Abstract
Aims. While previous studies have demonstrated that diabetic nephropathy is attributable to glucose-derived dicarbonyl compounds, methylglyoxal (MGO)-inducing apoptosis in renal mesangial cells, the molecular mechanism of upper stream redox signaling modulation, has not been fully elucidated. Methods: Rat mesangial cells pretreated with or without superoxide dismutase, diphenyloniodium, SB203580, and manumycin A were cultured in methylglyoxal stress-induced apoptosis. Signaling protein expression, flow cytometry, and morphological features of apoptotic cell death were assessed. Results: Methylglyoxal decreased cell viability in mesangial cells. Superoxide mediated methylglyoxal-induced caspase 3 cleavage. Pretreatment with diphenyloniodium, SB203580, and manumycin A reduced methylglyoxal augmentation of superoxide synthesis and caspase-3 activation. Methylglyoxal rapidly enhanced Ras activation and progressively increased cytosolic P38 and nuclear c-Jun activation. Scavenging of superoxide by superoxide dismutase or diphenyloniodium, inhibiting P38 by SB203580, and inhibiting Ras with manumycin A successfully reduced the promoting effect of methylglyoxal on P38 and c-Jun phosphorylation (activation). Furthermore, pretreatment with superoxide dismutase, diphenyloniodium, SB203580, and manumycin A significantly attenuated methylglyoxal induction of apoptosis on the basis of Annexin-V assay and terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate-biotin nick end-labelling (TUNEL) staining. Conclusions. This study has shown that methylglyoxal increased Ras modulation of superoxide-mediated P38 activation and c-Jun activation, which resulted in increased apoptosis.
INTRODUCTION
Glomerular mesangial cells play a control role in the pathogenesis of diabetic nephropathy. A loss of glomerular cellular components such as mesangial cells is characteristic of the advanced stages of glomerulosclerosis.Citation[1] A clinical study in patients with diabetic nephropathy reported a loss of glomerular mesangial cells that correlates with progression to diabetic glomerulosclerosis.Citation[2] High glucose that induces proapoptotic signaling has been found to induce renal mesangial cell apoptosis that results in glomerulosclerosis.Citation[3] Increased mesangial cell depletion coinciding with extracellular matrix accumulation is noted in hyperglycemia-induced segmentation of glomerular tuft, suggesting that mesangial cell apoptosis plays a crucial role in the pathogenesis of diabetic induction of glomerulosclerosis and diabetic nephropathy.Citation[4]
Reactive dicarbonyl compounds such as glyoxal and methylglyoxal (MGO) are major intermediates in Maillard reactions. It is well known that glucose-derived dicarbonyl intermediates such as MGO are potent precursors of advanced glycation end-products (AGEs),Citation[5] and the levels of these dicarbonyl compounds are elevated in the blood and tissues of diabetic subjects.Citation[6] MGO was also reported to increase the susceptibility of mesangial cells to apoptosis. Therefore, recent studies have raised the possibility that MGO-mediated Millard reactions directly affect cellular function to cause diabetic complications.Citation[7] Liu et al. have demonstrated that MGO induces apoptosis through the activation of P38 mitogen-activation protein kinase (MAPK) pathway in mesangial cells.Citation[8] However, the more detailed mechanism of upper stream signaling modulation in MGO-induced apoptosis has not been fully elucidated.
Oxidative stress is a condition in which cells are exposed to excessive levels of oxygen and derivatives of O2, especially superoxide (O2-). With regard to the kidneys, our recent studies have demonstrated enhanced superoxide production ex vivo by renal cortex from streptozotocin (STZ)-induced diabetic rats.Citation[9] We have also demonstrated increases in superoxide production in glomerular mesangial cells in response to a high concentration of glucose and AGEs,Citation[9] which are two factors clearly implicated in the pathogenesis of diabetic nephropathy. Recent studies have shown that the administration of antioxidant to diabetic rats is associated with activation of caspase-3, nuclear transcriptional factor (NF-KB), and P38 MAPK.Citation[10] Thus, it is a reasonable implication that O2-, as a primary signaling molecule, promotes the production of ROS downstream, which acts as a signaling transmitter to cause additional apoptosis through a redox-sensitive pathway. We therefore hypothesize that modulating the redox-sensitive pathway may develop methods to not only rescue MGO-induced apoptosis but also to influence many other cellular events, such as apoptotic transcription factor and gene expression.
Ras GTPases are small monomeric intracellular signaling molecules that act as molecular on/off switches in a range of crucial signal networks. Their functions include the control of cellular proliferation, apoptosis, migration and adhesion.Citation[11] Thus, we attempted to identify whether or not diabetic milieu-like high MGO levels involved in the redox-signaling cascade are propagated by activating Ras.
In this study, we will evaluate whether or not O2- scavenger and Ras inhibitor could rescue MGO-induced apoptosis in rat mesangial cells. We also investigate which species of oxygen radicals could be induced by MGO and how the oxygen radicals transmitted the signal cascade. In addition, we attempted to clarify the intracellular molecular mechanisms responsible for the MGO-induced apoptosis, especially Ras modulation of superoxide, that have not yet been indicated as mediators between intracellular stress and the execution of apoptosis in diabetic milieu-like MGO-induced renal injuries.
MATERIALS AND METHODS
Cell Cultures
A rat kidney mesangial cell line immortalized with psv3-Neo, CRL-2573 (American Type Culture Center, Gaithersburg, Maryland, USA), was used for the studies. The cells were maintained in 85% Dulbecco's-modified Eagle's medium 4 mM L-glutamine, adjusted to contain 1.5 g/L sodium bicarbonate and 4.5 g/L glucose, and supplemented with 0.4 mg/mL G418 + 15% fetal bovine serum at 37°C in a 5% carbon dioxide (CO2) atmosphere. Cells were confirmed as mesangial cells based on their typical morphology, positive immunofluorescence stain for desmin and vimentin, and negative stain for cytokeratin 8. Cells were harvested with trypsinization and resuspended in phosphate-buffered saline (PBS) for studies. Cell viability was determined using trypan blue exclusion.
Cell Proliferation Assay
Cell proliferation assay was spectrophotometrically measured by using the Cell Proliferation Kit (Boehringer Mannheim GmbH, Germany) according to the manufacturer's instructions. Briefly, cells (2 × 104 cells/well, 96-well) with or without high concentration MGO in the presence or absence of redox signaling modulators were cultured for 48 hours before adding 10 μl/well 3-(4,5-dimethythiazol-2-yl)-2,5-diphenyl tetrazolium brimide for an additional four-hour culture. Formazan synthesis in each well was resolved by 10% SDS-0.01 M HCl and colorimetrically measured at 550 nm. In addition, LC50 was also used as an index of cytotoxicity and determination of MGO treatment concentration.
MGO Treatment
When rat mesangial cells reached approximately 80% confluence, they were starved in DMEM with serum-free conditions overnight, followed by two washes in PBS. To examine the effects of MGO on the induction of diabetic caspase-3 activation, the cells were incubated with various concentrations for the indicated periods in each experiment. Cells (1 × 106 cells/dish, 100 mm culture dish) cultured in DMEM with 1% FBS without or with various concentration (10, 50, 100, and 200 μM) of MGO (Calbiochem, La Jolla, California, USA) for 48 hours were selected for the following experiments. To investigate the role of bio-active radicals in MGO (100 μM) promotion of apoptosis, subconfluent cell cultures were pretreated with or without 500 U/mL polyethylene glycol (PEG)-coupled bovine erythrocyte superoxide dismutase (SOD) and PEG-catalase (Sigma Chemical Inc., St. Louis, Missouri, USA) to scavenge superoxide and hydrogen peroxide. To elucidate the role of nitric oxide in the MGO-promoted apoptosis, cells were pretreated with or without 100 μM L-NAME (N-nitro-L-arginine methyl ester; Sigma Chemicals Inc., Missouri, USA) to inhibit nitric oxide production. To differentiate which oxidase was responsible for MGO-induced superoxide production, cells were pre-treated with 20 μM DPI (diphenyloniodium, a NADPH oxidase inhibitor), 30 μM allopurinol (a xanthine oxidase inhibitor) or 30 μM rotenone (a mitochondrial oxidase inhibitor; Sigma Chemical Inc., St. Louis, Missouri, USA). In some experiments, subconfluent cell cultures were pre-treated with 10 μM PD98059 (an ERK inhibitor, Calbiochem, La Jolla, California, USA), 10 μM SB203580 (a p38 inhibitor, Calbiochem, La Jolla, California, USA), and 10 μM SP600125 (a JNK inhibitor, Calbiochem, La Jolla, California, USA), or 2.5 μM manumycin A (Sigma-Aldrich Co.) to inhibit Ras activity, respectively, for two hours before MGO treatment.
Determination of Superoxide Production
Rat mesangial cell cultures (1 × 105 cells well, 96-well plate) with or without MGO treatment were subjected to assessment of superoxide. Superoxide synthesis was determined using horse heart cytochrome C reduction assay in the absence and presence of SOD and calculated from the molar extinction coefficient of 0.0282 μM−1 cm−1, as previously described.Citation[9]
Western Blotting
Cytosolic and nuclear extracts of cell cultures were prepared as previously described. Citation[9] Protein concentrations in cytosolic and nuclear extracts were determined by Bio-Rad assay kit (Bio-Rad Laboratories, Hercules, California, USA). Aliquots of cytosolic and nuclear extracts were subjected to Western blot assay. After total P38, nuclear c-Jun and caspase-3 on the blots were recognized by anti-P38, nuclear c-Jun, and caspase-3 antibodies (Cell Signaling Technology Inc, Beverly, Massachusetts, USA), followed by horseradish peroxidase-conjugated IgG as the second antibody and visualized with chemiluminescence agents. The cleaved caspase-3, phosphorylated P38 and nuclear c-Jun were recognized by stripping the membrane in a buffer containing 62.5 mM Tris-HCl (pH 6.7), 2% SDS, and 100 mM mercaptoethanol for 30 min at 50°C and then reprobed with mouse anti-cleaved caspase-3, phospho-P38, and nuclear c-Jun using a similar procedure. The relative intensities of the immunoblotting bands were measured by densitometry using Image-Pro Plus version 4.5 software (Media Cybernetics) and were expressed as intensities relative to vehicle.
Ras Activation Assay
Ras activation was assessed using a Ras activation assay kit (Upstate Biotechnology), according to the manufacturer's instructions. Briefly, cell lysates were pre-cleared with glutathione-agarose, followed by incubation with specific Raf Ras-binding domain conjugate. The immunoprecipitates were subjected to immunoblotting. Activated Ras proteins on the blots were recognized using Ras antibodies as primary antibody, goat anti-mouse horseradish peroxidase as secondary antibody, and visualized using chemiluminescence agents.
Flow Cytometry to Quantify Apoptosis
The serum-free mesangial cells were cultured without or with 100 μM MGO for 48 hours in the presence or absence of SOD, DPI, SB203580, or Ras inhibitor (manumycin A). The treated cells were harvested by trypsinization and then washed three times with 1× PBS. The cell pellet was then suspended in 200 μl of binding buffer containing annexin-V (10 μl) and propidium iodide (PI: 5 μl, BD Biosciences, San Jose, California, USA) and then incubated in the dark at room temperature for 15min. The volume of each cell suspension was increased to 500 μl PBS, PH7.4 and the percentage of viable, apoptotic, and necrotic cells were analyzed with a flow cytometer. Fluorescence intensity was determined by FACS can flow cytometry and analyzed by CellQuest software (Becton Dickinson).
Cytocentrifuge Preparation and Terminal Deoxynucleotidyl Transferase (TdT)-Mediated Deoxyuridine Triphosphate-Biotin Nick End-Labeling (TUNEL)
RMC cells treated with various treatments were harvested and washed by PBS before receiving cytospin using Cytospin-2 cytocentrifuge (500 rpm, 5 min) at a concentration of 1 × 104 cells/slide. These slides were fixed by methanol and cryoserved at –20°C until TUNEL analysis. Cryoreserved cytospin slides were rehydrated and followed with apoptosis assay using in situ cell death detection kits (Roche Diagnostics GmbH, Mannheim, Germany) and were performed according to the manufacturer's instructions. Apoptosis of mesangial cells was counted using the ratio of TUNEL positive-stained mesangial cell number to total cell number under 200× magnifications.
Statistical Analysis
All numeric data are expressed as means ± standard errors. The Wilcoxon test was used to evaluate differences between each sample of interest and its respective control. For analysis of time course, a multiple range of analysis of variance and Bonferroni post hoc tests were used. A value of p < 0.05 was considered statistically significant.
RESULTS
MGO-Induced Decrement of Cell Viability
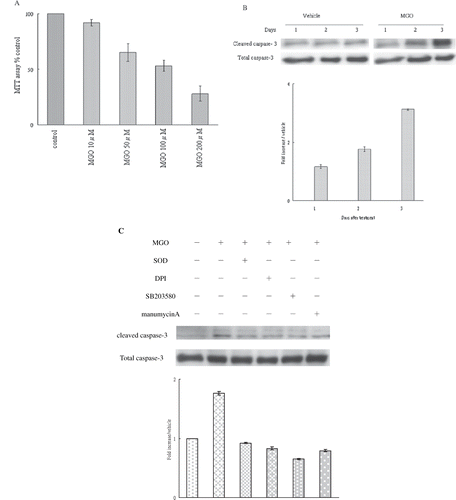
Raising the ambient MGO concentration for 48 hours significantly decreased cell proliferation. Cell proliferation was significantly reduced following exposure to various MGO concentrations (50 μM, 100 μM, and 200 μM) when compared to the control group (see ). This decrease in cell proliferation was in a dose-dependent manner, with an optimal response achieved at a MGO concentration of 100 μM causing the most significant affects on rat mesangial cells through LC50 calculation (see ).
Figure 1. (A) Effect of various MGO concentrations (10 μM, 50 μM, 100 μM, 200 μM) on the viability of rat renal mesangial cells. (B) MGO increased caspase-3 activation of mesangial cells. (C) Inhibition of superoxide by SOD or DPI, suppression of P38 activity by SB203580, and inhibition of Ras activity manumycin A rescued the promoting effect of MGO on cleaved caspase 3 protein expression. Total extract of cell cultures were subjected to Western blot assays. The relative intensities of the immunoblotting bands were measured by densitometry normalized to each vehicle. Experimental results are presented as means ± standard errors calculated from three triplicate experiments.
Optimal Dose and Time of MGO Treatment Induced Caspase-3 Activation in Rat Mesangial Cells
Mesangial cells were cultured in 100 μM for 72 hours. Immunoblotting showed that MGO significantly increased cleaved caspase-3 levels in one day. High MGO elicited significant promoting effects on caspase-3 activation after treatment for two days (see ). Therefore, a MGO concentration of 100 μm for 48 hours was tested in all subsequent experiments. We further investigated which reactive radical might be responsible for mediating MGO-induced caspase-3 activation. Scavenging of hydrogen peroxide with 500 units/mL PEG catalase and inhibition of nitric oxide synthase activity with 100 μm L-NAME did not affect MGO-promoted caspase-3 protein expression (data not shown). Nevertheless, SOD-PEG pretreatment (500 units/mL) significantly reduced MGO enhancement of caspase-3 activation (see ). These findings suggest that superoxide is involved in the MGO promotion of caspase-3 activation.
DPI Pretreatment Reduced MGO-Induced Superoxide and Caspase-3 Cleavage
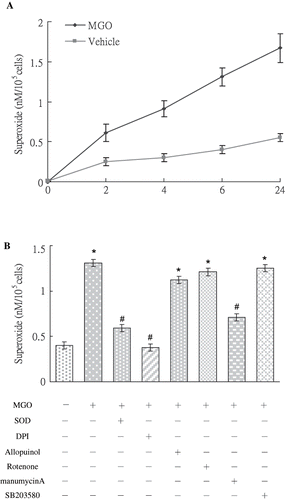
Results of SOD-inhibitable cytochrome c reduction assays showed that high MGO (see ) rapidly increased superoxide synthesis in two hours. The increased production of superoxide persisted for 24 hours (see ). We determined whether MGO promotion of superoxide production was linked to mitochondrial oxidase, xanthine oxidase, or NADPH oxidase. Pre-treatment with DPI (an NADPH oxidase inhibitor), but not with other oxidase inhibitors, significantly reduced MGO-promoted superoxide production (see ) and caspase-3 activation (see ). This suggests that NADPH oxidase was responsible for MGO-augmented superoxide production linked to caspase-3 cleavage.
Figure 2. (A) MGO increased superoxide synthesis by mesangial cells in 2 hours to 24 hours. (B) Inhibition of superoxide by DPI and manumycin A, but not by rotenone, allopurinol, or SB203580, reduced MGO augmented superoxide production at 6 hour. Results are presented with mean values ± standard errors calculated from six paired triplicate experiments. * and # respectively show the difference from vehicle and MGO group, p < 0.05.
MGO-Induced Membranous Ras, Cytosolic P38, and Nuclear c-Jun Activation
MGO rapidly increased Ras activation in one hour (see ). Pretreatment of SOD did not significantly affect MGO promotion of Ras activation (see ). We sought to investigate whether or not MGO-induced apoptosis was linked to MAPK activation. The caspase-3 protein activation under MGO treatment is significantly reversed by 10 μm SB 203580 (see ) but not PD98059 and SP600125 (data not shown). It suggests that caspase-3 activation in MGO treatment is dependent on p38 phosphorylation. Moreover, immunoblotting results showed that MGO treatments promoted cytosolic P38 (see ) in two and four hours and nuclear c-Jun activation in six hours (see ). We further sought to examine whether MGO-induced superoxide production could be associated with Ras activation. Inhibiting Ras activity with manumycin A significantly reduced the promoting effect of MGO on superoxide synthesis (see ) and caspase-3 cleavage (see ).
Figure 3. (A) MGO rapidly increased Ras activation in rat renal mesangial cells. (B) Inhibition of Ras activity by manumycin A but not inhibition of superoxide by SOD reduced the promoting effect of high MGO on Ras activation at 1 hour. Cell cultures (1 × 106 cells/dish, 100mm culture dish) were pretreated with 2.5μM manumycin A and subjected to high MGO treatment. The relative intensities of the immunoblotting bands were measured by densitometry normalized to each vehicle. Experimental results are presented as means ± standard errors calculated from three triplicate experiments.
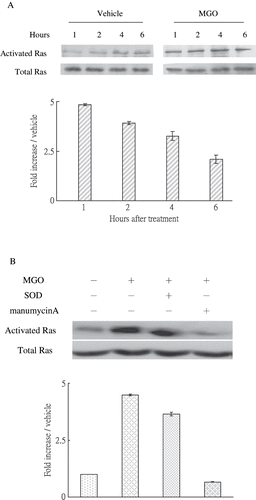
Figure 4. (A) High MGO increased cytosolic P38 activation. (B) Inhibition of superoxide by SOD or DPI or suppression of P38 activity by SB203580 and inhibition of Ras activity by manumycin A reduced the promoting effect of MGO on cytosolic P38 activation in four hours. Cytosolic fractions of cell lysate from cultures were subjected to Western blot assay. Immunoblotting of P38 showed equal loadings and transfer for all lanes. The relative intensities of the immunoblotting bands were measured by densitometry normalized to each vehicle. Experimental results are presented as means ± standard errors calculated from three triplicate experiments.
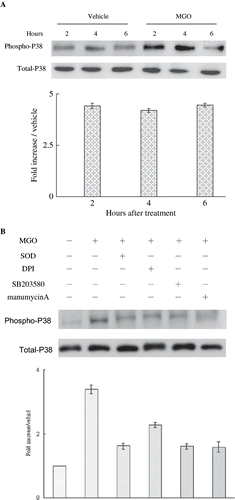
Figure 5. (A) MGO-induced nuclear c-Jun activation. Inhibition of superoxide by SOD or DPI, suppression of P38 activity by SB203580, and inhibiting Ras activity by manumycin A reduced the promoting effect of MGO on nuclear c-Jun activation. Cytosolic and nuclear fractions of cell lysate from cultures were subjected to Western blot assay. Immunoblotting of total c-Jun showed equal loadings and transfer for all lanes. The relative intensities of the immunoblotting bands were measured by densitometry normalized to each vehicle. Experimental results are presented as means ± standard errors calculated from three triplicate experiments.
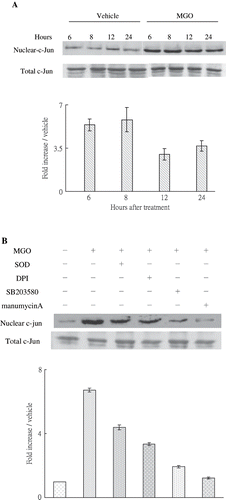
Ras Modulating Superoxide Mediated MGO-Increased Cytosolic P38 and Nuclear c-Jun Phosphorylation
The inhibition of Ras activity with manumycin A, pretreatment with SOD-PEG (500 units/mL) or DPI (5 μm), or suppression of P38 activity with SB203580 (10 μm) significantly reduced the enhancing effect of MGO on caspase-3 (see ), cytosolic P38 (see ), and nuclear c-Jun activation (see ). These findings suggest that MGO-induced cytosolic P38 and nuclear c-Jun phosphorylation and caspase-3 cleavage are modulated by membranous Ras expression, which subsequently accentuates superoxide anion synthesis dependent on NADPH oxidase.
SOD, DPI, SB203580, or Manumycin A Pretreatment Rescued MGO-Induced Mesangial Cell Apoptosis
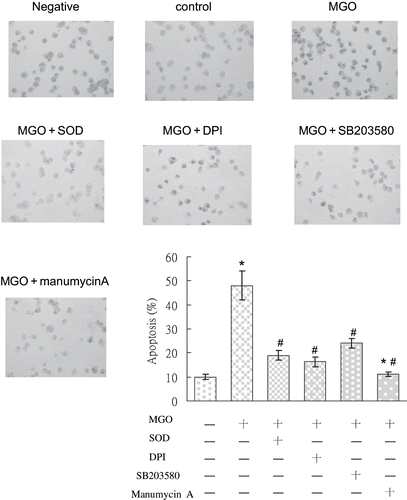
Rat mesangial cells treated with MGO and various signaling inhibitors were assessed for apoptosis using flow cytometry and TUNEL. The FACS was used to determine the degree of apoptosis by measuring the percentage of cells with positive Annexin-V assay. Increased apoptosis of MGO-exposed renal mesangial cells was confirmed using flow cytometric analysis of cells stained with annexin-V and popidium iodide. In the control group, only 6.2 ± 0.4 % of cells were annexin-V-positive. In contrast, annexin-V-positive cells in the MGO-treated group comprised 45.2 ±11.2% of the total cell population. Treatment with SOD, DPI, SB 203580, and manumycin A significantly reduced the percentage of apoptotic cells to 16.8 ± 2.1, 19.4 ± 2.3, 29.4 ± 4.6, and 22.5 ± 1.5 % (p < 0.05, n = 3; see and ). In the absence of TdT, no TUNEL staining was observed. Cells positive for TUNEL exhibited red staining in the nucleus. In comparison with the control group, MGO significantly increased cell apoptosis in 48 hours (see ). We examined whether MGO promotion of cell apoptosis was linked to activation of NADPH oxidase, phosphorylated p38, and Ras. Results of cytospin TUNEL staining showed that MGO significantly increased TUNEL staining (see ). Inhibiting diverse activities by SOD, DPI, SB203580, and manumycin A evidently reduced the promoting effect of MGO on cell apoptosis. These observations demonstrate that NADPH oxidase, phosphorylated p38, and Ras activation are associated with MGO-induced apoptosis in rat renal mesangial cells.
Figure 6. The profile in vehicle group (A) and the percentage of cells undergoing apoptosis (annexin V-positive, lower panel) under various conditions (B) were shown. Role of NADPH oxidase, P38 MAP kinase, and Ras signaling pathways was in MGO-induced mesangial cell apoptosis. SOD, DPI, SB203580, and manumycinA significantly reduced the percentage of apoptosis. The results shown represent the means ± SD of three experiments. * and # respectively show the difference from the vehicle and MGO group, p < 0.05.
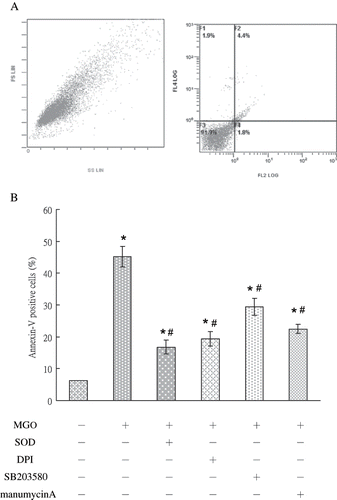
Figure 7. TUNEL staining photography of MGO-treated RMC with or without various treatments, including SOD, DPI, SB203580, and manumycin A. In comparison with the control group, mesangial cells in MGO treatment expressed strong TUNEL immunostaining. Decreased TUNEL staining was observed after pretreatment with SOD, DPI, SB203580, and manumycin A, individually. Specimens were observed in magnification = 200×.
DISCUSSION
In this study, apoptotic cell death coincided with caspase 3 activation in renal mesangial cells and was found to follow MGO stressors, which induced membranous-bound Ras and NADPH oxidase-dependent superoxide. This reactive oxygen bio-molecule plays a critical role in regulating cytosolic P38 MAPK and nuclear c-Jun activation. Mechanisms underlying high MGO levels induction of apoptosis in renal mesangial cells are not fully understood. Our findings provide evidence that renal mesangial cells respond to MGO by raising intracellular signal transduction and apoptotic expression through Ras modulation of superoxide-mediated P38 MAPK and c-Jun activation, which provide a clear molecular explanation for diabetes induced renal injuries. While previous studies have demonstrated that MGO caused sufficient cellular stress to induce apoptosis through the activation of P38 MAPK in rat mesangial cells,Citation[8] little has been done to define the role of pharmacological modulation of Ras in NADPH oxidase-dependent superoxide in the promotion of MGO in renal mesangial injuries. Our findings provide the first indication that modulation of Ras on NADPH oxidase-dependent superoxide treatment alleviated MGO-promoted oxidative damage and apoptosis in mesangial cells, which provides an alternative redox modulation strategy for rescuing diabetic renal injuries.
Previous research has demonstrated that AGEs-mediated cellular oxidant stress would trigger the Ras/MAPK pathway, implying that renal mesangial cells respond to AGEs through the initiation of oxidant stress and then subsequently activate the Ras pathway.Citation[12] In contrast to this AGEs-related finding, the current study found that MGO initially activated Ras protein and then elevated superoxide production. The small G protein Ras appears to be important molecular switches integrating diverse stimuli and transducing key signaling functions, such as superoxide production.Citation[13] Ras was also reported to control the activity of the key enzyme complex NADPH oxidase to control superoxide production in nonphagocytic cells.Citation[14] It is compatible with our finding that inhibiting Ras activity with manumycin A reduced the stimulatory effect of MGO on superoxide production and subsequently P38 phosphorylation, c-Jun expression, and apoptosis. We therefore suggest that the Ras pathway is responsible for superoxide production in MGO-induced apoptosis and imply that membrane-bound Ras acts as a crucial regulator to transmit to extra-cellular stress by increasing redox reaction and progressively activating intracellular signal transduction pathways.
With regard to the mechanism by which MGO induced apoptosis, we found that MGO-induced apoptosis was closely related to the activation of P38 MAPK. Recent investigation has indicated that superoxide plays a pivotal role in mediating between cellular stresses and activating P38 MAPK signaling.Citation[15] In our studies, SB203580, a specific inhibitor of P38 MAPK, efficiently inhibited MGO-induced apoptosis without any effect on superoxide production, implying that NADPH oxidase-dependent superoxide is a common upstream molecule of P38 MAPK. On the other hand, inhibitory effects of Ras by manumycinA on MGO-induced superoxide production may reverse activation of P38 MAPK. Therefore, we suggest that Ras modulation of NADPH oxidase plays an important role in mediating cellular stresses and apoptosis through the activation of the P38 MAPK pathways.
C-Jun is a critical transcriptional factor in response to cellular stress. Whitfield et al. have demonstrated that c-Jun can trigger apoptosis through transcription activation of some pro-apoptotic genes.Citation[16] Previous studies also found that genetic deletion of c-Jun or application of neutralizing antibodies against c-Jun leads to the resistance of cells to apoptotic stimuli,Citation[17] suggesting that c-Jun plays an important role in the generation of apoptosis. Furthermore, over-expression of the dominant-negative mutant of c-Jun could attenuate hydrogen peroxide-induced cellular injury, indicating that oxidative stress-induced cellular apoptosis is associated with c-Jun expression.Citation[18] On the basis of our demonstration in this study that both c-Jun expression and apoptosis are inhibited by superoxide scavengers such as SOD and DPI, we propose that NADPH oxidase-dependent superoxide is a key step in the signaling pathway in which MGO induces the activation of c-Jun and subsequently apoptosis. Another new observation is that manumycin A, a Ras inhibitor, can rescue MGO-induced apoptosis via reduction of c-Jun expression. These findings favor activation of Ras signaling, which seems to be a key signal for the activation of c-Jun and apoptosis by MGO treatment.
Kidney cells convert diabetic stimuli into biochemical responses after gene expression and cellular adaptation. MGO rapidly activated membrane-bound Ras proteins of mesangial cells, indicating that bioactive molecules located at the cell surface, such as G-proteins, may also be activated by MGO. Further studies are needed to explore the role of G protein and its receptor in regulating MGO-induced apoptotic response in mesangial cells.
In summary, we have provided evidence that Ras modulation by manumycin A and regulation of redox reaction by SOD may provide a promising regimen for regulating P38 MAPK signal transduction and activating the apoptotic transcription factor, c-Jun, resulting in a significant decrease in caspase-3 activation and apoptosis in MGO-treated mesangial cells. By using the exogenous Ras inhibitor- or SOD-attenuated signal transduction for diabetic renal injuries, the biopharmacological modulation of rescuing diabetic renal injuries may also be possible.
ACKNOWLEDGMENT
This work was supported in part by grant NSC95-2320-B-255-004-MY2 from the National Science Council, Taiwan, and CMRPF650011 from Chang Gung Memorial Hospital, Taiwan.
Related Research Data
REFERENCES
- Saito Y, Kida H, Takeda S, Yoshimura M, Yokoyama H, Koshino Y, Hattori N. Mesangiolysis in diabetic glomeruli: Its role in the formation of nodular lesions. Kidney Int. 1988; 34: 389–396
- Lemley KV, Abdullah I, Myers BD, Meyer TW, Blouch K, Smith WE, Bennett PH, Nelson RG. Evolution of incipient nephropathy in type 2 diabetes mellitus. Kidney Int. 2000; 58: 1228–1237
- Mishra R, Emancipator SN, Kern T, Simonson MS. High glucose evokes an intrinsic proapoptotic signaling pathway in mesangial cells. Kidney Int. 2005; 67: 82–93
- Dalla Vestra M, Saller A, Mauer M, Fioretto P. Role of mesangial expansion in the pathogenesis of diabetic nephropathy. J Nephrol. 2001; 14: S51–S57
- Frye EB, Degenhardt TP, Thorpe SR, Baynes JW. Role of the Maillard reaction in aging of tissue proteins. Advanced glycation end product-dependent increase in imidazolium cross-links in human lens proteins. J Biol Chem. 1998; 273: 18714–19719
- Kusunoki H, Miyata S, Ohara T, Liu BF, Uriuhara A, Kojima H, Suzuki K, Miyazaki H, Yamashita Y, Inaba K, Kasuga M. Relation between serum 3-deoxyglucosone and development of diabetic microangiopathy. Diabetes Care. 2003; 26: 1889–1894
- Fukunaga M, Miyata S, Liu BF, Miyazaki H, Hirota Y, Higo S, Hamada Y, Ueyama S, Kasuga M. Methylglyoxal induces apoptosis through activation of p38 MAPK in rat Schwann cells. Biochem Biophys Res Commun. 2004; 320: 689–695
- Liu BF, Miyata S, Hirota Y, Higo S, Miyazaki H, Fukunaga M, Hamada Y, Ueyama S, Muramoto O, Uriuhara A, Kasuga M. Methylglyoxal induces apoptosis through activation of p38 mitogen-activated protein kinase in rat mesangial cells. Kidney Int. 2003; 63: 947–957
- Lin CL, Wang FS, Kuo YR, Huang YT, Huang HC, Sun YC, Kuo YH. Ras modulation of superoxide activates ERK-dependent fibronectin expression in diabetes-induced renal injuries. Kidney Int. 2006; 69: 1593–1600
- Qanungo S, Wang M, Nieminen AL. N-acetyl-L-cysteine enhances apoptosis through inhibition of nuclear factor-kappaB in hypoxic murine embryonic fibroblasts. J Biol Chem. 2004; 279: 50455–50464
- Bokoch GM, Der CJ. Emerging concepts in the Ras superfamily of GTP-binding proteins. FASEB J. 1993; 7: 750–759
- Lander HM, Tauras JM, Ogiste JS, Hori O, Moss RA, Schmidt AM. Activation of the receptor for advanced glycation end products triggers a p21(ras)-dependent mitogen-activated protein kinase pathway regulated by oxidant stress. J Biol Chem. 1997; 272: 17810–17814
- Thannickal VJ, Day RM, Klinz SG, Bastien MC, Larios JM, Fanburg BL. Ras-dependent and -independent regulation of reactive oxygen species by mitogenic growth factors and TGF-beta1. FASEB J. 2000; 14: 1741–1748
- Seru R, Mondola P, Damiano S, Svegliati S, Agnese S, Avvedimento EV, Santillo M. HaRas activates the NADPH oxidase complex in human neuroblastoma cells via extracellular signal-regulated kinase 1/2 pathway. J Neurochem. 2004; 91: 613–622
- Chan SH, Hsu KS, Huang CC, Wang LL, Ou CC, Chan JYH. NADPH Oxidase-derived superoxide anion mediates angiotensin II-induced pressor effect via activation of p38 mitogen-activated protein kinase in the rostral ventrolateral medulla. Circ Res. 2005; 97: 772–780
- Wang FS, Wang CJ, Chen YJ, Chang PR, Huang YT, Sun Y C, Huang HC, Yang YJ, Yang KD. Ras induction of superoxide activates ERK-dependent angiogenic transcription factor HIF-1alpha and VEGF-A expression in shock wave-stimulated osteoblasts. J Biol Chem. 2004; 279: 10331–10337
- Watson A, Eilers A, Lallemand D, Kyriakis J, Rubin LL, Ham J. Phosphorylation of c-Jun is necessary for apoptosis induced by survival signal withdrawal in cerebellar granule neurons. J Neurosci. 1998; 18: 751–762
- Wang N, Verna L, Hardy S, Zhu Y, Ma KS, Birrer MJ, Stemerman MB. c-Jun triggers apoptosis in human vascular endothelial cells. Circ Res. 1999; 85: 387–393