

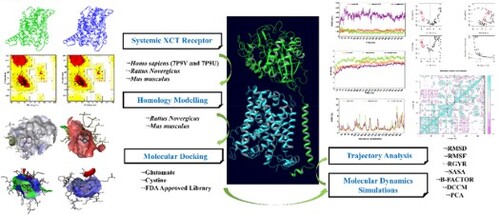
ABSTRACT
The heterodimeric amino acid transporter systemic XCT plays a crucial role in redox balance regulation by facilitating L-cystine and L-glutamate import/export in a 1:1 ratio. Translating findings from preclinical to clinical contexts poses challenges due to interspecies receptor variations. To understand these differences and achieve success during translating studies, in silico analysis of FDA-approved drugs was performed against three species for systemic XCT receptors with UniProt-ids D4ADU2 (Rattus norvegicus), Q9WTR6 (Mus musculus), and Q9UPY5 (Homo sapiens). Homology modelling produced validated 3D structures for D4ADU2 and Q9WTR6, achieving an excellent model quality score (∼84%). Virtual screening identified common hits in Rat and Mouse Homology Modelled Structures (RHMS and MHMS), including Avodart, Rolapitant, and Olmesartan. In Homo sapiens (PDBs 7P9U and 7P9 V), common hits Dihydroergotamine and Ergotamine showed binding affinities of −12.3 to −10.1 kcal/mol as compared to glutamate and cystine (−4.9 to −5.9 kcal/mol) within the same binding pocket. A 500 ns MD simulation confirmed structural stability, positive correlations and limited atomic displacements. Free energy calculations highlighted the top compounds highly favourable binding affinities across all receptors. These results shed light on structural and dynamical variations among the interspecies receptors-ligands interactions which provide valuable insights into transitioning from preclinical to clinical settings.
GRAPHICAL ABSTRACT
Disclosure statement
All the authors declare no conflict of interest.
Author’s contribution
NS – conducted the study, analyzed data, and wrote the manuscript, JS, AC – Analyzed the study, KLK, PKA – Guided, Conceptualised, Wrote, and revised the manuscript.
Data availability statement
The FASTA sequences and accession no. of SLC7A11-XCT wild types, i.e. Q9UPY5 (Homo sapiens), D4ADU2 (Rattus norvegicus), and Q9WTR6 (Mus musculus) were extracted from UniProt Database (https://www.uniprot.org/). The X-RAY-based 3D crystalline protein structure of PDB IDs 7P9U and 7P9V was retrieved from the RCSB Protein Data Bank (https://www.rcsb.org/). The FDA-approved ligand library set was extracted from the ZINC Database (https://zinc.docking.org/). The molecular docking and molecular dynamic simulation were conducted using AutoDock vina PyRx software & Schrodinger Maestro, respectively. MD trajectory analysis of DCCM and PCA was conducted utilising the VMD and Bio3d v.2.4-1.9000 package of R studio v1.4.1103 (http://thegrantlab.org/bio3d/). On request, the original data can be shared if needed.