
Abstract
Objectives: Fabry's disease is an X-linked inherited, rare, progressive, lysosomal storage disorder, affecting multiple organs due to the deficient activity of α-galactosidase A (α-Gal A) enzyme. The prevalence has been reported to be 0.15–1% in hemodialysis patients; however, the information on the prevalence in chronic kidney disease not on dialysis is lacking. This study aimed to determine the prevalence of Fabry’s disease in chronic kidney disease.
Methods: The patients older than 18 years, enclosing KDIGO 2012 chronic kidney disease definitions, not on dialysis, were enrolled. Dried blood spots on Guthrie papers were used to analyze α-Gal A enzyme and genetic analysis was performed in individuals with enzyme activity ≤1.2 μmol/L/h.
Results: A total of 1453 chronic kidney disease patients not on dialysis from seven clinics in Turkey were screened. The mean age of the study population was 59.3 ± 15.9 years. 45.6% of patients were female. The creatinine clearance of 77.3% of patients was below 60 mL/min/1.73 m2, 8.4% had proteinuria, and 2.5% had isolated microscopic hematuria. The mean value of patients’ α-Gal A enzyme was detected as 2.93 ± 1.92 μmol/L/h. 152 patients had low levels of α-Gal A enzyme activity (≤1.2 μmol/L/h). In mutation analysis, A143T and D313Y variants were disclosed in three male patients. The prevalence of Fabry’s disease in chronic kidney disease not on dialysis was found to be 0.2% (0.4% in male, 0.0% in female).
Conclusion: Fabry’s disease should be considered in the differential diagnosis of chronic kidney disease with unknown etiology even in the absence of symptoms and signs suggestive of Fabry’s disease.
Introduction
Fabry’s disease (OMIM #301500) is an X-linked inherited, rare disorder. The disease is progressive and affects multiple organs, particularly the heart, the kidney, and the central nervous system. It causes varying degrees of organ dysfunction due to lysosomal glycosphingolipid accumulation. The gene involved in Fabry’s disease is located on X chromosome in the position Xq22 and called as GLA gene. Mutations in GLA gene give rise to decreased α-galactosidase A enzyme (α-Gal A) activity, resulting in accumulation of glycosphingolipids, in particular, globotriaosylceramide (Gb3) intracellularly.Citation1 Accumulation of Gb3 in renal podocytes, tubular cells, mesangial cells, renal interstitial cells, cardiomyocytes, cardiac fibroblasts, nerve cells, and endothelial cells causes structural and functional abnormalities. Globotriaosylceramide accumulation, especially in capillary endothelial cells, leads to microvascular obstruction, resulting in ischemic tissue injury, which has been advocated as the main pathogenic mechanism.Citation1,Citation2
Fabry’s disease presents with variable clinical phenotypes in a broad spectrum of progressive disease.Citation1,Citation3,Citation4 Three phenotypes such as the classic, cardiac, and renal variants have been described.
The classic phenotype usually becomes symptomatic in childhood and characterized by angiokeratoma, hypohidrosis, corneal and lenticular opacities, acroparesthesia, abdominal pain, and hearing impairments. Multi-organ involvement develops in the course of the disease. The renal disease often begins with microalbuminuria and proteinuria in the second to third decades of life and its severity increases with age. The end-stage renal disease usually ensues in the fourth to fifth decades of life.Citation1,Citation5 The cardiac involvement typically presents before the age of 30 years and characterized by concentric symmetrical left ventricular hypertrophy, arrhythmias, valvular abnormalities, and premature coronary artery disease.Citation5,Citation6 Transient ischemic attack and ischemic stroke are frequently observed, and its prevalence is ∼13% owing to small vessel events.Citation7 The life expectancy in the classic phenotype is decreased by ∼20 years due to end-stage renal disease and life-threatening cardiovascular or cerebrovascular complications.Citation5,Citation8
In classic phenotype of Fabry’s disease, the level of α-Gal A activity is below 1%, which is the major determinant of the ultimate pathological and clinical phenotypes. Cardiac and renal phenotypes usually have 2–20% a-galactosidase A activity. Therefore, the disease manifestations present during the sixth to eighth decades of life with predominantly one organ system involvement.Citation1,Citation9 Since this is an X-linked recessive disease, the affected heterozygous females were reported presumably due to the pattern of X-chromosome inactivation. Vital organ involvement in females occurs at a later age when compared to the affected males.Citation10
The worldwide prevalence of the classic phenotype of Fabry’s disease has been estimated approximately as 1 in 117,000 to 1 in 476,000.Citation11,Citation12 The actual prevalence is probably higher than the existing data, particularly when late-onset phenotypes are taken into consideration. Fabry’s disease is usually diagnosed approximately with a delay of 20 years due to its rarity, the lack of awareness among clinicians, the diversity of findings, and the absence of specific symptoms.Citation5 Prevalence studies have often been performed in specific sub-groups, such as dialysis patients and the prevalence in hemodialysis patients has been reported to be 0.15–1%.Citation4,Citation5,Citation13,Citation14 Although there is no benefit from the point of renal outcomes in end-stage renal disease, the early diagnosis of the disease prior to the development of end-stage renal disease can yield beneficial renal outcomes. Additionally, the establishment of the diagnosis can be beneficial for managing involvements of other organs. In the case of timely diagnosis, treatment using available recombinant enzyme replacement improves outcome. Therefore the early diagnosis and initiation of therapy are the mainstays of an effective treatment.Citation15
There are lack of data concerning the prevalence of Fabry’s disease in chronic kidney disease patients not on dialysis. This study aimed to determine the prevalence of Fabry’s disease in chronic kidney disease population.
Methods
The present study is a cross-sectional, analytic, multicenter study. The patients older than 18 years, enclosing the kidney disease definitions, not on dialysis of “Kidney Disease: Improving Global Outcomes (KDIGO) Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease 2012 Guidelines”Citation16 were enrolled into the study. Patients who had been transfused within the last 6-month period were excluded. Totally, 1453 patients were screened in seven nephrology clinics located at Aydın, Denizli, İzmir, and Muğla in Turkey. The objectives, methods, targets and the potential hazards of the study were explained to all individuals. All patients were informed and gave their informed consents before participating in the study. General physical examination was carried out. Age and gender were recorded, and arterial blood pressure was measured in all patients. Demographic data, patients’ history, basic laboratory results such as serum creatinine, urinalysis, and daily urinary protein losses were recorded from the patients’ medical charts.
Blood collection
Two milliliters of venous blood sample was obtained from every patient. Blood samples were impregnated to four chambers of Guthrie paper, allowed to dry at room temperature, and stored at room temperature avoiding direct sun exposure until the analysis.
Measurement of α-Gal A activity and mutation analysis
Dried venous blood spots on Guthrie papers were used to analyze enzyme activity and genetic testing when required. The α-Gal A enzyme activity was determined by a fluorescence-based high throughput microplate method using the method by Chamoles et al.Citation17 The enzyme activities were calculated in μmol/L/h, and the patients with the enzyme activity ≤1.2 μmol/L/h were considered to have low α-Gal A activity and referred for genetic analysis.Citation18
Genetic analysis was performed by DNA Sanger sequence analysis in individuals with equal-lower than 1.2 μmol/L/h enzyme activity. Genomic DNA was isolated from dried blood spots. In this assay, to detect mutations in GLA gene, polymerase chain reaction amplification was followed by Sanger DNA sequencing. The 1.45-kb GLA gene consists of seven exons and encodes a polypeptide of 429 amino acids, including the first 31 amino acid residues forming a lysosomal signal peptide. Purified genomic DNA of exons 1–7 of the GLA gene were isolated by polymerase chain reaction amplification. Coding sequences and flanking intronic sequences were amplified to each of the seven exons and sequenced in the forward and reverse directions. Sequencing of a single exon is available for targeted mutation analysis. All α-Gal A enzyme activities and genetic testing were performed by ARCHIMED Life Science GmbH Laboratories Vienna, Austria.
Statistical analysis
Statistical evaluations were performed using the Statistical Package for Social Sciences for Windows, version 17 (SPSS Inc., Chicago, IL) software package. Quantitative variables were expressed as mean ± standard deviation and qualitative variables as numbers and percentages (n, %). The accordance of quantitative data with normal distribution was examined with the Kolmogorov–Smirnov test.
Approval of the ethics committee
The study was presented to the local ethics committee of clinical research at Adnan Menderes University, School of Medicine and received approval with the decision number of 56989545/050.04–26, dated 14.02.2014.
Results
A total of 1453 chronic kidney disease patients, not on dialysis, from seven nephrology clinics located at Aydın, Denizli, Izmir, and Muğla in Turkey were screened. The mean age of the study population was 59.3 ± 15.9 years. 45.6% of patients were female. In the study population, decreased creatinine clearance (below 60 mL/min/1.73 m2), proteinuria and isolated microscopic hematuria were the main criteria of enrollment in the study in 77.3%, 8.4% and 2.5% of patients, respectively. Hypertension, diabetes mellitus, history of cardiac disease and history of stroke were present in 75.5%, 36.8%, 27.1%, and 7.4% patients, respectively.
The mean serum creatinine and 24-h protein excretion rates were 2.1 ± 1 mg/dL and 1543 ± 2678 mg/day, respectively (). The mean α-Gal A enzyme activity was 2.93 ± 1.92 μmol/L/h. In 152 (17%; 86 males, 64 females) out of 1453 patients, α-Gal A enzyme activity was ≤1.2 μmol/L/h, which was considered as a positive screening test. The genetic mutation testing was performed in all 152 patients with depressed enzyme activity. Mutations in the GLA gene unique to Fabry’s disease were observed in three male patients (). In mutation analysis using sequential DNA analysis technique in exons and introns, A143T and D313Y variants were documented. All three patients were in older ages: Case 1 was at the age of 70 years and had a hemizygote [937G > T] p.[D313Y]) mutation, Case 2 was 83 years old and had a hemizygote [427G > A] p.[A143T] mutation and Case 3 was 66 years old and had a hemizygote [937G > T] p.[D313Y]) mutation. All three patients had moderate renal insufficiency, serum creatinine clearance values being 33.3 mL/min/1.73 m2 (creatinine 1.95 mg/dL), 38.2 mL/min/1.73 m2 (creatinine 1.81 mg/dL), 30.3 mL/min/1.73 m2 (creatinine 2.30 mg/dL), respectively. The patients in whom the mutation was detected were clinically evaluated at Adnan Menderes University, School of Medicine, Nephrology Department. The multidisciplinary evaluation of the patients was carried out by the same physicians from each discipline.
Table 1. Demographic and laboratory features of patients.
Table 2. Patients α-gal a enzyme activity and mutation results.
In the present study, which identified that mutations were present in three patients, the prevalence of Fabry’s disease mutations in chronic kidney disease patients not on dialysis was found to be 0.2% (0.4% in males, 0.0% in females).
Cases 1 and 2 were admitted for further investigation. Detailed information on demographic and clinical features and laboratory findings were already given in . Kidney biopsy specimen of index Case 1 was displayed in . The third patient (Case 3) rejected further evaluation.
Figure 1. Kidney biopsy specimen of index Case 1. (Representative histological section hematoxylin and eosin stain, ×100; Tubular thyroidization, glomerular global sclerosis, interstitial chronic inflammation, arterial medial thickening was observed. Vacuolization of the glomerular tuft and tubular cells was not detected.)
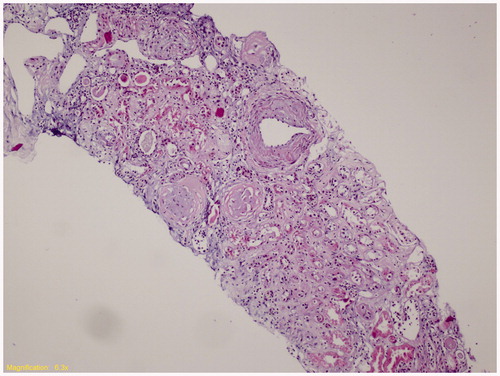
Table 3. Demographic, clinical, laboratory findings of two cases with Fabry disease.
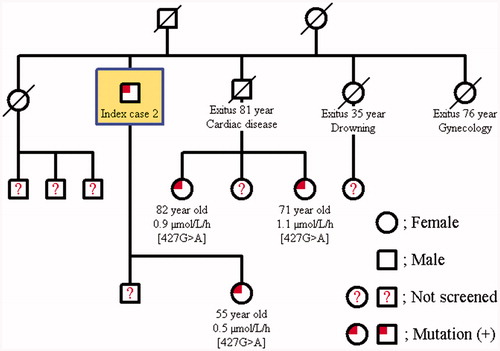
The family screening was performed in these two cases with Fabry’s disease. The same mutation was detected in the younger brother of Case 1; 49-year-old male in whom the α-Gal A enzyme activity was found to be 2.9 μmol/L/h and his only daughter was determined to be a carrier with an enzyme activity of 2.8 μmol/L/h. The other two younger brothers of Case 1 at 60 and 64 years of age had normal enzyme activities, and no mutation was detected (). Further examination of the younger brother with mutation failed to document renal, cardiac, cranial, ocular, auditory, or dermatological involvement. The daughter of Case 2 had a heterozygous [427G > A] p.[A143T] mutation, and her enzyme activity was lower (0.5 μmol/L/h). The two daughters of the grand brother of case 2 at the age of 82 years and 71 years also had the mutation of a carrier; their enzyme activities were 0.9 μmol/L/h and 1.1 μmol/L/h, respectively (). 82-year-old grand brother’s daughter revealed only cornea verticillata in further evaluation.
Figure 2. Pedigree family chart of Case 1.
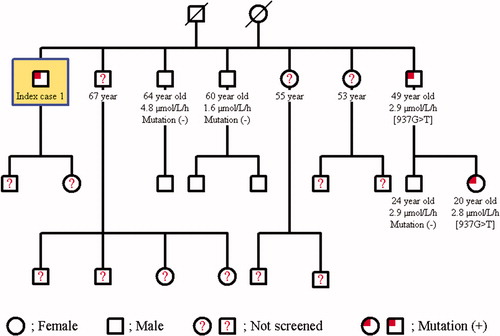
Figure 3. Pedigree family chart of Case 2.
Discussion
Fabry’s disease is a genetic disorder that can affect a broad range of organs, and it is likely to be undiagnosed in patients suffering from kidney disease, heart disease, or early stroke. The findings in studies, as in the present study, may contribute to not only achieving the correct diagnosis and improving medical care in these patients, but also to the early diagnosis of their relatives. In low renal involvement (the urine protein to creatinine ratio ≤0.5 g/g and <50% glomerulosclerosis), treatment with enzyme replacement provides a positive response.Citation15 In patients with mild-to-moderate renal involvement, estimated glomerular filtration rate, serum creatinine, and proteinuria remained stable through long-term enzyme replacement therapy. Baseline proteinuria over 1 g per day, advanced nephropathy (>50% glomerulosclerosis, creatinine >1.5 g/dL) and age >40 years on treatment baseline are associated with poor prognosis.Citation19
A systematic review was conducted to calculate the overall prevalence of Fabry’s disease in patients undergoing dialysis, having left ventricular hypertrophy, or stroke. Data revealed that the prevalence of Fabry’s disease in dialysis patients was 0.33% for males and 0.10% for females, in patients with left ventricular hypertrophy ranging from 0.9% to 3.9% in males and 1.1% to 11.8% in females due to the selection of study population and differences in the screening methods, and in premature strokes 4.2% in males and 2.1% in females.Citation20
The prevalence of Fabry’s disease in hemodialysis patients was 0.17% in Turkey, and it has also been reported as 0.02% in Japan in recent publications.Citation14,Citation21 In the present study, Fabry’s disease mutations were identified in three male patients out of 1453 patients, yielding a prevalence of 0.2% (0.4% in male); this is the first data in the literature concerning the prevalence of Fabry’s disease in chronic kidney disease population not on dialysis.
Various screening methods such as a-galactosidase activity in plasma or leukocytes, plasma Gb3, urinary Gb3, lysosomal α-Gal A protein assay, and dried blood spots on the filter were used in case findings-prevalence studies.Citation5,Citation14,Citation21,Citation22 The measurement of α-Gal A enzyme activity in dried blood spots, carried out by the fluorometric method, provides facilities for the handling of specimens and measurement in comprehensive (large-scale) prevalence studies due to stable enzyme activity up to six months.Citation5,Citation23 The present study was performed with dried blood spots for the reasons mentioned above.
The activity of α-Gal A enzyme in dried blot spots was found to be 6.12 ± 2.28 μmol/L/h and 5.6 ± 2.0 μmol/L/h in healthy subjects, 0–0.08 μmol/L/h in Fabry’s patients, below 1.1 and 1.56 μmol/L/h in hemizygous males, values ranging from very low to normal values in heterozygous females; one-third of the females were not identified using the enzymatic assay, due to significant residual enzyme activity.Citation17,Citation23,Citation24 In cardiac variants of men with left ventricular hypertrophy, values ranged from 0.4 to 1.2, ∼4 to 14% of the mean value in the controls was also detected.Citation18
It has been recommended in males that screening of Fabry’s disease is performed by measuring α-Gal A enzyme activity, followed by genetic analysis.Citation25 The values of enzyme activity in heterozygous females are very variable due to the residual enzyme activity, and enzyme assays fail to detect about one-third of heterozygous females. Therefore, genetic analysis of GLA gene has been suggested as a primary tool in females.Citation24–26 However, genetic testing is expensive, and it costs about 1000 Euro.Citation25 In this study, 656 females were screened preferably by enzyme levels due to the higher costs of genetic testing.
Missense mutations, nonsense mutations, single amino acid deletions and insertions are the common mutations in Fabry’s disease.Citation5,Citation25,Citation27 To date, 642 mutations have been identified in the GLA gene.Citation28 The present study revealed A143T and D313Y variants in three patients. Both variants have been reported in the “European newborn studies” as the most common variants.Citation29
Yasuda et al.Citation30 indicated that expression of the D313Y variant in COS-7 cells resulted in ∼60% wild-type residual enzyme activity and that the enzyme was localized to lysosomes. Also, the D313Y variant was found at a frequency of 0.45% in normal X chromosomes. The mutation D313Y causes low plasma enzyme activity in vitro due to neutral plasma; however, in the acid lysosomes, high residual lysosomal enzyme activity was demonstrated, and no significant accumulation of Gb3 and lysosomal Gb3, and no pathologic excretion of urinary Gb-3 has been reported.Citation25,Citation30–32 Therefore, D313Y mutation was named as pseudo-deficiency.Citation25,Citation30 There was no accumulation in kidney biopsy of our patient in compliance with the previously reported data.
A143T is a missense mutation due to a guanine-to-adenine substitution in codon 143 and has been reported to have 30–49% residual enzyme activity.Citation33 By using electron and light microscopy, no accumulation of Gb3 was observed in skin and kidney biopsy specimens, therefore it is considered to be nonpathogenic.Citation34 In contrast, A143T variant was reported to cause left ventricular hypertrophy and renal type Fabry’s disease.Citation35,Citation36 Our A143T mutation patient had advanced heart valve involvement and left ventricular hypertrophy, so it was thought to be a cardiac phenotype. The symptoms of patients with cardiac phenotype emerged after the sixth decade of life. In patients with a cardiac phenotype, left ventricular hypertrophy and/or cardiomyopathy may develop with proteinuria, and renal failure has also been reported.Citation1,Citation9
In the present study, the prevalence of Fabry’s disease was found to be 0.2% in chronic kidney disease patients not on dialysis. Fabry’s disease should be taken into consideration in the differential diagnosis of chronic kidney disease with unknown etiology even in the absence of symptoms and signs suggestive of Fabry’s disease such as sweating disorders, acroparesthesia, and angiokeratoma. Early diagnosis and treatment using enzyme replacement can be of use in the prevention of progressive disease.
Acknowledgements
This study was supported by Genzyme Corporation, a Sanofi company.
Disclosure statement
The authors report no conflicts of interest.
Related Research Data
References
- Germain DP. Fabry disease. Orphanet J Rare Dis. 2010;5:30.
- Tuttolomondo A, Pecoraro R, Simonetta I, Miceli S, Pinto A, Licata G. Anderson-Fabry disease: A multiorgan disease. Curr Pharm Des. 2013;19:5974–5996.
- Mehta A, Ricci R, Widmer U, et al. Fabry disease defined: Baseline clinical manifestations of 366 patients in the Fabry outcome survey. Eur J Clin Invest. 2004;34:236–242.
- Nakao S, Kodama C, Takenaka T, et al. Fabry disease: Detection of undiagnosed hemodialysis patients and identification of a “renal variant” phenotype. Kidney Int. 2003;64:801–807.
- Mehta A, Beck M, Eyskens F, et al. Fabry disease: A review of current management strategies. QJM. 2010;103:641–659.
- Anastasakis A, Papatheodorou E, Steriotis AK. Fabry disease and cardiovascular involvement. Curr Pharm Des. 2013;19:5997–6008.
- Ginsberg L, Manara R, Valentine AR, Kendall B, Burlina AP. Magnetic resonance imaging changes in Fabry disease. Acta Paediatr Suppl. 2006;95:57–62.
- Schiffmann R, Warnock DG, Banikazemi M, et al. Fabry disease: Progression of nephropathy, and prevalence of cardiac and cerebrovascular events before enzyme replacement therapy. Nephrol Dial Transplant. 2009;24:2102–2111.
- Ishii S, Chang HH, Kawasaki K, et al. Mutant alpha-galactosidase A enzymes identified in Fabry disease patients with residual enzyme activity: Biochemical characterization and restoration of normal intracellular processing by 1-deoxygalactonojirimycin. Biochem J. 2007;406:285–295.
- Deegan PB, Baehner AF, Barba Romero MA, Hughes DA, Kampmann C, Beck M. Natural history of Fabry disease in females in the Fabry outcome survey. J Med Genet. 2006;43:347–352.
- Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA. 1999;281:249–254.
- Poorthuis BJ, Wevers RA, Kleijer WJ, et al. The frequency of lysosomal storage diseases in The Netherlands. Hum Genet. 1999;105:151–156.
- Kotanko P, Kramar R, Devrnja D, et al. Results of a nationwide screening for Anderson-Fabry disease among dialysis patients. J Am Soc Nephrol. 2004;15:1323–1329.
- Okur I, Ezgu F, Biberoglu G, et al. Screening for Fabry disease in patients undergoing dialysis for chronic renal failure in Turkey: Identification of new case with novel mutation. Gene. 2013;527:42–47.
- Germain DP, Charrow J, Desnick RJ, et al. Ten-year outcome of enzyme replacement therapy with agalsidase beta in patients with Fabry disease. J Med Genet. 2015;52:353–358.
- Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney Int Suppl. 2013;3:1–150.
- Chamoles NA, Blanco M, Gaggioli D. Fabry disease: Enzymatic diagnosis in dried blood spots on filter paper. Clin Chim Acta. 2001;308:195–196.
- Nakao S, Takenaka T, Maeda M, et al. An atypical variant of Fabry's disease in men with left ventricular hypertrophy. N Engl J Med. 1995;333:288–293.
- Germain DP, Waldek S, Banikazemi M, et al. Sustained, long-term renal stabilization after 54 months of agalsidase beta therapy in patients with Fabry disease. J Am Soc Nephrol. 2007;18:1547–1557.
- Linthorst GE, Bouwman MG, Wijburg FA, Aerts JM, Poorthuis BJ, Hollak CE. Screening for Fabry disease in high-risk populations: A systematic review. J Med Genet. 2010;47:217–222.
- Saito O, Kusano E, Akimoto T, et al. Prevalence of Fabry disease in dialysis patients: Japan Fabry disease screening study (J-FAST). Clin Exp Nephrol. 2016;20:284–293.
- Andrade J, Waters PJ, Singh RS, et al. Screening for Fabry disease in patients with chronic kidney disease: Limitations of plasma α-galactosidase assay as a screening test. Clin J Am Soc Nephrol. 2008;3:139–145.
- Ceci R, Francesco P, Mucci J, Cancelarich L, Fossati C, Rozenfeld P. Reliability of enzyme assays in dried blood spots for diagnosis of 4 lysosomal storage disorders. Adv Biol Chem. 2011;1:58–64.
- Caudron E, Germain DP, Prognon P. Fabry disease: Enzymatic screening using dried blood spots on filter paper. Rev Med Interne. 2010;31:263–269.
- Terryn W, Cochat P, Froissart R, et al. Fabry nephropathy: Indications for screening and guidance for diagnosis and treatment by the European Renal Best Practice. Nephrol Dial Transplant. 2013;28:505–517.
- Kes P, Furic-Curko V, Basic-Jukic N. Anderson-Fabry disease in females. BANTAO J. 2014;12:20–26.
- Saito S, Ohno K, Sakuraba H. Fabry-database.org: Database of the clinical phenotypes, genotypes and mutant α-galactosidase A structures in Fabry disease. J Hum Genet. 2011;56:467–468.
- Fabry mutants list. Available at: http://fabry-database.org/mutants/. Accessed 26 December 2015.
- van der Tol L, Smid BE, Poorthuis BJ, et al. A systematic review on screening for Fabry disease: Prevalence of individuals with genetic variants of unknown significance. J Med Genet. 2014;51:1–9.
- Yasuda M, Shabbeer J, Benson SD, Maire I, Burnett RM, Desnick RJ. Fabry disease: Characterization of alpha-galactosidase A double mutations and the D313Y plasma enzyme pseudodeficiency allele. Hum Mutat. 2003;22:486–492.
- Niemann M, Rolfs A, Giese A, et al. Lyso-Gb3 indicates that the alpha-galactosidase A mutation D313Y is not clinically relevant for Fabry disease. JIMD Rep. 2013;7:99–102.
- Froissart R, Guffon N, Vanier MT, Desnick RJ, Maire I. Fabry Disease: D313Y is an alpha-galactosidase A sequence variant that causes pseudodeficient activity in plasma. Mol Genet Metab. 2003;80:307–314.
- Lukas J, Giese AK, Markoff A, et al. Functional characterisation of alpha-galactosidase a mutations as a basis for a new classification system in fabry disease. PLoS Genet. 2013;9:e1003632.
- Nance CS, Klein CJ, Banikazemi M, et al. Later-onset Fabry disease: An adult variant presenting with the cramp-fasciculation syndrome. Arch Neurol. 2006;63:453–457.
- Spada M, Pagliardini S, Yasuda M, et al. High incidence of later-onset Fabry disease revealed by newborn screening. Am J Hum Genet. 2006;79:31–40.
- Terryn W, Vanholder R, Hemelsoet D, et al. Questioning the pathogenic role of the GLA p.Ala143Thr mutation in Fabry disease: Implications for screening studies and ERT. JIMD Rep. 2012;8:101–108.