reactively hyperplastic and acidophilic. Interstitium was sclerosed and infiltrated with lymphocytes and plasma cells, a feature characterizing the tubular epithelium, too, giving the appearance of tubulointerstitial nephritis. The intima of some arterioles showed hyaline deposition (). Immunohistochemical investigation revealed IgM and C3 depositions in the areas of sclerosis and the mesangium, and IgG along the glomerular base membrane. In short, the above-described morphology and immunohistochemistry were consistent with FSGS.
Figure 2 Renal biopsy of the older brother (case 1) revealing lesions compatible with focal segmental glomerulosclerosis (Silver-methenamine stain, magnification ×100).
The patient was treated with antihypertensive medication and corticosteroids (32 mg methylprednisolone daily for 12 weeks, gradually decreased because of lack of clinical improvement). Renal function deteriorated steadily, resulting, 3 months after commencement of therapy, in the patient starting dialysis.
Case 2
The younger of the two brothers, 38 years old, had developed lower extremity spasticity and gait disorder at the age of 12. Eight years ago, he developed edemas of the lids and ankles, hypertension, proteinuria of unknown gravity, and renal failure, which progressed to ESRD, despite antihypertensive medication and proper diet. Five years ago he entered a regular dialysis program, and a year later he received a renal transplant from his mother. The histocompatibility antigens of the recipient were: A11, A30, B13, B51, DR4, DR7; that is, identical to his brother's; and of the donor: A1, A11, B8, B51, DR3, DR4. On admission, the patient had a cushingoid appearance, gait disturbance, and blood pressure of 150/95 mmHg. His immunosuppressive regimen included methylprednisolone 8 mg and cyclosporin A 125 mg daily. He was also receiving atenolol, 100 mg per day, and an angiotensin receptor blocker. Ophthalmologic examination revealed bilateral cataract due to corticosteroid treatment. The abnormal findings of the neurological examination were the following: spasticity, hyperactive tendon reflexes with clonus of the lower, and less so, of the upper extremities, and positive Babinski sign bilaterally; fine finger movements were mildly impaired on the left side; disturbed, spastic gait; and, finally, unobtainable somatosensory EPs from the lower limbs. Laboratory investigation was unremarkable, and renal function, 5 years after transplantation, remained normal: urea 67 mg/dL, serum creatinine 1.2 mg/dL; normal urine analysis and sediment.
DISCUSSION
The diagnosis of Strumpell's disease in both brothers was established on the basis of clinical findings and EPs and through exclusion of other possible disorders (subacute spinal cord degeneration, multiple sclerosis, cervical spondylosis, and intraspinal tumors). Regarding the pattern of inheritance followed in the particular case, it was probably not of the autosomal dominant type, because the mother and, according to the history, the father had no neurological manifestations of any kind. Autosomal recessive inheritance seems more likely, in agreement with which are the clinical findings of the two brothers, in particular pes cavus and hyperactive reflexes of the upper and lower extremities, which especially characterize the disorder in the case of recessive inheritance.Citation[2]
The renal biopsy of the older brother revealed lesions compatible with FSGS, but glomerulosclerosis could have been secondary to other diseases; for example, Alport syndrome.Citation[6] Indeed, the histological findings from the biopsy could have been attributed to this syndrome, if the IgM and C3 depositions weren't so evident. Moreover, no other findings typical of Alport syndrome were confirmed, either ophthalmological or auditory; neither hematuria was found. Therefore, we accept the diagnosis of FSGS regarding the older brother and, taking into consideration the renal transplantation history of the younger brother and the ESRD history of the father, we conclude that the family was affected with familial FSGS, as the Duke criteria for familial FSGS are fulfilled;Citation[5] according to the Duke classification of individual status, all three male members (father and two sons) are considered “affected.” An autosomal dominant mode of inheritance is likely, since the transmission is from father to son and two consecutive generations are affected. Other arguments supporting the familial FSGS hypothesis are the following: 1) a strong association of familial FSGS with histocompatibility antigen DR4 has been found, an association even stronger in Caucasian individuals and patients over 18 years of age;Citation[7] 2) the absence of relapse in the renal transplant of the younger brother 5 years after transplantation is against sporadic FSGS (relapse risk 20% or moreCitation[8]), but consistent with familial FSGS, where an extremely small risk of recurrence is reported: none out of 13 transplant-recipients reported by Rana et al.,Citation[9] none out of 9 by Nyberg et al.,Citation[10] and only one recurrence in 48 transplants in the Duke cohort.Citation[5]
There have been reports of FSGS combined with Charcot-Marie-Tooth, another degenerative disease of the nervous system.Citation[11],Footnote[12] The combination, however, of familial FSGS with Strumpell's disease has, to our knowledge, never been reported before and is extremely challenging from a genetic point of view. Up to now, familial FSGS has been attributed to four specific genes: the NPHS1,Citation[13] NPHS2,Citation[14] ACTN4,Citation[15] and, lately, TRPC6 gene,Citation[16] which encode, respectively, nephrin, podocin, a-actinin 4, and the transient receptor potential cation channel protein 6. These genes are located on genes 19q13, 1q25–31, 11q21–22, and 11q (again, respectively). Based on current knowledge, familial FSGS and Strumpell's disease do not share common genes, making it difficult to hypothesize gene linkage between the two disorders. Research, however, on the genetics of both diseases is not over, and novel genes are discovered (the TRPC6 gene was only recently found), while in some families with FSGS no specific locus can be identified.Citation[9] Still, the possibility exists that two genetic loci, one responsible for each disease and situated in proximity with each other, could have been the cause of this unique clinical combination. Unfortunately, the two affected brothers have been lost-to-follow up, and genetic studies on them and their kindred are impossible at this point.
In conclusion, we reported the cases of two brothers with a rare neurological degenerative disease, Strumpell's familial spastic paraplegia, who also suffered from a glomerulopathy, most probably familial FSGS, the combination of the two being unique in world literature. From the study of these two cases and the current knowledge on the genetics of the two diseases, an etiologic relationship between the two disorders cannot be confirmed, but should be taken into consideration in future research.
Notes
12. Lennert T, Hanefeld F, Bernstein J. Charcot-Marie-Tooth disease and chronic nephropathy. 10th Meeting of the European Society of Pediatric Nephrology; 1976.
REFERENCES
- Holmes G, Shaywitz B. Strumpell's pure familial spastic paraplegia: case study and review of the literature. J. Neurosurg. Psychiatr 1977; 40: 1003–1008, [CSA]
- Rup T. Disorders of upper and lower motor neuron. Neurology in Clinical Practice, W Bradley, R Daroff, D Fenichel, D Marsden. Butterworth-Heinemann, Boston, MA 1991; 1688–1717
- Bruyn RP, Van Veen MM, Kremer H, et al. Familial spastic paraplegia: evidence for a fourth locus. Clin. Neurol. Neurosurg 1997; 99: 87–90, [INFOTRIEVE], [CSA], [CROSSREF]
- Kobayashi H, Garcia C, Alfonso G, et al. Molecular genetics of familial spastic paraplegia: a multitude of responsible genes. J. Neurol. Sci 1996; 137: 131–138, [INFOTRIEVE], [CSA], [CROSSREF]
- Winn MP. Approach to the evaluation of heritable diseases and update on familial focal segmental glomerulosclerosis. Nephrol. Dial. Transplant 2003; 18([Suppl 6])vi14–vi20, [INFOTRIEVE], [CSA]
- Glassock R, Cohen A, Adler S, . Secondary glomerular diseases. The Kidney, B Brenner, F Rector, et al. WB Saunders, London 1991; 1280–1368
- Schnaper W. Focal segmental glomerulosclerosis. Immunologic Renal Diseases, EG Neilson, WG Couser. Lippincott-Raven, Philadelphia–New York 1997; 1003–1026
- Newstead CG. Recurrent disease in renal transplants. Nephrol. Dial. Transplant 2003; 18(Suppl 6)vi68–vi74, [INFOTRIEVE], [CSA]
- Rana K, Isbel N, Buzza M, et al. Clinical, histopathologic, and genetic studies in nine families with focal segmental glomerulosclerosis. Am. J. Kidney Dis 2003; 41(6)1170–1178, [INFOTRIEVE], [CSA], [CROSSREF]
- Nyberg G, Friman S, Svalander C, Norden G. Spectrum of hereditary renal disease in a kidney transplant population. Nephrol. Dial. Transplant 1995; 10: 859–865, [INFOTRIEVE], [CSA]
- Lemieux G, Neemeh JA. Charcot-Marie-Tooth disease and nephritis. Can. Med. Assoc. J 1967; 97: 1193–1198, [INFOTRIEVE], [CSA]
- Mathis B, Kim S, Calabrese K, et al. A locus for inherited focal segmental glomerulosclerosis maps to chromosome 19q13. Kidney Int 1998; 53: 282–286, [INFOTRIEVE], [CSA], [CROSSREF]
- Boute N, Gribouva O, Roselli S, et al. NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nature Genet 2000; 24: 349–354, [INFOTRIEVE], [CSA], [CROSSREF]
- Kaplan JM, Kim SH, North KN, et al. Mutations in ACTN4, encoding alpha-actinin 4, cause familial focal segmental glomerulosclerosis. Nature Genet 2000; 24: 251–256, [INFOTRIEVE], [CSA], [CROSSREF]
- Winn MP, Conlon PJ, Lynn KL, et al. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science 2005; 308(5729)1801–1804, [INFOTRIEVE], [CSA], [CROSSREF]