
Abstract
All the stereoisomers of butanol type 1,7-seco-2,7′-cyclolignane were stereoselectively synthesized by employing (S)- and (R)-Evans’ auxiliaries to construct the stereochemistry. (+)- and (−)-Kadangustin J and their diastereomers were also prepared. The optical purity of the synthesized butanol type 1,7-seco-2,7′-cyclolignane was more than 99%ee.
Graphical Abstract
All the stereoisomers of butanol type 1,7-seco-2,7'-cyclolignane with more than 99%ee were stereoselectively synthesized. (+)- and (-)-Kadangustin J and their diastereomers were also prepared.
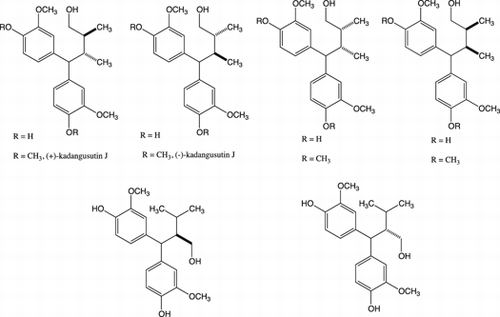
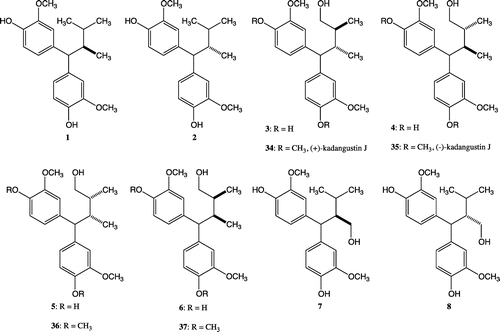
Lignans, which are biosynthesized as secondary metabolites in many plant families, are constructed from two phenylpropanoid units. The biological activity of many kinds of classical lignan that are bonded at the 8 and 8′ positions of phenyl propane has been clarified.Citation1) Neolignans with combinations other than the 8-8′ bond have also been reported.Citation2,Citation3) 1,7-Seco-2,7′-cyclolignane, in which the shift of the 7-aryl group to the 7′ position has been observed, are types of neolignans.Citation3) Some 1,7-seco-2,7′-cyclolignanes bearing a lactoneCitation4–Citation6) or butanolCitation7,Citation8) structure have been isolated and the cytotoxic activity and anti-HIV activity have been found. Stimulated by the unique lignan bearing diphenyl methyl structure and its biological activity, we turned our attention to synthesizing all the stereoisomers of butane type 1,7-seco-2,7′-cyclolignanes 1, 2 and butanol type 1,7-seco-2,7′-cyclolignanes 3–8, 34–37 (Fig. ). Although many kinds of biological activity have been discovered in the course of lignan and neolignan research, we have been unable to find information on the effect of stereochemistry on this biological activity in many cases. Our efforts to establish the relationship between the stereochemistry and biological activity of lignans and neolignans have been continuing.Citation9–Citation13) We began this synthetic research to develop a study on the effects of the stereochemistry of butane or butanol on their biological activity. The collation of stereoisomer libraries of natural products should contribute to the process for determination of the receptor. In our previous research, a synthetic route to compounds 1 and 2 was developed.Citation14) The syntheses of all the stereoisomers of butanol type 1,7-seco-2,7′-cyclolignanes 3–8 and kadangustin J 34–37 are described in this article. The success of this project would enable us to estimate the biological activity of all the stereoisomers of butanol type 1,7-seco-2,7′-cyclolignane containing kadangustin J.
Fig. 1. All the stereoisomers of butane-type 1,7-seco-2,7′-cyclolignane 1 and 2, Butanol-type 1,7-seco-2,7′-cyclolignane 3–8, and Kadangustin J 34–37.
Results and discussion
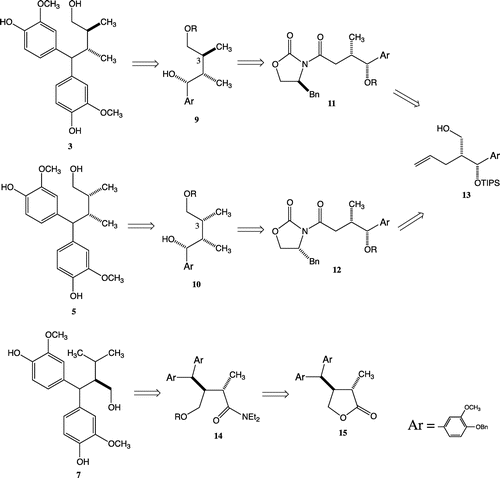
As illustrated in Scheme , the diphenylmethyl structures of target compounds 3 and 5 can be, respectively, obtained by the Friedel-Crafts reaction from benzyl alcohols 9 and 10. The stereochemistry at C-3 of benzyl alcohols 9 and 10 can be constructed by methylations to 11 and 12, respectively, bearing S- and R-Evans’ auxiliaries. Requisite intermediates 11 and 12 can be transformed from alcohol 13Citation15) by reductive elimination of the primary hydroxy group and oxidative cleavage of the olefin to carboxylic acid, introduction of the auxiliary. Enantiomers 4 and 6 can be obtained from the enantiomer of 13 by the same route. Positional isomer 7 can be converted from amide 14, which can be obtained from protected lactone type 1,7-seco-2,7′-cyclolignane 15, which had previously been synthesized in our laboratory.Citation14) Compound 8 can be obtained from the enantiomer of 15.
Scheme 1. Proposed general strategy for synthesis of butanol-type 1,7-seco-2,7′-cyclolignane.
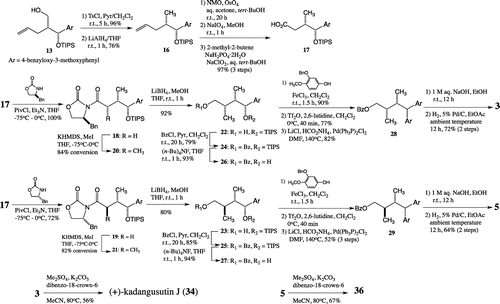
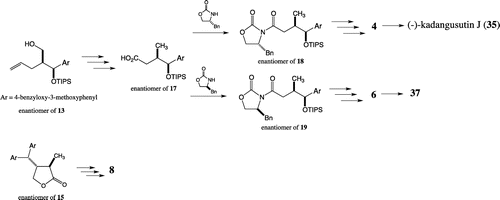
Syntheses of stereoisomers 3 and 5 were started with alcohol 13Citation15) (Scheme ). After the reduction of the hydroxy methyl group to the methyl group via a tosylate, resulting compound 16 was converted to carboxylic acid 17 by oxidative cleavage of the alkene which was, respectively, combined with (S)- and (R)-Evans’ auxiliaries to give 18 and 19. Stereoselective methylation followed by the removal of auxiliary gave primary alcohols 22 and 23, whose hydroxy groups were, respectively, protected as benzoyl esters 24 and 25. To obtain the substrates for the Friedel-Crafts reaction, the silyl ethers were cleaved, giving benzyl alcohols 26 and 27. The Friedel-Crafts reactions of 26 and 27 with 3-benzyloxy-4-methoxyphenol in the presence of FeCl3Citation16) gave diphenylmethyl compounds with diastereomeric ratios of 1:1. After conversion of the resulting phenolic hydroxy groups to triflates, removal of the resulting aromatic triflates was performed by using LiCl, HCO2NH4, and Pd(Ph3P)2Cl2Citation17), respectively, giving 28 and 29. Finally, deprotection of 28 and 29, respectively, gave stereoisomers 3 and 5. (+)-Kadangustin JCitation8) (34) and 36 were, respectively, obtained from 3 and 5 by selective methylation of the phenolic hydroxy groups using dimethyl sulfate and K2CO3.
Scheme 2. Synthesis of butanol-type 1,7-seco-2,7′-cyclolignane (I).
Protected lactone type 1,7-seco-2,7′-cyclolignane 15Citation14) was selected as a starting material for the synthesis of 7 (Scheme ). Ring opening of the lactone to amide 30 was accomplished by reaction with diethylamine in the presence of AlMe3, whose hydroxy group was protected as a silyl ether. After a two-steps reduction of the silyloxy amide 31 to the corresponding primary alcohol, mesylation followed by treatment with Super-hydride gave 32. The reduction of the mesylate was not successful by NaBH4 reduction in HMPA. Wolff–Kishner reduction of the aldehyde prepared from silyloxy amide 31 by DIBAL-H reduction gave 33 in a very low yield. Finally, deprotection of 33 afforded 7.
Scheme 3. Synthesis of butanol type 1,7-seco-2,7′-cyclolignane (II).
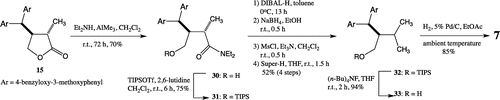
Enantiomers 4 and 6 were obtained from the enantiomer of 13 by applying the same method. Enantiomer 8 was obtained by the same synthetic method as that described for 7 from the enantiomer of 15 (Scheme ). The enantiomeric excess of all synthesized compounds was more than 99%. (−)-Kadangustin JCitation18) (35) and 37 were also, respectively, obtained from 4 and 6.
Scheme 4. Syntheses of the enantiomers.
Experimental setup
Optical rotations were measured on a Horiba SEPA-200 instrument. IR data were measured with a Horiba FT-720 instrument. NMR data were obtained using a JNM-EX400 spectrometer. EIMS data were measured with a JMS-MS700V spectrometer. IR data were measured with a Horiba FT-720 instrument. The silica gel used was Wakogel C-300 (Wako, 200–300 mesh). The purity of each compound containing enantiomers was confirmed by 1H NMR. HPLC analysis was performed with Shimadzu LC-6AD and SPD-6AV instruments. The chiral column used for HPLC analysis of enantiomeric excess was a 250 mm × 4.6 mm i.d., 5 μM, CHIRALCEL AD-H (DAICEL Chemical Industries, Ltd, Tokyo, Japan).
(4S,5R)-5-(4-Benzyloxy-3-methoxyphenyl)-4-methyl-5-(triisopropylsilyloxy)-1-pentene (16)
To an ice-cooled solution of alcohol 13 (16.5 g, 34.0 mmol) and pyridine (9.52 mL, 0.12 mol) in CH2Cl2 (80 mL) was added p-TsCl (12.5 g, 65.6 mmol). The reaction solution was stirred at room temperature for 5 h before additions of H2O and CH2Cl2. The organic solution was separated and washed with 1 M aq. HCl solution. After further washing with sat. aq. NaHCO3 solution, the organic solution was dried (Na2SO4). After concentration, the residue was applied to silica gel column chromatography (EtOAc/hexane = 1/9) to give unstable tosylate (20.3 g, 32.8 mmol, 96%) as a colorless oil; +24 (c 1.2, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 0.90–0.95 (21H, m), 1.59 (1H, m), 2.08 (1H, m), 2.20 (1H, m), 2.44 (3H, s), 3.85 (3H, s), 3.93 (1H, dd, J = 9.6, 8.3 Hz), 4.13 (1H, dd, J = 9.6, 4.8 Hz), 4.79 (1H, d, J = 6.1 Hz), 4.84 (1H, d, J = 17.5 Hz), 4.91 (1H, d, J = 10.0 Hz), 5.12 (2H, s), 5.55 (1H, m), 6.62 (1H, d, J = 8.2, 2.0 Hz), 6.75 (1H, d, J = 8.2 Hz), 6.86 (1H, d, J = 2.0 Hz), 7.26–7.38 (5H, m), 7.43 (2H, d, J = 8.2 Hz), 7.77 (2H, d, J = 8.2 Hz); 13C NMR (100 MHz, CDCl3) δ: 12.4, 17.95, 18.02, 21.6, 30.1, 46.3, 55.9, 69.6, 71.1, 73.6, 110.7, 113.4, 117.1, 119.4, 127.4, 127.8, 128.0, 128.5, 129.8, 133.1, 134.9, 135.6, 137.1, 144.7, 147.5, 149.4, 158.0. A solution of tosylate (20.3 g, 31.8 mmol) in THF (20 mL) was added to an ice-cooled suspension of LiAlH4 (1.21 g, 31.9 mmol) in THF (10 mL). The reaction mixture was stirred at room temperature for 1 h before additions of sat. aq. MgSO4 solution (5 mL) and K2CO3. The mixture was filtered, and then the filtrate was concentrated. The residue was applied to silica gel column chromatography (EtOAc/hexane = 1/9) to give 16 (11.3 g, 24.1 mmol, 76%) as a colorless oil;
+40 (c 0.3, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 0.87 (3H, d, J = 6.8 Hz, CH3), 0.89–1.00 (21H, m), 1.57 (1H, m, 4-H), 1.83 (1H, m, 3-H), 2.32 (1H, m, 3-H), 3.87 (3H, s, CH3), 4.59 (1H, d, J = 5.2 Hz, 5-H), 4.94 (1H d, J = 16.7 Hz, 1-H), 4.95 (1H, d, J = 16.5 Hz, 1-H), 5.12 (2H, s, OCH2Bn), 5.72 (1H, m, 2-H), 6.69 (1H, dd, J = 8.7, 1.8 Hz, ArH), 6.79 (1H, d, J = 8.7 Hz, ArH), 6.91 (1H, d, J = 1.8 Hz, ArH), 7.25–7.37 (3H, m, ArH), 7.43–7.45 (2H, m, ArH); 13C NMR (100 MHz, CDCl3) δ: 12.5, 15.4, 18.05, 18.12, 36.2, 41.8, 55.9, 71.2, 78.8, 111.0, 113.20, 113.23, 115.5, 119.2, 119.4, 127.4, 127.8, 128.5, 136.8, 137.4, 138.0, 147.1, 149.1; IR (CHCl3) cm−1: 2945, 2868, 1510, 1463, 1261, 1090. MS (EI) m/z: 468 (M+, 3), 399 (100). Anal. Calcd for C29H44O3Si: C, 74.31; H, 9.48%. Found: C, 74.34; H, 9.50%. (4R,5S)-(16). 81% yield,
−40 (c 1.1, CHCl3).
(3S,4R)-4-(4-Benzyloxy-3-methoxyphenyl)-3-methyl-4-(triisopropylsilyloxy)butanoic acid (17)
A reaction solution of 16 (9.94 g, 21.2 mmol), 4-methylmorpholine N-oxide (2.95 g, 25.2 mmol), and OsO4 (2% aq. solution, 1.60 mL) in acetone (90 mL), tert-BuOH (20 mL), and H2O (20 mL) was stirred at room temperature for 20 h before the addition of sat. aq. Na2S2O3 solution. After concentration of the mixture, the residue was dissolved in EtOAc and H2O. The organic solution was separated, washed with brine, and dried (Na2SO4). Concentration gave crude glycol. A reaction mixture of crude glycol and NaIO4 (5.40 g, 25.2 mmol) in MeOH (200 mL) was stirred at room temperature for 1 h before concentration to give crude aldehyde. To an ice-cooled solution of crude aldehyde, 2-methyl-2-butene (8.90 mL, 84.0 mmol), and NaH2PO4·2H2O (32.8 g, 0.21 mol) in tert-BuOH/H2O (3/1, 100 mL) was added a solution of NaClO2 (11.4 g, 0.13 mol) in tert-BuOH/H2O (3/1, 50 mL). The reaction mixture was stirred at room temperature for 1 h before additions of sat. aq. NH4Cl solution and CHCl3. The organic solution was separated, washed with H2O, and dried (Na2SO4). Concentration followed by silica gel column chromatography (EtOAc/hexane = 1/5) gave carboxylic acid 17 (10.0 g, 20.5 mmol, 97%) as a colorless oil; +34 (c 0.7, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 0.92 (3H, d, J = 6.9 Hz, CH3), 0.95–1.01 (21H, m, TIPSi), 1.90 (1H, dd, J = 15.3, 9.1 Hz, 2-H), 2.41 (1H, m, 3-H), 2.61 (1H, dd, J = 15.3, 4.8 Hz, 2-H), 3.88 (3H, s, OCH3), 4.68 (1H d, J = 4.8 Hz, 4-H), 5.14 (2H, s, OCH2Bn), 6.69 (1H, d, J = 7.2 Hz, ArH), 6.80 (1H, d, J = 7.2 Hz, ArH), 6.93 (1H, s, ArH), 7.26–7.38 (3H, m, ArH), 7.43–7.45 (2H, m, ArH), 7.26–7.45 (1H, overlapped, CO2H); 13C NMR (100 MHz, CDCl3) δ: 12.4, 16.3, 18.0, 18.1, 36.3, 38.4, 55.9, 71.1, 78.0, 110.9, 113.2, 119.4, 127.4, 127.8, 128.5, 135.2, 137.2, 147.3, 149.1, 179.8; IR (CHCl3) cm−1: 3500, 2945, 1707, 1508, 1464, 1261, 1093. MS (EI) m/z: 486 (M+, 6), 399 (100). Anal. Calcd. for C28H42O5Si: C, 69.10; H, 8.70%. Found: C, 69.22; H, 8.78%. (2R,3S)-(17). 87% yield,
−34 (c 2.1, CHCl3).
(S)-4-Benzyl-3-[(3S,4R)-4-(4-benzyloxy-3-methoxyphenyl)-3-methyl-4-(triisopropylsilyloxy)butanoyl]-2-oxazolidinone (18)
To a solution of carboxylic acid 17 (4.66 g, 9.57 mmol) in THF (100 mL) were added Et3 N (1.34 mL, 9.61 mmol) and PivCl (1.18 mL, 9.58 mmol) added at −75 °C. After the mixture was stirred at 0 °C for 1 h, lithium salt of (S)-4-benzyl-2-oxazolidinone, which was prepared from (S)-4-benzyl-2-oxazolizinone (1.70 g, 9.59 mmol) and n-BuLi (3.70 mL, 2.70 M in hexane, 9.99 mmol) in THF (80 mL) at −75 °C, was added at −75 °C. The reaction mixture was stirred at 0 °C for 1 h before the addition of sat. aq. NH4Cl solution. The organic solution was separated, washed with brine, and dried (Na2SO4). Concentration followed by silica gel column chromatography (EtOAc/hexane = 1/5) gave alkanoly oxazolidinone 18 (6.18 g, 9.57 mmol, 100%) as a colorless oil; +33 (c 1.9, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 0.95–1.01 (24H, m, TIPSi, CH3), 2.53 (1H, m, CHCH3), 2.61–2.72 (2H, m, C = OCH2), 3.21–3.26 (2H, m, CH2Bn), 3.91 (3H, s, OCH3), 4.08 (2H, d, J = 5.1 Hz, 5-CH2), 4.54 (1H, m, 4-H), 4.67 (1H, d, J = 5.4 Hz, TIPSOCHAr), 5.12 (2H, s, OCH2Bn), 6.72 (1H, d, J = 8.2 Hz, ArH), 6.80 (1H, d, J = 8.2 Hz, ArH), 6.98 (1H, s, ArH), 7.16–7.18 (2H, m, ArH), 7.28–7.36 (6H, m, ArH), 7.42–7.43 (2H, m, ArH); 13C NMR (100 MHz, CDCl3) δ: 12.5, 16.5, 18.0, 18.1, 37.77, 37.83, 38.0, 55.1, 55.8, 66.0, 71.1, 78.4, 110.8, 113.0, 119.6, 127.3, 127.4, 127.8, 128.5, 128.9, 129.4, 135.4, 136.1, 137.2, 147.2, 149.2, 153.3, 172.8; IR (CHCl3) cm−1: 2945, 1783, 1699, 1508, 1456, 1384, 1261,1090. MS (EI) m/z: 645 (M+, 7), 602 (100); HRMS (M+) m/z calcd for C38H51O6NSi: 645.3485, found: 645.3480. (4R)-3-[(3R,4S)]-(18). 97% yield,
−34 (c 1.6, CHCl3).
(4S)-4-Benzyl-3-[(2S,3S,4R)-4-(4-benzyloxy-3-methoxyphenyl)-2,3-dimethyl-4-(triisopropylsilyloxy)butanoyl]-2-oxazolidinone (20)
To a solution of KHMDS (14.4 mL, 0.50 M in toluene, 7.20 mmol) in THF (20 mL) was added a solution of 18 (3.10 g, 4.80 mmol) in THF (10 mL) at −75 °C. After the mixture was stirred at −75 °C for 20 min, MeI (12.0 mL, 0.19 mmol) was added. The reaction solution was stirred at −70 °C for 1 h, and then warmed to −15 °C. After 1.5 h at −15 °C, sat. aq. NH4Cl solution and EtOAc were added. The organic solution was separated, washed with brine, and dried (Na2SO4). Concentration followed by silica gel column chromatography (EtOAc/hexane = 1/8) gave 20 (1.00 g, 1.52 mmol, 32%) as a colorless oil along with recovered 18 (1.92 g, 2.97 mmol, 62%); +46 (c 1.5, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 0.91–1.05 (21H, m, TIPSi), 1.08 (3H, d, J = 6.9 Hz, CH3), 1.09 (3H, d, J = 6.8 Hz, CH3), 2.51 (1H, m, C = OCHCHCH3), 2.63 (1H, dd, J = 13.3, 9.5 Hz, CH2Bn), 3.08 (1H, dd, J = 13.3, 2.0 Hz, CH2Bn), 3.63 (1H, m, C = OCHCH3), 3.87 (1H, d, J = 8.7 Hz, 5-H), 3.90 (3H, s, OCH3), 3.98 (1H, d, J = 8.7 Hz, OCH3), 4.20 (1H, m, 4-H), 4.47 (1H, d, J = 6.7 Hz, ArCHOTIPS), 5.09 (2H, s, OCH2Bn), 6.76 (2H, s, ArH), 6.95 (1H, s, ArH), 7.13–7.17 (2H, m, ArH), 7.26–7.31 (6H, m, ArH), 7.37–7.38 (2H, m, ArH); 13C NMR (100 MHz, CDCl3) δ: 12.7, 13.3, 15.5, 18.0, 18.1, 37.8, 38.5, 42.4, 55.3, 55.8, 65.7, 71.1, 79.7, 111.6, 113.0, 120.3, 127.2, 127.4, 127.9, 128.5, 128.9, 129.4, 135.4, 136.5, 137.1, 147.1, 148.9, 152.6, 177.0; IR (CHCl3) cm−1: 2945, 1781, 1700, 1508, 1466, 1382, 1093. MS (EI) m/z: 659 (M+, 7), 617 (100); HRMS (EI) m/z calcd for C39H53O6NSi: 659.3642, found: 659.3632. (4R)-3-[(2R,3R,4S)]-(20). 33% yield,
−46 (c 1.1, CHCl3).
(2S,3S,4R)-4-(4-Benzyloxy-3-methoxyphenyl)-2,3-dimethyl-4-(triisopropylsilyloxy)-1-butanol (22)
To an ice-cooled solution of LiBH4 (0.32 g, 14.7 mmol) and MeOH (0.65 mL, 16.0 mmol) in THF (50 mL) was added a solution of alkanoyl oxazolidinone 20 (2.20 g, 3.33 mmol) in THF (20 mL). The reaction solution was stirred at room temperature for 1 h, and then sat. aq. NH4Cl solution was added. After concentration, the residue was dissolved in EtOAc and H2O. The organic solution was separated, washed with brine, and dried (Na2SO4). Concentration followed by silica gel column chromatography (EtOAc/hexane = 1/3) gave alcohol 22 (1.50 g, 3.08 mmol, 92%) as a colorless oil; +41 (c 1.3, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 0.68 (3H, d, J = 6.8 Hz, CH3), 0.93–0.99 (21H, m, TIPSi), 0.97 (3H, d, J = 6.1 Hz, CH3), 1.47 (1H, m, 3-H), 1.57 (1H, br. s, OH), 1.96 (1H, m, 2-H), 3.40 (2H, d, J = 6.7 Hz, 1-H), 3.86 (3H, s, OCH3), 4.53 (1H, d, J = 7.4 Hz, 4-H), 5.12 (2H, s, OCH2Bn), 6.72 (1H, d, J = 8.1 Hz, ArH), 6.78 (1H, d, J = 8.1 Hz, ArH), 6.95 (1H, s, ArH), 7.27–7.37 (3H, m, ArH), 7.42–7.44 (2H, m, ArH); 13C NMR (100 MHz, CDCl3) δ: 11.60, 11.63, 12.5, 18.0, 18.1, 35.3, 42.3, 56.0, 67.5, 71.1, 78.8, 110.7, 113.3, 119.6, 127.4, 127.8, 128.5, 137.2, 137.3, 147.3, 149.5; IR (CHCl3) cm−1: 3566, 2945, 1505, 1452, 1259, 1139, 1059, 1025. MS (EI) m/z: 486 (M+, 6), 400 (100); HRMS (EI) m/z calcd for C29H46O4Si: 486.3166, found: 486.3167. (2R,3R,4S)-(22). 95% yield,
−41 (c 1.2, CHCl3).
(2S,3S,4R)-4-(4-Benzyloxy-3-methoxyphenyl)-2,3-dimethyl-4-(triisopropylsilyloxy)butyl benzoate (24)
To an ice-cooled solution of alcohol 22 (1.50 g, 3.08 mmol) and pyridine (0.49 mL, 6.06 mmol) in CH2Cl2 (5 mL) was added BzCl (0.54 mL, 4.65 mmol). The reaction mixture was stirred at room temperature for 20 h before additions of H2O and CH2Cl2. The organic solution was separated and washed with 1 M aq. HCl solution. After further washing with sat. aq. NaHCO3 solution, the organic solution was dried (Na2SO4). Concentration followed by silica gel column chromatography (EtOAc/hexane = 1/8) gave benzoate 24 (1.43 g, 2.42 mmol, 79%) as a colorless oil; +69 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 0.80 (3H, d, J = 7.0 Hz, CH3), 0.92–0.99 (21H, m, TIPSi), 1.05 (3H, d, J = 6.9 Hz, CH3), 1.82 (1H, m, 3-H), 2.01 (1H, m, 2-H), 3.80 (3H, s, OCH3), 4.08 (1H, dd, J = 10.7, 6.3 Hz, 1-H), 4.17 (1H, dd, J = 10.7, 7.5 Hz, 1-H), 4.55 (1H, d, J = 7.8 Hz, 4-H), 5.13 (2H, s, OCH2Bn), 6.72 (1H, dd, J = 8.2, 2.0 Hz, ArH), 6.78 (1H, d, J = 8.2 Hz, ArH), 6.92 (1H, d, J = 2.0 Hz, ArH), 7.25–7.56 (7H, m, ArH), 7.54 (1H, m, ArH), 7.96–7.97 (2H, m, ArH); 13C NMR (100 MHz, CDCl3) δ: 11.1, 11.5, 12.6, 18.0, 18.2, 32.1, 42.5, 55.8, 68.7, 71.1, 78.3, 110.4, 113.3, 119.5, 127.4, 127.8, 128.3, 128.5, 129.5, 130.4, 132.8, 137.2, 137.6, 147.4, 149.6, 166.5; IR (CHCl3) cm−1: 2945, 1711, 1504, 1460, 1279, 1137, 1116, 1063. MS (EI) m/z: 590 (M+, 13), 547 (100); HRMS (EI) m/z calcd for C36H50O5Si: 590.3428, found: 590.3412. (2R,3R,4S)-(24). 80% yield,
−69 (c 1.4, CHCl3).
(2S,3S,4R)-4-(4-Benzyloxy-3-methoxyphenyl)-4-hydroxy-2,3-dimethylbutyl benzoate (26)
A reaction solution of silyl ether 24 (1.43 g, 2.42 mmol) and (n-Bu)4NF (3.60 mmol, 1 M in THF, 3.60 mmol) in THF (10 mL) was stirred at room temperature for 1 h before the addition of sat. aq. NH4Cl solution and EtOAc. The organic solution was separated and washed with sat. aq. CuSO4 solution. After further washing with sat. aq. NaHCO3 solution followed by brine, the organic solution was dried (Na2SO4). Concentration followed by silica gel column chromatography (EtOAc/hexane = 1/2) gave alcohol 26 (0.98 g, 2.26 mmol, 93%) as a colorless oil; +60 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 0.92 (3H, d, J = 7.0 Hz, CH3), 1.02 (3H, d, J = 6.8 Hz, CH3), 1.85 (1H, m, 3-H), 1.92–2.12 (1H, br., OH), 2.04 (1H, m, 2-H), 3.79 (3H, s, OCH3), 4.17–4.23 (2H, m, 1-H2), 4.55 (1H, d, J = 7.4 Hz, 5-H), 5.13 (2H, s, OCH2Bn), 6.76 (1H, d, J = 8.1 Hz, ArH), 6.81 (1H, d, J = 8.1 Hz, ArH), 6.88 (1H, s, ArH), 7.25–7.44 (7H, m, ArH), 7.55 (1H, m, ArH), 7.99–8.00 (2H, m, ArH); 13C NMR (100 MHz, CDCl3) δ: 10.0, 12.1, 33.5, 40.9, 55.9, 68.1, 71.1, 76.7, 109.6, 113.6, 118.8, 127.3, 127.8, 128.4, 128.5, 129.5, 130.3, 132.9, 137.0, 137.2, 147.6, 149.8, 166.5; IR (CHCl3) cm−1: 3619, 1976, 1716, 1520, 1452, 1277, 1138, 1026. MS (EI) m/z: 434 (M+, 25), 243 (100); HRMS (EI) m/z calcd for C27H30O5: 434.2093, found: 434.2094. (2R,3R,4S)-(26). 93% yield,
−60 (c 1.0, CHCl3).
(2S,3R)-4,4-Bis(4-benzyloxy-3-methoxyphenyl)-2,3-dimethylbutyl benzoate (28)
A reaction mixture of 3-benzyloxy-4-methoxyphenol (0.42 g, 1.82 mmol), benzyl alcohol 26 (0.71 g, 1.63 mmol), and FeCl3 (13 mg, 0.080 mmol) in CH2Cl2 (30 mL) was stirred at room temperature for 1.5 h before the addition of sat. aq. NaHCO3 solution. The organic solution was separated and dried (Na2SO4). Concentration followed by silica gel column chromatography (EtOAc/hexane = 1/5) gave bisphenol compound (0.95 g, 1.47 mmol, 90%) as a diastereomeric mixture of 1/1; HRMS (EI) m/z calcd for C41H42O7: 646.2931, found: 646.2911. To a solution of bisphenol compound (0.78 g, 1.21 mmol) and 2,6-lutidine (0.48 mL, 4.11 mmol) in CH2Cl2 (5 mL), Tf2O (0.27 mL, 1.60 mmol) was added at 0 °C. After stirring at 0 °C for 40 min, H2O and CH2Cl2 were added. The organic solution was separated and washed with sat. aq. CuCO4 solution. After further washing with sat. aq. NaHCO3 solution, the organic solution was dried (Na2SO4). Concentration followed by silica gel column chromatography (5% EtOH in EtOAc/hexane = 1/3) gave triflate (0.97 g, 1.25 mmol, 77%) as a diastereomeric mixture of 1/1. The diastereomeric isomers could be partially separated; Rf = 0.24, +31 (c 0.4, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 0.77 (3H, d, J = 6.6 Hz), 0.92 (3H, d, J = 6.8 Hz), 1.26 (1H, m), 2.12 (1H, m), 3.80 (3H, s), 3.81 (3H, s), 4.10 (1H, d, J = 11.4 Hz), 4.13 (1H, dd, J = 10.7, 6.6 Hz), 4.24 (1H, dd, J = 10.7, 7.9 Hz), 5.04 (1H, d, J = 12.3 Hz), 5.09 (2H, s), 5.10 (1H, d, J = 12.3 Hz), 6.72 (1H, s), 6.78 (1H, d, J = 8.4 Hz), 6.80 (1H, d, J = 8.4 Hz), 6.86 (1H, s), 6.87 (1H, s), 7.20–7.46 (11H, m), 7.58 (1H, m), 7.65 (1H, m), 8.03–8.05 (2H, m); 13C NMR (100 MHz, CDCl3) δ: 9.70, 11.3, 24.4, 32.8, 55.9, 56.3, 58.3, 68.7, 71.1, 71.5, 107.3, 112.4, 114.2, 119.4, 119.8 (q, J = 278.7 Hz), 122.9, 124.1, 127.3, 127.4, 127.8, 128.3, 128.4, 128.46, 128.52, 128.7, 129.5, 129.8, 130.4, 133.0, 134.9, 136.0, 137.2, 137.3, 140.0, 144.2, 147.0, 147.1, 149.6, 149.7, 166.5; MS (EI) m/z: 778 (M+, 18), 587 (100); HRMS (EI) m/z calcd for C42H41O9F3S: 778.2423, found: 778.2431. Rf = 0.29,
+21 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 0.81 (3H, d, J = 6.5 Hz), 0.94 (3H, d, J = 6.8 Hz), 2.07 (1H, m), 2.59 (1H, m), 3.72 (3H, s), 3.87 (3H, s), 4.01 (1H, d, J = 11.9 Hz), 4.18 (1H, dd, J = 10.8, 6.5 Hz), 4.26 (1H, dd, J = 10.8, 6.3 Hz), 5.06 (1H, d, J = 12.2. Hz), 5.10 (1H, d, J = 12.2 Hz), 5.11 (2H, s), 6.73 (1H, s), 6.80 (1H, d, J = 8.4 Hz), 6.82 (1H, d, J = 8.4 Hz), 6.88 (1H, s), 6.98 (1H, s), 7.29–7.50 (12H, m), 7.60 (1H, m), 8.04–8.06 (2H, m); 13C NMR (100 MHz, CDCl3) δ: 10.0, 11.9, 33.1, 36.7, 47.4, 55.9, 56.2, 68.5, 71.1, 71.5, 107.4, 110.6, 112.9, 114.1, 119.3, 119.7 (q, J = 74.4 Hz), 127.3, 127.4, 127.8, 128.2, 128.46, 128.50, 128.7, 129.48, 129.51, 133.1, 135.4, 136.0, 137.3, 140.4, 146.8, 149.4, 149.5, 149.6, 166.4; MS (EI) m/z: 778 (M+, 43), 587 (100); HRMS (EI) m/z calcd for C42H41O9F3S: 778.2424, found: 778.2410. A reaction mixture of triflate (0.26 g, 0.33 mmol), LiCl (0.29 g, 6.84 mmol), HCO2NH4 (1.71 g, 27.1 mmol), and Pd(Ph3P)2Cl2 (0.13 g, 0.19 mmol) in DMF (2.3 mL) was stirred at 140 °C under N2 gas for 20 h. After additions of H2O and EtOAc, the organic solution was separated, washed with brine, and dried (Na2SO4). Concentration followed by silica gel column chromatography (EtOAc/hexane = 1/5) gave bisphenyl compound 28 (0.17 g, 0.27 mmol, 82%) as a colorless oil;
+38 (c 0.1, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 0.75 (3H, d, J = 6.6 Hz, CH3), 0.87 (3H, d, J = 6.9 Hz, CH3), 2.08 (1H, m, 3-H), 2.55 (1H, m, 2-H), 3.53 (1H, d, J = 11.7 Hz, 4-H), 3.79 (3H, s, OCH3), 3.85 (3H, s, OCH3), 4.15 (1H, dd, J = 10.7, 6.6 Hz, 1-H), 4.22 (1H, dd, J = 10.7, 8.0 Hz, 1-H), 5.07 (2H, s, OCH2Bn), 5.08 (2H, s, OCH2Bn), 6.75–6.81 (6H, m, ArH), 7.26–7.47 (12H, m, ArH), 7.57 (1H, m, ArH), 8.03–8.05 (2H, m, ArH); 13C NMR (100 MHz, CDCl3) δ: 9.9, 12.1, 32.9, 37.0, 55.8, 56.0, 56.1, 68.8, 71.1, 111.8, 112.1, 114.2, 119.7, 127.3, 127.8, 128.4, 128.5, 128.6, 129.48, 129.52, 130.5, 132.9, 137.4, 137.9, 146.7, 146.8, 149.5, 149.7, 166.5; IR (CHCl3) cm−1: 3012, 2960, 1714, 1510, 1277. MS (EI) m/z: 630 (M+, 5), 204 (100); HRMS (EI) m/z calcd for C41H42O6: 630.2981, found: 630.3001. (2R,3S)-(28). 61% yield, two steps,
−37 (c 0.6, CHCl3).
(2S,3R)-4,4-Bis(4-hydroxy-3-methoxyphenyl)-2,3-dimethyl-1-butanol (3)
A reaction solution of benzoate 28 (0.23 g, 0.36 mmol) in EtOH (15 mL) and 1 M aq. NaOH solution (15 mL) was stirred at room temperature for 12 h before additions of H2O and CHCl3. The organic solution was separated and dried (Na2SO4). Concentration gave crude alcohol. A reaction mixture of crude alcohol and 5% Pd/C (0.20 g) in EtOAc (5 mL) was stirred under H2 gas at ambient temperature for 12 h before filtration. The filtrate was concentrated, and then the residue was applied to silica gel column chromatography (EtOAc/hexane = 1/1) to give 3 (90 mg, 0.26 mmol, 72%) as a colorless oil; +10 (c 0.7, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 0.68 (3H, d, J = 6.8 Hz, CH3), 0.74 (3H, d, J = 6.9 Hz, CH3), 1.24 (1H, br. s, 1-OH), 1.75 (1H, m, 3-H), 2.58 (1H, m, 2-H), 3.45 (1H, dd, J = 10.7, 6.6 Hz, 1-H), 3.49 (1H, d, J = 11.8 Hz, 4-H), 3.49 (1H, dd, J = 10.7, 8.3 Hz, 1-H), 3.83 (3H, s, OCH3), 3.84 (3H, s, OCH3), 5.56 (1H, s, OH), 5.57 (1H, s, OH), 6.76 (1H, s, ArH), 6.78 (1H, d, J = 1.3 Hz, ArH), 6.80–6.82 (4H, m, ArH); 13C NMR (100 MHz, CDCl3) δ: 9.6, 11.9, 35.98, 36.04, 55.9, 56.1, 67.0, 110.5, 114.3, 114.5, 120.3, 120.4, 136.7, 137.2, 143.8, 146.5, 146.6; IR (CHCl3) cm−1 3630, 3017, 2974, 1512, 1269, 1219, 1036. MS (EI) m/z: 346 (M+, 59), 260 (100), 229 (70); HRMS (EI) m/z calcd for C20H26O5: 346.1780, found: 346.1780. >99%ee (AD-H, iso-PrOH/hexane = 1.7/1, 1 mL/min, 280 nm, tR6.6 min). (2R,3S)-(4). 71% yield,
−10 (c 1.0, CHCl3),>99%ee (AD-H, iso-PrOH/hexane = 1.7/1, 1 mL/min, 280 nm, tR9.4 min).
(4R)-4-Benzyl-3-[(3S,4R)-4-(4-benzyloxy-3-methoxyphenyl)-3-methyl-4-(triisopropylsilyloxy)butanoyl]-2-oxazolidinone (19)
The title compound was obtained from 17 by the same synthetic method as that of compound 18 by using (R)-4-benzyloxazolidinone in 72% yield as a colorless oil; −7 (c 1, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 0.93 (3H, d, J = 6.7 Hz, CH3), 0.97–1.02 (21H, m, TIPSi), 2.50 (1H, m, CHCH3), 2.61–2.74 (2H, m, C = OCH2), 3.24 (2H, d, J = 14.0 Hz, CH2Bn), 3.91 (3H, s, OCH3), 4.08–4.15 (2H, m, 5-H), 4.62 (1H, m, 4-H), 4.73 (1H, d, J = 4.9 Hz, ArCHOTIPS), 5.12 (2H, s, OCH2Bn), 6.73 (1H, d, J = 8.2 Hz, ArH), 6.80 (1H, d, J = 8.2 Hz, ArH), 7.01 (1H, s, ArH), 7.19–7.25 (2H, m, ArH), 7.27–7.36 (6H, m, ArH), 7.42–7.43 (2H, m, ArH); 13C NMR (100 MHz, CDCl3) δ: 12.5, 16.0, 18.05, 18.12, 37.6, 37.9, 38.1, 55.2, 55.9, 66.1, 71.1, 78.2, 110.9, 113.1, 119.4, 127.3, 127.4, 127.7, 128.5, 128.9, 129.4, 135.4, 136.0, 137.3, 147.2, 149.2, 153.3, 172.8; IR (CHCl3) cm−1: 2940, 1782, 1698, 1508, 1456, 1384, 1261,1090. MS (EI) m/z: 645 (M+, 4), 399 (100), 290 (57); HRMS (M+) m/z calcd for C38H51O6NSi: 645.3485, found: 645.3456. (4S)-3-[(3R,4S)]-(19). 75% yield,
+7 (c 1, CHCl3).
(4R)-4-Benzyl-3-[(2R,3S,4R)-4-(4-benzyloxy-3-methoxyphenyl)-4-(triisopropylsilyloxy)-2,3-dimethylbutanoyl]-2-oxazolidinone (21)
The title compound was obtained from 19 by the same synthetic method as that of compound 20 in 32% yield as a colorless oil along with 61% recovery of 19; −10 (c 1.3, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 0.98–1.01 (21H, m, TIPSi), 1.08 (3H, d, J = 6.9 Hz, CH3), 1.31 (3H, d, J = 7.0 Hz, CH3), 2.08 (1H, m, C = OCHCHCH3), 2.67 (1H, dd, J = 13.3, 9.6 Hz, CH2Bn), 3.14 (1H, dd, J = 13.3, 2.8 Hz, CH2Bn), 3.82 (1H, dd, J = 8.7, 7.9 Hz, 5-H), 3.87 (3H, s, OCH3), 3.95 (1H, dd, J = 8.7, 7.9 Hz, 5-H), 4.02 (1H, m, C = OCH), 4.32 (1H, m, 4-H), 4.86 (1H, d, J = 4.3 Hz, ArCHOTIPS), 5.10 (2H, s, OCH2Bn), 6.71 (1H, dd, J = 8.2, 1.4 Hz, ArH), 6.77 (1H, d, J = 8.2 Hz, ArH), 6.93 (1H d, J = 1.4 Hz, ArH), 7.15–7.17 (2H, m, ArH), 7.25–7.32 (6H, m, ArH), 7.38–7.40 (2H, m, ArH); 13C NMR (100 MHz, CDCl3) δ: 12.7, 13.2, 16.3, 18.1, 37.8, 38.8, 45.5, 55.3, 56.0, 65.8, 71.2, 75.9, 111.0, 113.4, 119.2, 127.3, 127.8, 128.5, 128.9, 129.4, 135.4, 137.3, 147.2, 149.2, 152.7, 176.5; IR (CHCl3) cm−1: 2944, 1780, 1701, 1508, 1466, 1382, 1090. MS (EI) m/z: 659 (M+, 4), 400 (100), 290 (57); HRMS (EI) m/z calcd for C39H53O6NSi: 659.3642, found: 659.3638. (4S)-3-[(2S,3R,4S)]-(21). 43% yield,
+11 (c 0.9, CHCl3).
(2R,3S,4R)-4-(4-Benzyloxy-3-methoxyphenyl)-2,3-dimethyl-4-(triisopropylsilyloxy)-1-butanol (23)
The title compound was obtained from 21 by the same synthetic method as that of compound 22 in 80% yield as a colorless oil; +40 (c 1.4, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 0.94–0.99 (27H, m, TIPSi, CH3 × 2), 1.29 (1H, m, 3-H), 1.61 (1H, br. s, OH), 1.71 (1H, m, 2-H), 3.29 (1H, dd, J = 10.4, 7.3 Hz, 1-H), 3.48 (1H, dd, J = 10.4, 5.9 Hz, 1-H), 3.87 (3H, s, OCH3), 4.74 (1H, d, J = 6.2 Hz, 4-H), 5.12 (2H, s, OCH2Bn), 6.73 (1H, dd, J = 8.1, 1.4 Hz, ArH), 6.79 (1H, d, J = 8.1 Hz, ArH), 6.94 (1H, d, J = 1.4 Hz, ArH), 7.27–7.37 (3H, m, ArH), 7.42–7.44 (2H, m, ArH); 13C NMR (100 MHz, CDCl3) δ: 11.5, 12.7, 16.7, 18.1, 18.2, 36.1, 45.9, 56.0, 65.4, 71.2, 77.9, 110.7, 113.5, 119.3, 127.4, 127.8, 128.5, 137.3, 138.1, 147.2, 149.4; IR (CHCl3) cm−1: 3560, 2945, 1505, 1452, 1258, 1140, 1059, 1025. MS (EI) m/z: 486 (M+, 9), 400 (100); Anal. Calcd for C29H46O4Si: C, 71.56; H, 9.53%. Found: C, 71.43; H, 9.59%. (2S,3R,4S)-(23). 74% yield,
−40 (c 0.9, CHCl3).
(2R,3S,4R)-4-(4-Benzyloxy-3-methoxyphenyl)-2,3-dimethyl-4-(triisopropylsilyloxy)butyl benzoate (25)
The title compound was obtained from 23 by the same synthetic method as that of compound 24 in 85% yield as a colorless oil; +3 (c 1.4, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 0.94–0.99 (21H, m. TIPSi), 1.04 (3H, d, J = 6.8 Hz, CH3), 1.08 (3H, d, J = 7.0 Hz, CH3), 1.79 (1H, m, 3-H), 1.87 (1H, m, 2-H), 3.83 (3H, s, OCH3), 4.06 (1H, dd, J = 10.9, 7.5 Hz, 1-H), 4.22 (1H, dd, J = 10.9, 4.9 Hz, 1-H), 4.74 (1H, d, J = 6.8 Hz, 4-H), 5.11 (2H, s, OCH2Bn), 6.75 (1H, dd, J = 8.2, 1.4 Hz, ArH), 6.80 (1H, d, J = 8.2 Hz, ArH), 6.94 (1H, d, J = 1.4 Hz, ArH), 7.25–7.36 (3H, m, ArH), 7.40–7.44 (4H, m, ArH), 7.54 (1H, m, ArH), 8.01–8.03 (2H, m, ArH); 13C NMR (100 MHz, CDCl3) δ: 11.6, 12.7, 17.4, 18.1, 18.2, 32.8, 46.3, 55.9, 67.3, 71.2, 77.8, 110.7, 113.6, 119.3, 127.4, 127.8, 128.3, 128.5, 129.5, 130.5, 132.8, 137.3, 137.9, 147.3, 149.4, 166.6; IR (CHCl3) cm−1: 2945, 1711, 1504, 1460, 1279, 1137, 1116, 1063. MS (EI) m/z: 590 (M+, 16), 547 (100); Anal. Calcd for C36H50O5Si: C, 73.18; H, 8.53%. Found: C, 72.95; H, 8.63%. (2S,3R,4S)-(25). 87% yield,
−3 (c 0.9, CHCl3).
(2R,3S,4R)-4-(4-Benzyloxy-3-methoxyphenyl)-4-hydroxy-2,3-dimethylbutyl benzoate (27)
The title compound was obtained from 25 by the same synthetic method as that of compound 26 in 94% yield as a colorless oil; −22 (c 1.1, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 0.99 (3H, d, J = 7.0 Hz, CH3), 1.11 (3H, d, J = 6.8 Hz, CH3), 1.82 (1H, m, 3-H), 1.97 (1H, m, 2-H), 3.79 (1H, br. s, OH), 3.83 (3H, s, OCH3), 4.14 (1H, dd, J = 10.9, 7.3 Hz, 1-H), 4.40 (1H, dd, J = 10.9, 5.2 Hz, 1-H), 4.77 (1H, d, J = 4.7 Hz, 4-H), 5.12 (2H, s, OCH2Bn), 6.79 (1H, dd, J = 8.2, 1.4 Hz, ArH), 6.83 (1H, d, J = 8.2 Hz, ArH), 6.89 (1H, d, J = 1.4 Hz, ArH), 7.26–7.36 (3H, m, ArH), 7.41–7.45 (4H, m, ArH), 7.55 (1H, m, ArH), 8.01–8.03 (2H, m, ArH); 13C NMR (100 MHz, CDCl3) δ:10.3, 16.4, 34.8, 43.5, 56.0, 67.5, 71.1, 75.6, 109.92, 109.95, 109.96, 113.8, 113.89, 113.91, 118.2, 127.3, 127.8, 128.4, 128.5, 129.5, 130.4, 132.9, 137.2, 137.4, 147.4, 149.7, 166.6; IR (CHCl3) cm−1: 3619, 1976, 1716, 1520, 1452, 1277, 1138, 1026. MS (EI) m/z: 434 (M+, 20), 243 (100); HRMS (EI) m/z calcd for C27H35O5: 434.2093, found: 434.2094. (2S,3R,4S)-(27). 96% yield,
+23 (c 1.2, CHCl3).
(2R,3R)-4,4-Bis(4-benzyloxy-3-methoxyphenyl)-2,3-dimethylbutyl benzoate (29)
The title compound was obtained from 27 by the same synthetic method as that of compound 28 in 52% yield as a colorless oil through three steps; −5 (c 0.6, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 0.83 (3H, d, J = 7.0 Hz, CH3), 1.09 (3H, d, J = 6.9 Hz, CH3), 2.07 (1H, m, 3-H), 2.33 (1H, m, 2-H), 3.71 (1H, d, J = 11.5 Hz, 4-H), 3.80 (3H, s, OCH3), 3.85 (3H, s, OCH3), 4.10 (1H, dd, J = 10.9, 7.9 Hz, 1-H), 4.37 (1H, dd, J = 10.9, 5.1 Hz, 1-H), 5.07 (2H, s, OCH2Bn), 5.08 (2H, s, OCH2Bn), 6.75 (1H, dd, J = 8.3, 1.9 Hz, ArH), 6.77–6.80 (5H, m, ArH), 7.26–7.36 (6H, m, ArH), 7.39–7.45 (6H, m, ArH), 7.55 (1H, m, ArH), 8.01–8.03 (2H, m, ArH); 13C NMR (100 MHz, CDCl3) δ: 13.3, 17.2, 33.1, 41.2, 55.99, 56.03, 56.1, 66.4, 71.1, 112.0, 112.1, 114.1, 114.3, 119.5, 119.8, 127.25, 127.27, 127.29, 127.7, 128.4, 128.45, 128.49, 129.5, 130.5, 132.9, 137.37, 137.40, 137.5, 137.9, 146.66, 146.70, 149.5, 149.6, 166.7; IR (CHCl3) cm−1: 3012, 2960, 1714, 1510, 1277. MS (EI) m/z: 630 (M+, 7), 204 (100); HRMS (EI) m/z calcd for C41H42O6: 630.2981, found: 630.3001. (2S,3S)-(29). 49% yield,
+6 (c 0.7, CHCl3).
(2R,3R)-4,4-Bis(4-hydroxy-3-methoxyphenyl)-2,3-dimethyl-1-butanol (5)
The title compound was obtained from 29 by the same synthetic method as that of compound 3 in 64% yield as a colorless oil through two steps; +24 (c 0.8, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 0.72 (3H, d, J = 6.9 Hz, CH3), 0.92 (3H, d, J = 6.9 Hz, CH3), 1.20 (1H, br. s, 1-OH), 1.80 (1H, m, 3-H), 2.28 (1H, m, 2-H), 3.34 (1H, dd, J = 10.4, 7.8 Hz, 1-H), 3.66 (1H, dd, J = 10.4, 5.4 Hz, 1-H), 3.70 (1H, d, J = 11.7 Hz, 4-H), 3.826 (3H, s, OCH3), 3.834 (3H, s, OCH3), 5.56 (1H, s, ArOH), 5.58 (1H, s, ArOH), 6.72 (1H, d, J = 1.8 Hz, ArH), 6.75 (1H, d, J = 1.4 Hz, ArH), 6.78 (1H, dd, J = 8.2, 1.8 Hz, ArH), 6.80 (1H, d, J = 8.2 Hz, ArH), 6.80–6.84 (2H, m, ArH); 13C NMR (100 MHz, CDCl3) δ: 13.3, 16.6, 36.5, 40.9, 55.9, 56.1, 64.6, 110.6, 110.7, 114.4, 114.5, 120.1, 120.2, 136.9, 137.2, 143.8, 143.9, 146.4, 146.6; IR (CHCl3) cm−1: 3560, 3019, 2980, 1511, 1269, 1219, 1036. MS (EI) m/z: 346 (M+, 53), 260 (100), 229 (60); HRMS (EI) m/z calcd for C20H26O5: 346.1780, found: 346.1770. >99%ee (AD-H, iso-PrOH/hexane = 1.7/1, 1 mL/min, 280 nm, tR4.2 min). (2S,3S)-(6). 62% yield, two steps,
−24 (c 0.6, CHCl3), >99%ee (AD-H, iso-PrOH/hexane = 1.7/1, 1 mL/min, 280 nm, tR4.7 min).
(2S,3S)-4,4-Bis(4-benzyloxy-3-methoxyphenyl)-3-hydroxymethyl-2-methyl-N,N-diethylbutanamide (30)
To an ice-cooled solution of AlMe3 (1.40 mL, 0.98 M in hexane, 1.37 mmol) in CH2Cl2 (2 mL) were added Et2NH (0.15 mL, 1.45 mmol) and lactone 15 (0.36 g, 0.67 mmol) in CH2Cl2 (1mL) under N2 gas. The resulting reaction solution was stirred at room temperature for 72 h before pouring into an ice-cooled H2O. The mixture was dissolved in EtOAc and H2O. The organic solution was separated and washed with sat. aq. NaHSO4 solution. After further washing with brine, the organic solution was dried (Na2SO4). Concentration followed by silica gel column chromatography (EtOAc/hexane = 1/1) gave hydroxyamide 30 (0.29 g, 0.47 mmol, 70%) as a colorless oil; −30 (c 1.4, CHCl3); 1H NMR (400 MHz, C5D5 N) δ: 0.64 (3H, t, J = 6.9 Hz, CH2CH3), 1.11 (3H, t, J = 6.9 Hz, CH2CH3), 1.33 (3H, d, J = 7.3 Hz, CH3), 2.49 (1H, m, CHHCH3), 2.57 (1H, m, CHHCH3), 2.74 (1H, m, 3-H), 3.14 (1H, m, CHHCH3), 3.36 (1H, m, 2-H), 3.51 (1H, m, CHHCH3), 3.75 (3H, s, OCH3), 3.75–3.84 (1H, overlapped, CHHOH), 3.84 (3H, s), 4.15 (1H, dd, J = 11.6, 1.4 Hz, CHHOH), 4.61 (1H, d, J = 12.4 Hz, 4-H), 5.00 (1H, br. s, OH), 5.11 (2H, s, OCH2Bn), 5.22 (2H, s, OCH2Bn), 7.08 (1H, d, J = 8.3 Hz, ArH), 7.14 (1H, d, J = 8.3 Hz, ArH), 7.18 (1H, dd, J = 8.2, 1.9 Hz, ArH), 7.25 (1H, dd, J = 8.3, 1.9 Hz, ArH), 7.28–7.40 (5H, m, ArH), 7.44 (1H, d, J = 1.9 Hz, ArH), 7.55–7.61 (6H, m, ArH); 13C NMR (100 MHz, CDCl3) δ: 12.8, 13.7, 18.2, 36.8, 41.0, 42.2, 47.5, 50.2, 56.0, 56.2, 57.7, 70.9, 71.0, 112.2, 112.8, 114.0, 114.5, 119.5, 119.9, 127.09, 127.14, 127.6, 127.8, 128.4, 128.5, 136.5, 137.2, 137.4, 146.56, 146.62, 149.3, 149.9, 175.8; IR (CHCl3) cm−1: 3629, 3021, 1625. MS (EI) m/z: 611 (M+, 9), 129 (100); HRMS (EI) m/z calcd for C38H45O6 N: 611.3247, found: 611.3252. (2R,3R)-(30). 76% yield,
+30 (c 0.5, CHCl3).
(2S,3S)-4,4-Bis(4-benzyloxy-3-methoxyphenyl)-2-methyl-3-(triisopropylsilyloxy)methyl-N,N,-diethylbutanamide (31)
To an ice-cooled solution of amide 30 (94 mg, 0.12 mmol) and 2,6-lutidine (0.045 mL, 0.39 mmol) in CH2Cl2 (5 mL) was added TIPSOTf (0.050 mL, 0.19 mmol). After the resulting reaction solution was stirred at room temperature for 6 h, sat. aq. NaHCO3 solution was added. The organic solution was separated and washed with sat. aq CuCO4 solution. After further washing with sat. aq. NaHCO3 solution, the organic solution was dried (Na2SO4). Concentration followed by silica gel column chromatography (EtOAc/hexane = 1/3) gave silyl ether 31 (0.12 g, 0.16 mmol, 75%) as a colorless oil; −1 (c 2, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 0.78 (3H, t, J = 7.1 Hz, CH2CH3), 0.90–0.93 (21H, m, TIPSi), 1.03 (3H, t, J = 6.9 Hz, CH2CH3), 1.19 (3H, d, J = 7.1 Hz, CH3), 2.54 (1H, m, 3-H), 2.73 (2H, m, CH2CH3), 2.89 (1H, m, 2-H), 3.08 (1H, m, CHHCH3), 3.35 (1H, m, CHHCH3), 3.59 (1H, dd, J = 10.5, 2.8 Hz, TIPSOCHH), 3.83 (6H, s, OCH3 × 2), 3.85 (1H, d, J = 13.6 Hz, 4-H), 4.27 (1H, dd, J = 10.5, 5.6 Hz, TIPSOCHH), 5.06 (2H, s, OCH2Bn), 5.08 (2H, s, OCH2Bn), 6.72–6.77 (4H, m, ArH), 6.83–6.84 (2H, m, ArH), 7.24–7.27 (2H, m, ArH), 7.30–7.34 (4H, m, ArH), 7.38–7.40 (4H, m, ArH); 13C NMR (100 MHz, CDCl3) δ: 12.0, 13.1, 14.2, 17.1, 18.2, 35.6, 40.0, 41.6, 49.6, 52.4, 56.0, 56.2, 63.4, 71.1, 71.3, 112.5, 112.7, 114.2, 114.3, 120.0, 120.1, 127.2, 127.3, 127.75, 127.82, 128.6, 137.4, 137.6, 137.7, 137.9, 146.6, 146.7, 149.4, 149.7, 175.3; IR (CHCl3) cm−1: 2940, 1626, 1512, 1464, 1261, 1141. MS (EI) m/z: 768 (M+, 0.6), 131 (100). Anal. Calcd for C46H63O6NSi: C, 73.27; H, 8.42%. Found: C, 73.33; H, 8.48%. (2R,3R)-(31). 73% yield,
+1 (c 0.8, CHCl3).
(2R)-1,1-Bis(4-benzyloxy-3-methoxyphenyl)-3-methyl-2-(triisopropylsilyloxy)methylbutane (32)
To a solution of silyl ether 31 (0.12 g, 0.16 mmol) in toluene (5 mL) was added DIBAL-H (0.91 mL, 1 M in toluene, 0.91 mmol) at 0 °C. After the reaction solution was stirred at 0 °C for 13 h, sat. aq. NH4Cl solution was added. The organic solution was separated, washed with brine, and dried (Na2SO4). The concentration gave crude aldehyde. To an ice-cooled solution of aldehyde in EtOH (8 mL) was added NaBH4 (20 mg, 0.53 mmol), and then the reaction mixture was stirred at room temperature for 0.5 h before additions of sat. aq. NH4Cl solution and EtOAc. The organic solution was separated, washed with brine, and dried (Na2SO4). Concentration gave crude alcohol. To an ice-cooled solution of alcohol and Et3 N (36 μL, 0.26 mmol) in CH2Cl2 (5 mL) was added MsCl (20 μL, 0.26 mmol). After the reaction mixture was stirred at room temperature for 0.5 h, H2O and CH2Cl2 were added. The organic solution was separated and dried (Na2SO4). Concentration gave crude mesylate. To an ice-cooled solution of mesylate in THF (10 mL) was added Super-Hydride (5.00 mL, 1.00 M in THF, 5.00 mmol), and then the reaction solution was stirred at room temperature for 1.5 h before the addition of sat. aq, NH4Cl solution. After concentration, the residue was dissolved in CHCl3 and H2O. The organic solution was separated and dried (Na2SO4). Concentration followed by silica gel column chromatography (EtOAc/hexane = 1/6) gave 32 (57 mg, 0.083 mmol, 52%, four steps) as a colorless oil; −3 (c 0.8, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 0.88 (3H, d, J = 7.3 Hz, CH3), 0.90–0.94 (21H, m, TIPSi), 1.02 (3H, d, J = 6.9 Hz, CH3), 1.75 (1H, m, 3-H), 2.22 (1H, m, 2-H), 3.61 (1H, dd, J = 10.3, 6.4 Hz, TIPSOCHH), 3.65 (1H, dd, J = 10.3, 3.2 Hz, TIPSOCHH), 3.77 (1H, d, J = 11.5 Hz, 1-H), 3.84 (3H, s, OCH3), 3.86 (3H, s, OCH3), 5.08 (4H, s, OCH2Bn), 6.74 (1H, d, J = 8.2 Hz, ArH), 6.78–6.82 (5H, m, ArH), 7.26–7.30 (2H, m, ArH), 7.33–7.36 (4H, m, ArH), 7.39–7.42 (4H, m, ArH); 13C NMR (100 MHz, CDCl3) δ: 11.9, 17.2, 18.0, 18.1, 22.7, 27.7, 49.0, 51.6, 56.0, 56.1, 61.9, 71.1, 71.2, 112.1, 114.0, 114.3, 119.7, 120.0, 121.9, 127.2, 127.3, 127.7, 128.5, 137.5, 138.1, 146.6, 149.4; IR (CHCl3) cm−1: 2941, 2851, 1508, 1465, 1263, 1148. MS (EI) m/z: 682 (M+, 16), 439 (100); HRMS (EI) m/z calcd for C43H58O5Si: 682.4054, found: 682.4042. (2S)-(32). 63% yield, four steps,
+3 (c 1, CHCl3).
(R)-2-Bis(4-benzyloxy-3-methoxyphenyl)methyl-3-methy-1-butanol (33)
To a solution of silyl ether 32 (57 mg, 0.083 mmol) in THF (5 mL) was added (n-Bu)4NF (0.17 mL, 1 M in THF, 0.17 mmol). After stirring at room temperature for 2 h, sat. aq. NH4Cl solution and EtOAc were added. The organic solution was separated, washed with brine, and dried (Na2SO4). Concentration followed by silica gel column chromatography (EtOAc/hexane = 1/3) gave alcohol 33 (40 mg, 0.078 mmol, 94%) as a colorless oil, −43 (c 0.5, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 0.83 (3H, d, J = 6.9 Hz, CH3), 1.01 (3H, d, J = 6.9 Hz, CH3), 1.76 (1H, m, 3-H), 2.21 (1H, m, 2-H), 3.57–3.66 (2H, m, 1-H2), 3.79 (1H, d, J = 11.4 Hz, Ar2CH), 3.85 (3H, s, OCH3), 3.86 (3H, s, OCH3), 5.08 (2H, s, OCH2Bn), 5.09 (2H, s, OCH2Bn), 6.77–6.86 (6H, m, ArH), 7.25–7.26 (2H, m, ArH), 7.28–7.36 (4H, m, ArH), 7.39–7.42 (4H, m, ArH); 13C NMR (100 MHz, CDCl3) δ: 16.5, 22.1, 27.6, 49.8, 52.8, 55.9, 56.0, 61.7, 71.0, 111.7, 111.8, 113.9, 114.2, 119.6, 127.2, 127.67, 127.70, 128.4, 137.1, 137.2, 137.3, 146.6, 147.8, 149.5, 149.6; IR (CHCl3) cm−1: 3735, 3025, 3000, 1508, 1261, 1141, 1036. MS (EI) m/z: 526 (M+, 18), 439 (100); HRMS (EI) m/z calcd for C34H38O5: 526.2720, found: 526.2735. (2S)-(33). 97% yield,
+43 (c 0.8, CHCl3).
(R)-2-Bis(4-hydroxy-3-methoxyphenyl)methyl-3-methyl-1-butanol (7)
A reaction mixture of benzyl ether 33 (40 mg, 0.078 mmol) and 5% Pd/C (0.10 g) in EtOAc (10 mL) was stirred under H2 gas at ambient temperature for 2 h before filtration. After the filtrate was concentrated, the residue was applied to a silica gel column chromatography (EtOAc/hexane = 1/1) to give secocyclolignane 7 (23 mg, 0.066 mmol, 85%) as a colorless oil, −55 (c 0.3, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 0.82 (3H, d, J = 6.9 Hz, CH3), 1.02 (3H, d, J = 6.9 Hz, CH3), 1.78 (1H, m, 3-H), 2.22 (1H, m, 2-H), 3.64 (2H, s, 1-H2), 3.78 (1H, d, J = 11.5 Hz, Ar2CH), 3.85 (3H, s, OCH3), 3.86 (3H, s, OCH3), 5.49 (1H, s, ArOH), 5.52 (1H, s, ArOH), 6.77 (1H, s, ArH), 6.81 (1H, d, J = 1.9 Hz, ArH), 6.82 (1H, d, J = 8.2 Hz, ArH), 6.84 (2H, s, ArH), 6.87 (1H, dd, J = 8.2, 1.9 Hz, ArH); 13C NMR (100 MHz, CDCl3) δ: 16.6, 22.2, 27.7, 49.9, 53.0, 55.8, 55.9, 61.8, 110.46, 110.54, 114.5, 114.7, 120.1, 120.3, 136.2, 136.3, 143.9, 144.2, 146.5, 146.7; IR (CHCl3) cm−1: 3571, 3000, 1511, 1269, 1036. MS (EI) m/z: 346 (M+, 7), 259 (100); HRMS (EI) m/z calcd for C20H26O5: 346.1780, found: 346.1774. > 99%ee (AD-H, iso-PrOH/hexane = 1.7/1, 1 mL/min, 280 nm, tR6.5 min). (S)-8. 79% yield,
+55 (c 0.5, CHCl3),>99%ee (AD-H, iso-PrOH/hexane = 1.7/1, 1 mL/min, 280 nm, tR4.7 min).
(2S,3R)-4,4-Bis(3,4-dimethoxyphenyl)-2,3-dimethyl-1-butanol (34), (+)-kadangustin J
A reaction mixture of butanol type secocyclolignane 3 (14 mg, 40 μmol), dimethyl sulfate (50 μL, 0.53 mmol), K2CO3 (0.11 g, 0.80 mmol), and dibenzo-18-crown-6 (2 mg) in CH3CN (8 mL) was stirred at 80 °C for 24 h before additions of CHCl3 and H2O. The organic solution was separated, washed with brine, and dried (Na2SO4). Concentration followed by silica gel TLC (2% MeOH in CHCl3) gave 34, (+)-kadangustin J (9 mg, 24 μmol, 60%) as colorless powders. The NMR data agreed with those in the literature.Citation8) +21 (c 0.1, MeOH),
+4.9 (c 0.171, MeOH) in the literature.Citation8) (2R,3S)-35, (−)-kadangustin J. 65% yield,
−21 (c 0.2, MeOH),
−20.7 (c 1.19, MeOH) in the literature.Citation18)
(2R,3R)-4,4-Bis(3,4-dimethoxyphenyl)-2,3-dimethyl-1-butanol (36)
The title compound was obtained from 5 by the same synthetic method as that of compound 34 in 68% yield as colorless powders, +18 (c 0.1, MeOH); 1H NMR (400 MHz, CDCl3) δ: 0.78 (3H, d, J = 6.9 Hz, CH3), 0.99 (3H, d, J = 6.9 Hz, CH3), 1.57 (1H, br. s, OH), 1.82 (1H, m, 3-H), 2.31 (1H, m, 2-H), 3.37 (1H, dd, J = 10.5, 7.3 Hz, 1-H), 3.67 (1H, dd, J = 10.5, 6.0 Hz, 1-H), 3.75 (1H, d, J = 11.5 Hz, 4-H), 3.82 (6H, s, OCH3 × 2), 3.83 (6H, s, OCH3 × 2), 6.76 (1H, d, J = 8.3 Hz, ArH), 6.77–6.78 (1H, overlapped, ArH), 6.79 (1H, d, J = 8.3 Hz, ArH), 6.80 (1H, d, J = 2.3 Hz, ArH), 6.83 (1H, dd, J = 8.3, 1.9 Hz, ArH), 6.86 (1H, dd, J = 8.3, 2.3 Hz, ArH); 13C NMR (100 MHz, CDCl3) δ: 13.3, 16.6, 36.6, 40.9, 55.8, 55.87, 55.90, 56.0, 64.7, 111.2, 111.4, 119.6, 137.4, 137.8, 147.3, 147.4, 148.8, 149.0. MS (EI) m/z: 374 (M+, 11), 287 (100); HRMS (EI) m/z calcd for C22H30O5: 374.2094, found: 374.2088. (2S,3S)-37. 67% yield,
−18 (c 0.1, MeOH).
Enantiomeric excess of butane-type secocyclolignane 1 and 2
1: >99%ee, AD-H, iso-PrOH/hexane = 1/2, 1 mL/min, 271 nm, tR10 min. 2: >99%ee, AD-H, iso-PrOH/hexane = 1/2, 1 mL/min, 271 nm, tR8 min.
Acknowledgments
Part of this study was performed at INCS (Johoku station) of Ehime University. We are grateful to Marutomo Co. for financial support.
References
- Ayres DC, Loike JD. Lignans: chemical, biological and clinical properties. Cambridge: Cambridge University Press; 1990 A registry of the natural lignans; p. 12–84.
- Ward RS. Lignans, neolignans, and related compounds. Nat. Prod. Rep. 1999;16:75–96.
- Moss GP. Nomenclature of lignans and neolignans (IUPAC recommendations 2000). Pure Appl. Chem. 2000;72:1493–1523.
- Xu S, Li N, Ning M-M, Zhou C-H, Yang Q-R, Wang M-W. Bioactive compounds from Peperomia pellucida. J. Nat. Prod. 2006;69:247–250.
- Wu J-J, Li N, Hasegawa T, Sakai J, Mitsui T, Ogura H, Kataoka T, Oka S, Kiuchi M, Tomida A, Turuo T, Li M, Tang W, Ando M. Bioactive secolignans from Peperomia dindygulensis. J. Nat. Prod. 2006;69:790–794.
- Zhang G-L, Li N, Wang Y-H, Zheng Y-T, Zhang Z, Wang M-W. Bioactive secolignans from Peperomia heyneana. J. Nat. Prod. 2007;70:662–664.
- Xu L-J, Huang F, Chen S-B, Li L-N, Chen S-L, Xiao P-G. A cytotoxic neolignan from Schisandra propinqua (Wall.) Baill. J. Integr. Plant Biol. 2006;48:1493–1497.
- Gao X-M, Pu J-X, Huang S-X, Yang L-M, Huang H, Xiao W-L, Zheng Y-T, Sun H-D. Lignans from Kadsura angustifolia. J. Nat. Prod. 2008;71:558–563.
- Akiyama K, Yamauchi S, Maruyama M, Sugahara T, Kishida T, Koba Y. Antimicrobial activity of stereoisomers of morinol A and B, tetrahydropyran sesquineolignans. Biosci. Biotechnol. Biochem. 2009;73:129–133.
- Nakato T, Yamauchi S, Tago R, Akiyama K, Maruyama M, Sugahara T, Kishida T, Koba Y. Syntheses and antimicrobial activity of tetrasusbstituted tetrahydrofuran lignan stereoisomers. Biosci. Biotechnol. Biochem. 2009;73:1608–1617.
- Kawahara S, Iwata I, Fujita E, Yamawaki M, Nishiwaki H, Sugahara T, Yamauchi S, Akiyama K, Kishida T. IgE-suppressive activity of (-)-matairesinol and its structure-activity relationship. Biosci. Biotechnol. Biochem. 2010;74:1878–1883.
- Nishiwaki H, Hasebe A, Kawaguchi Y, Akamatsu M, Shuto Y, Yamauchi S. Larvicidal activity of (-)-dihydroguaiaretic acid derivatives against Culex pipiens. Biosci. Biotechnol. Biochem. 2011;75:1735–1739.
- Nishiwaki H, Kumamoto M, Shuto Y, Yamauchi S. Stereoselective syntheses of all stereoisomers of lariciresinol and their plant growth inhibition activities. J. Agric. Food Chem. 2011;59:13089–13095.
- Morita M, Nishiwaki H, Shingai Y, Fujimoto A, Masuda T, Yamauchi S. First diastereoselective construction of butane-type and butyrolactone-type secocyclolignane structures. Biosci. Biotechnol. Biochem. 2011;75:939–943.
- Yamauchi S, Hayashi Y, Nakashima Y, Kirikihira T, Yamada K, Masuda T. Effect of benzylic oxygen on the antioxidant activity of phenolic lignans. J. Nat. Prod. 2005;68:1459–1470.
- Stadler D, Bach T. Concise stereoselective synthesis of (-)-podophyllotoxin by an intermolecular iron (III)-catalyzed Friedel-Crafts alkylation. Angew. Chem. Int. Ed. 2008;47:7557–7559.
- England DB, Kerr MA. Synthesis and cross-coupling reactions of substituted 5-triflyloxyindoles. J. Org. Chem. 2005;70:6519–6522.
- Rye CE, Barker D. Asymmetric synthesis of (+)-galbelgin, (-)-kadangustin J, (-)-cyclogalgravin and (-)-pycnanthulignenes A and B, three structurally distinct lignan classes, using a common chiral precursor. J. Org. Chem. 2011;76:6636–6648.