
Abstract
Tissue factor pathway inhibitor-2 (TFPI-2) is a major inhibitor of extracellular matrix degradation. Decreases in TFPI-2 contribute to malignant tumor cell production, and TFPI-2 is a presumed tumor suppressor. TFPI-2 gene transcription is regulated by two epigenetic mechanisms: DNA methylation of the promoter and K4 methylation of histone 3 (H3). Lysine-specific demethylase 1 (LSD1) and LSD2 demethylate H3K4me2/1. LSD1 has been implicated in TFPI-2 regulation through both epigenetic mechanisms, but the involvement of LSD2 remains unknown. We prepared a monoclonal anti-LSD2 antibody that clearly distinguishes LSD2 from LSD1. Knockdown of LSD1 or LSD2 by siRNAs increased TFPI-2 protein and mRNA. Simultaneous knockdown of both LSD1 and LSD2 showed additive effects. Bisulfite sequencing revealed that CpG sites in the TFPI-2 promoter region were unmethylated. These results indicate that LSD2 also contributes to TFPI-2 regulation through histone modification, and that further studies of the involvement of LSD2 in tumor malignancy are warranted.
Graphical Abstract
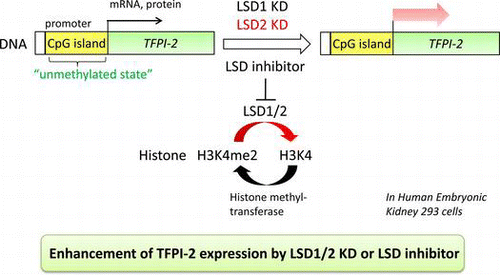
Knockdown of LSD1/2 or LSD inhibitor increased a presumed tumor suppressor TFPI-2. This study indicates that LSD2 also contributes to TFPI-2 regulation through histone modification.
Introduction
Methylation of DNA and modifications of histones are central cores of the epigenetic regulation of gene expression.Citation1) DNA cytosine-5 methyltransferase 1 (DNMT1) is responsible for methylating CpG sites in newly synthesized DNA strands. Hypermethylation of CpG islands in a gene promoter represses transcription, and is frequently seen in physiological conditions as well as diseases including cancer.Citation2) The cellular concentration of DNMT1 is controlled through methylation and demethylation of its lysine residue K142. SET7 methylates DNMT1 and the methylated DNMT1 is degraded through the ubiquitin-mediated pathway.Citation3) Lysine-specific demethylase 1 (LSD1), also known as KDM1a/AOF2, functions primarily as a demethylase of histone lysines (see below), but can also remove the methyl residue on DNMT1. Thus, LSD1-induced stabilization of DNMT1 is thought to stimulate DNA methylation.Citation3,4)
The H3 and H4 histones have amino-terminal tails that protrude from the nucleosome, and these tails undergo a variety of covalent modifications including lysine methylation.Citation5) There are two groups of histone lysine demethylases with different cofactor requirements,Citation5–9) namely KDM1 that requires FAD as a cofactor and JumonjiC (JmjC)-domain-containing proteins that use Fe(II) and α-ketoglutarate as cofactors. LSD1, which belongs to KDM1, demethylates di- and mono-methylated histone 3 (H3) at lysine 4 (H3K4me2/1).Citation6) Since H3K4me2 is a positive mark associated with the promoters of active genes,Citation7) LSD1-catalyzed demethylation of H3K4me2/1 usually represses transcription. Increased expression of LSD1 has been found in some types of cancer including bladder cancer, lung cancer, colorectal carcinoma,Citation10) and malignant neuroblastoma,Citation11) thus encouraging the development of LSD1 inhibitors for cancer therapy.
LSD2, also known as KDM1b/AOF1, is the only mammalian homolog of LSD1 and shows a sequence identity of 28%. Like LSD1, LSD2 also demethylates H3K4me2/1.Citation12–14) It has been shown that LSD2 is required for the establishment of maternal genomic imprints.Citation13)
Extracellular matrix (ECM) degradation is regulated by the balance of activities of various proteinases and their inhibitors. Deregulation of this process by over expressing proteinases or repressing the expression of their inhibitors gives rise to malignant tumor cells with high invasiveness.Citation15,16) Tissue factor pathway inhibitor-2 (TFPI-2), a secretory protein that belongs to the Kunitz-type serine proteinase inhibitor family, inhibits a wide variety of serine proteinases that are crucial for ECM degradation. Reduced expression of TFPI-2 has been implicated in the malignancy of some types of cancers.Citation17–19) In addition, TFPI-2 has been shown to inhibit new blood vessel formation,Citation20) one of the hallmarks for tumor growth.Citation21) Thus, a decrease in TFPI-2 is likely to be correlated with progression of tumorigenesis and metastasis of cancer cells. Expression of the TFPI-2 gene has been shown to undergo epigenetic regulations.Citation11,22,23)
Methylation of the TFPI-2 gene promoter has been proposed as a major mechanism that causes diminished expression of TFPI-2 in human glioma cells and choriocarcinoma cells.Citation22,23) In poorly differentiated neuroblastoma cells, however, it has been shown that small interfering RNA (siRNA)-mediated knockdown of LSD1 increases TFPI-2 expression, accompanied by an increase in the H3K4me2 marker in the TFPI-2 promoter.Citation11) These observations indicate that there may be two LSD1-mediated pathways for repression of the TFPI-2 gene in malignant tumor cells. One pathway is that the stabilization of DNMT1 through LSD1-mediated demethylation promotes methylation of the TFPI-2 promoter and the other is that the LSD1-mediated decrease in H3K4me2 in the TFPI-2 promoter represses transcriptional initiation.
Despite similarity of the catalytic mechanism between LSD1 and LSD2,Citation12) the involvement of LSD2 in the regulation of TFPI-2 expression has not been examined. It is noteworthy that the LSD2 gene is located at chromosome 6p22, where chromosomal disarrangements such as translocations, deletions, and amplifications occur in cancers with high frequencies.Citation24,25) Thus LSD2 may play a role for cancer development with malignancy, which makes it worthwhile to examine LSD2-mediated regulation of TFPI-2 gene expression.
In this paper, we show that TFPI-2 expression in HEK293 cells is induced by knockdown of not only LSD1, but also LSD2. Furthermore, we report that CpG sites in the TFPI-2 promoter are not methylated. These findings indicate that LSD2 represses TFPI-2 expression through histone methylation, and suggest that LSD2 is also involved in the malignancy of tumor cells.
Materials and methods
Cell culture
HEK293 cells were obtained from the RIKEN Cell Bank (Ibaraki, Japan; Cell No. RCB1637) and maintained in Dulbecco’s modified Eagle’s medium (DMEM) (D5796; Sigma, St. Louis, MO) supplemented with 10% (v/v) fetal bovine serum (FBS) (Nichirei, Tokyo, Japan), 100 μg/mL streptomycin, and 100 U/mL penicillin at 37 °C in a humidified 5% CO2 incubator. Other cells were cultured in RPMI1640 medium containing the supplements described above.
Production and purification of recombinant His-tagged human full-length LSD2 and His-tagged LSD2ΔN25
The AOF1 gene encoding human LSD2 (amino acids 1–822) was amplified by PCR using the cDNA from HeLa cells as a template with forward primer 5′-CAGTAATCATATGGCAACTCCACGGGGGAGGACAAAG-3′ and reverse primer 5′-TAGTCTCGAGTTAAAATGCTGCAATCTTGCTTG-3′. The underlines show restriction enzyme sites for Nde I and Xho I that were introduced into the forward and reverse primers, respectively. The PCR product was ligated into pET28b (Novagen Inc., Madison, WI) digested with Nde I and Xho I, and the resultant plasmid was designated pETLSD2. The gene encoding an LSD2 mutant with 25 amino acid residues deleted from the N-terminus (LSD2ΔN25) was amplified by PCR from pETLSD2. The PCR fragment was digested with Nde I and Xho I and ligated into the pET28b vector to generate a plasmid designated pETLSD2ΔN25.
Escherichia coli BL21(DE3) cells were transformed with pETLSD2 or pETLSD2ΔN25 for expression of full-length LSD2 or LSD2ΔN25 bearing a hexahistidine-tag at the N-terminus. Recombinant E. coli BL21(DE3) cells carrying pETLSD2 or pETLSD2ΔN25 were grown in 200 mL of NZCYM medium containing 50 μg/mL kanamycin at 37 °C with shaking at 200 rpm. When the absorbance at 600 nm reached 0.7, the temperature was lowered to 16 °C and isopropyl-β-D-thiogalactopyranoside was added to a final concentration of 0.5 mM. The culture was continued for a further 19 h with shaking at 110 rpm, and the cells were then harvested by centrifugation at 3,100 × g for 10 min at 4 °C. LSD2 was purified by chromatography through a HisTrap column using the same method previously described for purification of LSD1,Citation26) except that 50 mM sodium phosphate buffer (pH 8.0) was used instead of 50 mM sodium phosphate buffer (pH 7.5). The partially purified LSD2 was dissolved in 50 mM HEPES-NaOH (pH 8.0) containing 5% glycerol, 1 mM PMSF, 1.5 μg/mL pepstatin A, and 1.5 μg/mL leupeptin, and stored at −30 °C until use.
Assay of LSD2 activity
The histone demethylation reaction for assaying LSD2 activity was carried out for 4.5 h at 37 °C in 30 μL of 50 mM HEPES-NaOH (pH 8.0) containing 0.1% BSA, 0.16 mg/mL calf thymus histones, and 7 μg of His-tagged full-length LSD2. To assess the inhibitory effect of N-((1S)-3-(3-(trans-2-aminocyclopropyl)phenoxy)-1-(benzylcarbamoyl)propyl) benzamide (NCL-1)Citation27) on LSD2, NCL-1 was dissolved in DMSO. The final concentration of DMSO was 6.7%, which had no deteriorative effect on LSD2. The reactions were stopped with 6 μL of 6 × SDS sample buffer and the mixtures were boiled for 5 min at 95 °C. The reaction mixture (14 μL) was subjected to 15% SDS-PAGE and the separated proteins were transferred to a nitrocellulose membrane. The membrane was blocked with 3% skim milk in 25 mM Tris–HCl (pH 7.4) containing 0.15 M NaCl and 0.1% (v/v) Tween-20 (TBS-T) for 1 h at room temperature and then incubated overnight with a rabbit polyclonal anti-H3K4me2 antibody (07-030; Merck Millipore Inc., Billerica, MA; 1:3,000 dilution). The membrane was washed with TBS-T and incubated with an anti-rabbit secondary antibody. To determine the relative abundance of histone H3K4me2 to total histone H3, the same membrane was stripped by incubation at 50 °C for 30 min in 62.5 mM Tris–HCl (pH 6.7) containing 100 mM 2-mercaptoethanol and 2% SDS. After three 10-min washes with TBS-T, the membrane was incubated with a rabbit anti-human histone H3 antibody (#07-690; Upstate Biotechnology Inc., Lake Placid, NY; 1:25,000 dilution) for 2 h at room temperature, followed by incubation with an anti-rabbit secondary antibody. Total histone H3 and histone H3K4me2 were quantified by measuring chemiluminescent signals as described below (see Western blotting).
Expression of Flag-tagged full-length human LSD1 and LSD2 in HEK293 cells
Plasmids for expression of Flag-tagged full-length human LSD1 and LSD2 were constructed using pEGFP-N1 (Clontech Inc., Mountain View, CA). HEK293 cells were transfected with each plasmid in the presence of Lipofectamine 2000 (Invitrogen Inc., Carlsbad, CA) and cultured for 24 h.
Production of a monoclonal antibody (mAb) against human LSD2
The purified His-tagged full-length LSD2 was administered to mice as an antigen to prepare hybridoma cells producing an anti-LSD2 mAb. The hybridoma cells were prepared as described previously.Citation28) The cells were subcloned by the limiting dilution method, and a clone named mAb3E was established. The mAb3E antibody belonged to the IgG1 subclass. Ascites were generated by injection of 1 × 107 hybridoma cells into pristine-primed mice. The mAb3E antibody in the ascites was precipitated with 50% ammonium sulfate and purified with a protein G column.
Western blotting
HEK293 cells were lysed with 62.5 mM HEPES-NaOH (pH 7.5) containing 1% SDS, 10% glycerol, 5% 2-mercaptoethnol, and 1% proteinase inhibitor cocktail (25955-11; Nacalai Tesque Inc., Kyoto, Japan). The solutions were incubated on ice for 10 min, followed by incubation at 95 °C for 10 min. The cell lysates were clarified by centrifugation at 16,000 × g for 5 min and the supernatants were collected. The protein concentrations were determined with a Protein Assay Kit (Bio-Rad Laboratories Inc., Hercules, CA) using bovine gamma globulin as a standard. The cell lysates were subjected to 10 or 12.5% SDS-PAGE. The separated proteins were transferred to nitrocellulose membranes with a pore size of 0.2 μm (Protran BA83; Whatman Plc, Kent, UK) in transfer buffer (25 mM Tris, 192 mM glycine, 10% methanol, 0.01% SDS) using a Mini Trans-Blot Cell (Bio-Rad Laboratories Inc.). Transfer of proteins to the membranes was confirmed by staining with Ponceau S. The membranes were blocked with 5% skim milk in TBS-T for 2.5 h at room temperature, and then incubated overnight at 4 °C with rabbit polyclonal anti-LSD1 (ab17721; Abcam Inc., Cambridge, UK; 1:2,000 dilution), monoclonal anti-LSD2 (mAb3E; 1:3,000 dilution), anti-Flag (M185; MBL Inc., Nagoya, Japan; 1:2,000 dilution), anti-TFPI-2 (sc-48380; Santa Cruz Biotechnology Inc., Santa Cruz, CA; 1:1,000 dilution), or anti-β-actin (sc-47778; Santa Cruz Biotechnology Inc.; 1:1,000 dilution) antibodies. The membranes were washed in TBS-T and incubated for 1.5 h at room temperature with a species-specific secondary antibody, goat anti-rabbit IgG antibody conjugated to horseradish peroxidase (HRP) (458; MBL Inc.; 1:5,000 dilution) or goat anti-mouse IgG antibody conjugated to HRP (330; MBL Inc.; 1:5,000 dilution), and then washed again with TBS-T. In the case of His-tagged proteins, the membranes after transfer were blocked with 1.3% Block Ace solution (DS Pharma Biomedical Co. Ltd., Osaka, Japan) for 1 h at room temperature and then incubated with a rabbit polyclonal anti-His-probe antibody (sc-803; Santa Cruz Biotechnology Inc.; 1:500 dilution), followed by incubation with the anti-rabbit secondary antibody.
Chemiluminescent signals for the immunoreactive proteins were developed with Chemi-Lumi One (Nacalai Tesque Inc.) and captured using a luminescent image analyzer (LAS-4000; GE Healthcare Japan, Tokyo, Japan). The relative intensities of the bands were quantified using ImageQuantTL software.
Knockdown of LSDs by siRNAs
The siRNA duplex oligonucleotides used for knockdown of LSD1 and LSD2 were as follows and obtained from the indicated sources: 5′-AACACAAGGAAAGCTAGAAGA-3′ (LSD1siRNA2 from Integrated DNA Technology Inc., Coralville, IA); 5′-CACAGGGATCTGACCGCCCTA-3′ (LSD1siRNA10 from Qiagen K·K. Tokyo, Japan); 5′-ATCGATGCGGTATGAAACCAA-3′ (LSD2siRNA1 from Qiagen K·K.); and 5′-AAGACATTCAAGGAACCGTCT-3′ (LSD2siRNA2 from Integrated DNA Technology Inc.). AllStars Negative control siRNA was obtained from Qiagen K·K. HEK293 cells were suspended in DMEM containing 10% FBS and seeded at 1 × 104 cells/well (total volume: 0.1 mL) in 96-well plates for cell proliferation assays, 1 × 105 cells/well (total volume: 1 mL) in 12-well plates for RNA extraction, and 2.5 × 105 cells/well (total volume: 2.5 mL) in 6-well plates for protein extraction. After incubation for 24 h, Lipofectamine RNAiMAX transfection reagent (Invitrogen Inc.) was used to transfect siRNA duplexes into the cells according to the manufacturer’s instructions. Each of the siRNAs was added to a final concentration of 10 nM. At 72 h after transfection, the cells were used for extraction of protein or RNA.
RNA extraction and quantitative reverse transcription-PCR (qRT-PCR)
Total RNA was extracted using the RNAzol RT reagent (Molecular Research Center Inc., Cincinnati, OH). One microgram of total RNA was reverse-transcribed by ReverTra Ace-α – reverse transcriptase (Toyobo Co. Ltd., Osaka, Japan) with an oligo (dT)20 primer. For qRT-PCR, each reaction was carried out in a total volume of 20 μL consisting of 2 μL of cDNA solution (equivalent to 5 ng of reverse-transcribed RNA), 10 μL of THUNDERBIRD SYBR qPCR Mix (Toyobo Co. Ltd.), 0.6 μL of 5 μM forward primer, 0.6 μL of 5 μM reverse primer, and 6.8 μL of deionized and sterilized water in 96-well plates in a LightCycler 480 system (Roche Diagnostics K·K., Tokyo, Japan). The primers used were: LSD1 FWD, 5′-CAGGCTTGGCAGCAGCTCGA-3′; LSD1 REV, 5′-TCCACCCACACGATCCCTGGC-3′; LSD2 FWD, 5′-CCTGGCTTTGAGAAACCTCATC-3′; LSD2 REV, 5′-TCTCCACTTCCTGAACGCATC-3′; TFPI-2 FWD, 5′-GTCGATTCTGCTGCTTTTCC-3′; TFPI-2 REV, 5′-CAGCTCTGCGTGTACCTGTC-3′; GAPDH FWD, 5′-CAGCCTCAAGATCATCAGCA-3′; and GAPDH REV, 5′-TGTGGTCATGAGTCCTTCCA-3′. For standard curves, the concentration of the cDNA solution isolated from untreated cells was varied by fourfold serial dilution in the reaction mixture. The PCR program was 95 °C for 1 min for the first cycle, and then 50 cycles of 95 °C for 15 s, 55 °C for 30 s, and 72 °C for 30 s, followed by a standard dissociation protocol to ensure that each amplicon was a single product. The data were analyzed using Roche LightCycler® 480 software version 1.5.0.39, and the crossing point was calculated by the second derivative maximum method. A standard curve was generated to show the linear correlation between the log (relative concentration of cDNA) and the crossing point value. The relative amount of the target mRNA in each sample was calculated from the standard curves and normalized by that of GAPDH mRNA.
Bisulfite sequencing
DNA isolated from HEK293 cells was treated with bisulfite using an EZ DNA Methylation-Gold Kit (Zymo Research Inc., Irvine, CA) according to the manufacturer’s instructions. The TFPI-2 promoter region and differentially methylated region (DMR)Citation29,30) were amplified with TFPI-2 primers (FWD, 5′-AGGTAGGTTTAATTTTTTAATTTGG-3′; REV, 5′-CTATTAACTCCTAAACAACATC-3′) and DMR primers (FWD, 5′-GTGTTATGTTTTATAAATAGATAAG-3′; REV, 5′-AACTCATATACCTCTACAATTC-3′), respectively. The PCR products were cloned into the pGEM®-T Easy Vector (Promega, Madison, WI) and the vectors used to transform E. coli. DNA from white colonies were amplified with M13/pUC seq primer (5′-GCCAGGGTTTTCCCAGTCAGGA-3′) and M13 reverse primer (5′-GGAAACAGCTATGACCATG-3′). After the sizes of the amplified products were confirmed, the products were sequenced by a 3130 Genetic Analyzer (Applied Biosystems, Foster City, CA) using M13/pUC seq primer. DNA sequencing was performed with 25 colonies for the TFPI-2 promoter and seven colonies for the DMR.
Results and discussion
Specificities of antibodies against LSD1 and LSD2
His-tagged full-length human LSD2 was produced in E. coli and the purified LSD2 was administered to mice to prepare a hybridoma clone producing a mAb against human LSD2. The mAb3E antibody produced by one of the hybridoma clones reacted efficiently with the antigen. We examined the antigen specificities of mAbE3 and a commercially available polyclonal antibody against LSD1. Immunoblotting of HEK293 extracts with the mAb3E and anti-LSD1 antibodies revealed one clear band for each antibody (Fig. (A)). The size of the component recognized by the anti-LSD1 antibody was larger than that recognized by mAb3E. We expressed the Flag-tagged full-length LSD1 or LSD2 in HEK293 cells. When extracts from the cells expressing Flag-tagged LSD1 and LSD2 were mixed and the mixture was subjected to immunoprecipitation with an anti-Flag antibody, western blotting of the immunoprecipitates using an anti-Flag antibody detected two bands (Fig. (B)). In contrast, the extract from cells expressing Flag-tagged LSD1 or LSD2 showed one band with the anti-LSD1 antibody or mAb3E, respectively. The size difference was consistent with that found for endogenous LSD1 and LSD2. These results indicate that mAb3E and the anti-LSD1 antibody can clearly distinguish LSD2 and LSD1.
Fig. 1. Antigen specificities of anti-human LSD2 monoclonal antibody (mAb3E) and anti-human LSD1 polyclonal antibody.
Notes: (A) Western blotting of HEK293 cell lysates (20 μg of protein) was carried out using the indicated antibodies. (B) Extracts from HEK293 cells expressing Flag-tagged full-length LSD1 and LSD2 were mixed at 1:1 (v/v) and the mixture was subjected to immunoprecipitation with an anti-Flag antibody. Flag-tagged LSD1 and LSD2 in the immunoprecipitates were detected by immunoblotting with an anti-Flag antibody (left column). The extract from cells expressing Flag-tagged LSD1 was immunoprecipitated with an anti-Flag antibody and the immunoprecipitates were analyzed with an anti-LSD1 antibody (middle column). The extract from cells expressing Flag-tagged LSD2 were treated as described for the middle column and analyzed with mAb3E. (C) His-tagged full-length LSD2 or His-tagged LSD2ΔN25 purified from E. coli was detected by western blotting using an anti-His antibody and mAb3E. It should be noted that mAb3E does not react with LSD2ΔN25. D, LSD2 in human cell lines was detected by western blotting using mAb3E.
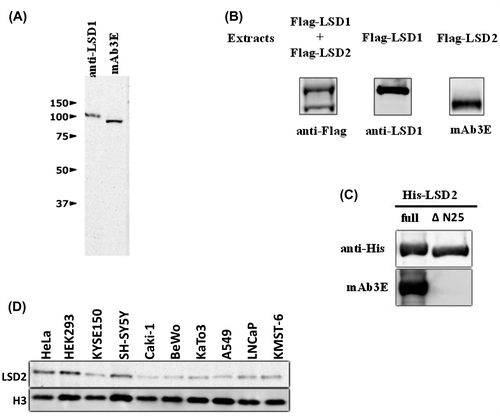
mAb3E reacted with His-tagged full-length human LSD2 produced in E. coli, but not with LSD2ΔN25 that lacks N-terminal amino acids 1–25 (Fig. (C)). Although the human and mouse LSD2 proteins are highly homologous (92% identity in the amino acid sequences), this N-terminal region contains a sequence with little homology.Citation12) Mouse LSD2 did not react with mAb3E (data not shown). Taken together, we conclude that the epitope for mAb3E is present in the N-terminal fragment containing amino acids 1–25 of human LSD2.
As shown in Fig. (D), several human cell lines expressed LSD2 protein to different extents. Of the cell lines tested, HEK293 cells showed the highest expression of LSD2. A high transfection efficiency of HEK293 cells has been widely accepted as of their properties, which may facilitate knockdown experiments. We, therefore, used HEK293 cells throughout this study.
Induction of TFPI-2 by inhibitors of LSDs
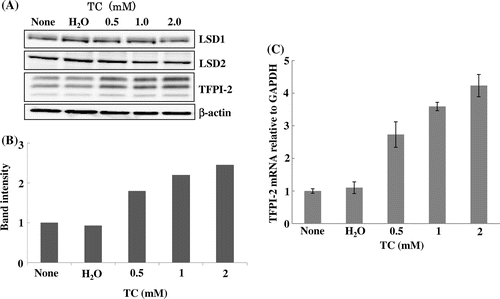
The catalytic domain of FAD-dependent LSD1 belongs to the family of amine oxidases including monoamine oxidases. Assessment of monoamine oxidase inhibitors for their ability to inhibit LSD1 revealed that trans-2-phenylcyclopropylamine (tranylcypromine; TC) was the most potent inhibitor.Citation31) It has been shown that LSD2 is also inhibited by this compound.Citation12,14) Incubation of TC with HEK293 cells increased the TFPI-2 protein level in a dose-dependent manner without any impact on the levels of LSD1 and LSD2 (Fig. (A) and (B)). The triplet bands reflected differentially glycosylated TFPI-2 proteins.Citation32) TC also elevated TFPI-2 mRNA (Fig. (C)), suggesting that the increase in TFPI-2 protein is caused by transcriptional activation, although the possibility that TC might stabilize TFPI-2 mRNA through an unknown mechanism has not been excluded.
Fig. 2. Induction of TFPI-2 protein and mRNA by the LSD inhibitor TC.
Notes: TC was dissolved in water and incubated with HEK293 cells at the indicated concentrations for 72 h. (A) Immunoblotting. The triplet bands of TFPI-2 reflect differentially glycosylated products. (B) Quantification of TFPI-2 protein bands by densitometry. TFPI-2 in the control cells was defined as 1. (C)TFPI-2 mRNA measured by qRT-PCR. The ratio of TFPI-2 mRNA to GAPDH mRNA in the control cells was defined as 1. Data represent the mean ± SD of duplicate samples.
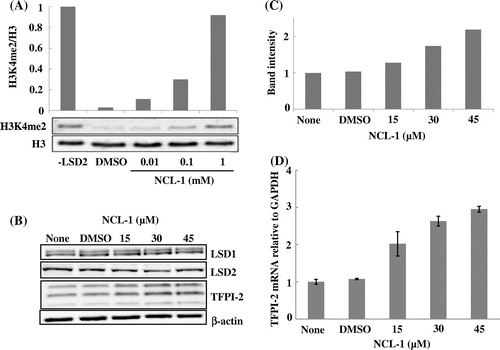
NCL-1 was synthesized as an inhibitor based on the crystal structure of LSD1,Citation26,27) and its therapeutic potential for breast cancer has been assessed using a preclinical model.Citation33) Inhibition of LSD1 by NCL-1 has been confirmed,Citation26) but it is not known whether this compound also inhibits LSD2. The activity of His-tagged full-length LSD2 purified from an E. coli extract was assayed using calf thymus histone as the substrate. As the calf thymus histone contains H3K4me2, LSD2 activity can be measured by monitoring a decrease of immunoreactive H3K4me2 with western blotting. As shown in Fig. (A), the H3K4me2 band was almost completely disappeared by LSD2 and the disappearance was prevented by NCL-1 in a dose-dependent manner, indicating that NCL-1 inhibited LSD2. Incubation of NCL-1 with HEK293 cells increased the TFPI-2 protein and mRNA levels in a dose-dependent manner (Fig. (B)–(D)). Thus, these two compounds inhibit both LSD1 and LSD2. To examine which LSD inhibition is responsible for the induction of TFPI-2, we performed knockdown of LSD1 and LSD2.
Fig. 3. Inhibition of LSD2 and induction of TFPI-2 protein and its mRNA by NCL-1.
Notes: NCL-1 was dissolved in DMSO. (A) Inhibition of LSD2 activity by NCL-1. The His-tagged full-length LSD2 activity was assessed by measuring the decrease in H3K4me2 by immunoblotting. The ratio of H3K4me2 to total H3 in the absence of LSD2 was defined as 1. (B) Induction of TFPI-2 protein in HEK293 cells treated with NCL-1 for 72 h. The triplet bands of TFPI-2 reflect differentially glycosylated products. (C) Quantification of TFPI-2 bands by densitometry. TFPI-2 in the control cells was defined as 1. (D) Induction of TFPI-2 mRNA in HEK293 cells treated with NCL-1. The ratio of TFPI-2 mRNA to GAPDH mRNA in the control cells was defined as 1. Data represent the mean ± SD of duplicate samples.
Induction of TFPI-2 by knockdown of LSD1 and LSD2
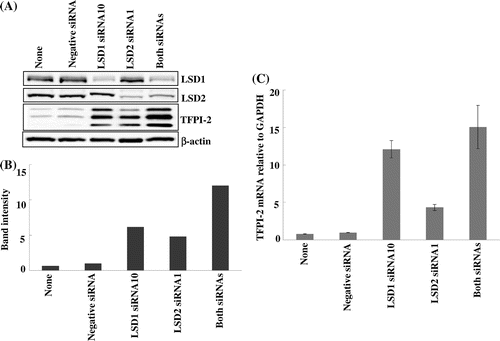
Two siRNAs each for LSD1 (LSD1siRNA2 and LSD1siRNA10) and LSD2 (LSD2siRNA1 and LSD2siRNA2) were examined to exclude off-target effects. LSD1siRNA2 reduced not only LSD1 mRNA, but also GAPDH mRNA, and inhibited cell proliferation, while LSD1siRNA10 decreased LSD1 mRNA significantly, but did not affect GAPDH mRNA (data not shown). LSD2siRNA1 and LSD2siRNA2 both decreased LSD2 mRNA significantly without any deteriorative effects on the cells. Therefore, LSD1siRNA10 and LSD2siRNA1 were used throughout the experiments.
Knockdown of LSD1 and LSD2 in HEK293 cells induced robust expression of TFPI-2 protein (Fig. (A) and (B)). The increase in TFPI-2 protein was probably attributable to an increase in its mRNA expression (Fig. (C)). Simultaneous knockdown of LSD1 and LSD2 showed the highest induction, which appeared to be additive effects of knockdown of the individual LSDs. These findings, together with those for the LSD inhibitors, indicate that inhibition of not only LSD1 but also LSD2 gives rise to increases in TFPI-2 protein and mRNA, suggesting transcriptional activation.
Fig. 4. Induction of TFPI-2 protein and mRNA by knockdown of LSD1 and LSD2.
Notes: Knockdown of LSD1 and LSD2 by siRNAs was performed as described in the Materials and Methods. (A) Immunoblotting. LSD1 and LSD2 were efficiently knocked down by LSD1siRNA10 and LSD2siRNA1, respectively. The triplet bands of TFPI-2 reflect differentially glycosylated products. (B) Quantification of TFPI-2 bands by densitometry. TFPI-2 from HEK293 cells treated with a negative siRNA was defined as 1. (C) TFPI-2 mRNA measured by qRT-PCR. The ratio of TFPI-2 mRNA to GAPDH mRNA in HEK293 cells treated with a negative siRNA was defined as 1. Data represent the mean ± SD of duplicate samples.
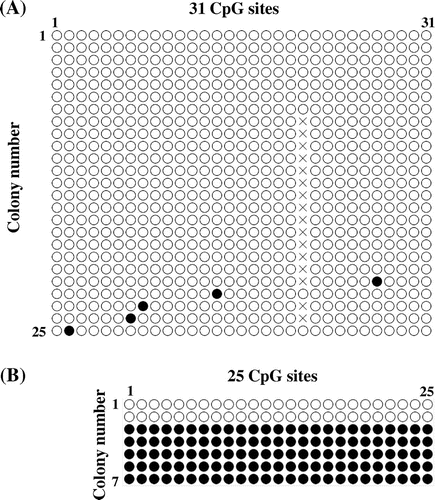
Methylation of the TFPI-2 promoter
As described above, LSD1 stabilizes DNMT1 through demethylation,Citation4) and thereby promotes DNA methylation including the TFPI-2 promoter. Induction of TFPI-2 expression by inhibition or knockdown of LSD1 and LSD2 may be derived from DNA demethylation. To examine this possibility, bisulfite sequencing was applied to assess the methylation of the TFPI-2 promoter region (−178 to +160), wherein 31 CpG sites exist. HEK293 cells almost completely lacked TFPI-2 promoter methylation (Fig. (A)). We also explored the methylation status of the DMR, which is located near the TFPI-2 gene.Citation29) The DMR region examined contains 25 CpG sites. As shown in Fig. (B), the DMR from two white E. coli colonies was fully unmethylated, whereas the DMR from five colonies was fully methylated. These results for the DMR are consistent with the observation that the typical pattern for the DMR consists of fully methylated and fully unmethylated alleles,Citation29) thereby validating our experimental procedures.
Fig. 5. Methylation status of the TFPI-2 promoter and DMR by bisulfite sequencing.
Notes: (A) Methylation status of the TFPI-2 promoter. The TFPI-2 promoter DNA containing 31 CpG sites was prepared from 25 white E. coli colonies and analyzed by bisulfite sequencing. ○, unmethylated; ●, methylated; X, unable to be determined. (B) Methylation status of the DMR. The DMR DNA containing 25 CpG sites was prepared from seven E. coli colonies and analyzed by bisulfite sequencing. ○, unmethylated; ●, methylated.
Taken together, it appears that both LSD1 and LSD2 contribute to repression of TFPI-2 expression in HEK293 cells through demethylation of H3K4me2, but not through methylation of the TFPI-2 promoter. Decreased expression of TFPI-2 is likely to contribute to the malignancy of cancer cells including metastasis,Citation17–19) and LSD1 may play a role in this process.Citation11) Although the involvement of LSD2 in cancer including metastasis remains unknown, our results suggest that LSD2 also plays a role.
LSD1 has been thought to repress the expression of active genes through demethylation of H3K4me2 in their promoter regions.Citation7) LSD2 also demethylates H3K4me2/1,Citation12–14) suggesting repressive effects on gene expression. Contrary to this expectation, Fang et al.Citation14) found that the majority (77%) of genes tested were downregulated after shRNA-mediated LSD2 depletion in HeLa cells. Interestingly, LSD2 predominantly associates with the intragenic regions of actively transcribed genes, and not with the promoter regions where LSD1 accumulates,Citation14) thus linking LSD2 to transcriptional regulation after initiation. Thus far, we are unable to provide reliable results of chromatin immunoprecipitation assays to identify the TFPI-2 chromatin regions where LSD1 and LSD2 function. Further studies of the mechanism by which TFPI-2 expression is repressed by LSD2 are warranted, as LSD2 is likely to be involved in tumorigenesis.
Acknowledgments
We thank Sayaka Nakagawa, Hiroka Ishisaka, Yuuya Yoshizawa, and Yoshiko Iwakura for their technical support.
Notes
Abbreviations: DMR, differentially methylated region; DNMT1, DNA cytosine-5 methyltransferase 1; ECM, extracellular matrix; LSD, lysine-specific demethylase; mAb, monoclonal antibody; NCL-1, N-((1S)-3-(3-(trans-2-aminocyclopropyl)phenoxy)-1-(benzylcarbamoyl)propyl) benzamide; qRT-PCR, quantitative reverse transcription-PCR; siRNA, small interfering RNA; TC, trans-2-phenylcyclopropylamine; TFPI-2, tissue factor pathway inhibitor-2.
Related Research Data
References
- Berger SL. The complex language of chromatin regulation during transcription. Nature 2007;447:407–412.10.1038/nature05915
- Baylin SB, Ohm JE. Epigenetic gene silencing in cancer – a mechanism for early oncogenic pathway addiction? Nat. Rev. Cancer 2006;6:107–116.10.1038/nrc1799
- Esteve PO, Chin HG, Benner J, Feehery GR, Samaranayake M, Horwitz GA, Jacobsen SE, Pradhan S. Abnormal development of the cerebral cortex and cerebellum in the setting of lamin B2 deficiency. Proc. Nat. Acad. Sci. USA 2010;106:5076–5081.10.1073/pnas.0810362106
- Wang J, Hevi S, Kurash JK, Lei H, Gay F, Bajko J, Su H, Sun W, Chang H, Xu G, Gaudet F, Li E, Chen T. The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation. Nat. Genet. 2009;41:125–129.10.1038/ng.268
- Marmorstein R, Trievel RC. Histone modifying enzymes: structures, mechanisms, and specificities. Biochim. Biophys. Acta. 2009;1789:58–68.10.1016/j.bbagrm.2008.07.009
- Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, Shi Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004;119:941–953.10.1016/j.cell.2004.12.012
- Shi Y. Histone lysine demethylases: emerging roles in development, physiology and disease. Nat. Rev. Genet. 2007;8:829–833.10.1038/nrg2218
- Klose RJ, Kallin EM, Zhang Y. JmjC-domain-containing proteins and histone demethylation. Nat. Rev. Genet. 2006;7:715–727.10.1038/nrg1945
- Pedersen MT, Helin K. Histone demethylases in development and disease. Trends Cell Biol. 2010;20:662–671.10.1016/j.tcb.2010.08.011
- Hayami S, Kelly JD, Cho HS, Yoshimatsu M, Unoki M, Tsunoda T, Field HI, Neal DE, Yamaue H, Ponder BA, Nakamura Y, Hamamoto R. Overexpression of LSD1 contributes to human carcinogenesis through chromatin regulation in various cancers. Int. J. Cancer 2011;128:574–586.10.1002/ijc.v128.3
- Schulte JH, Lim S, Schramm A, Friedrichs N, Koster J, Versteeg R, Ora I, Pajtler K, Klein-Hitpass L, Kuhfittig-Kulle S, Metzger E, Schule R, Eggert A, Buettner R, Kirfel J. Lysine-specific demethylase 1 is strongly expressed in poorly differentiated neuroblastoma: implications for therapy. Cancer Res 2009;69:2065–2071.10.1158/0008-5472.CAN-08-1735
- Karytinos A, Forneris F, Profumo A, Ciossani G, Battaglioli E, Binda C, Mattevi A. A novel mammalian flavin-dependent histone demethylase. J. Biol. Chem. 2009;284:17775–17782.10.1074/jbc.M109.003087
- Ciccone DN, Su H, Hevi S, Gay F, Lei H, Bajko J, Xu G, Li E, Chen T. KDM1B is a histone H3K4 demethylase required to establish maternal genomic imprints. Nature 2009;461:415–418.10.1038/nature08315
- Fang R, Barbera AJ, Xu Y, Rutenberg M, Leonor T, Bi Q, Lan F, Mei P, Yuan GC, Lian C, Peng J, Cheng D, Sui G, Kaiser UB, Shi Y, Shi YG. Human LSD2/KDM1b/AOF1 regulates gene transcription by modulating intragenic H3K4me2 methylation. Mol. Cell 2010;39:222–233.10.1016/j.molcel.2010.07.008
- Overall CM, López-Otín C. Strategies for MMP inhibition in cancer: innovations for the post-trial era. Nat. Rev. Cancer 2002;2:657–672.10.1038/nrc884
- Sato H, Takino T, Okada Y, Cao J, Shinagawa A, Yamamoto E, Seiki M. A matrix metalloproteinase expressed on the surface of invasive tumour cells. Nature 1994;370:61–65.10.1038/370061a0
- Rao CN, Lakka SS, Kin Y, Konduri SD, Fuller GN, Mohanam S, Rao JS. Expression of tissue factor pathway inhibitor 2 inversely correlates during the progression of human gliomas. Clin. Cancer Res. 2001;7:570–576.
- Lakka SS, Konduri SD, Mohanam S, Nicolson GL, Rao JS. In vitro modulation of human lung cancer cell line invasiveness by antisense cDNA of tissue factor pathway inhibitor-2. Clin. Exp. Metastasis 2000;18:239–244.10.1023/A:1006755223357
- Konduri SD, Tasiou A, Chandrasekar N, Rao JS. Overexpression of tissue factor pathway inhibitor-2 (TFPI-2), decreases the invasiveness of prostate cancer cells in vitro. Int. J. Oncol. 2001;18:127–131.
- Kondraganti S, Gondi CS, Gujrati M, McCutcheon I, Dinh DH, Rao JS, Olivero WC. Restoration of tissue factor pathway inhibitor inhibits invasion and tumor growth in vitro and in vivo in a malignant meningioma cell line. Int. J. Oncol. 2006;29:25–32.
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646–674.10.1016/j.cell.2011.02.013
- Konduri SD, Srivenugopal KS, Yanamandra N, Dinh DH, Olivero WC, Gujrati M, Foster DC, Kisiel W, Ali-Osman F, Kondraganti S, Lakka SS, Rao JS. Promoter methylation and silencing of the tissue factor pathway inhibitor-2 (TFPI-2), a gene encoding an inhibitor of matrix metalloproteinases in human glioma cells. Oncogene 2003;22:4509–4516.10.1038/sj.onc.1206695
- Hubé F, Reverdiau P, Iochmann S, Rollin J, Cherpi-Antar C, Gruel Y. Transcriptional silencing of the TFPI-2 gene by promoter hypermethylation in choriocarcinoma cells. Biol. Chem. 2003;384:1029–1034.
- Heidenblad M, Lindgren D, Jonson T, Liedberg F, Veerla S, Chebil G, Gudjonsson S, Borg Å, Månsson W, Höglund M. Tiling resolution array CGH and high density expression profiling of urothelial carcinomas delineate genomic amplicons and candidate target genes specific for advanced tumors. BMC Med. Genet. 2008;1:3.10.1186/1755-8794-1-3
- Orlic M, Spencer CE, Wang L, Gallie BL. Expression analysis of 6p22 genomic gain in retinoblastoma. Genes, Chromosomes and Cancer 2006;45:72–82.10.1002/(ISSN)1098-2264
- Ueda R, Suzuki T, Mino K, Tsumoto H, Nakagawa H, Hasegawa M, Sasaki R, Mizukami T, Miyata N. Identification of cell-active lysine specific demethylase 1-selective inhibitors. J. Am. Chem. Soc. 2009;131:17536–17537.10.1021/ja907055q
- Ogasawara D, Suzuki T, Mino K, Ueda R, Khan MN, Matsubara T, Koseki K, Hasegawa M, Sasaki R, Nakagawa H, Mizukami T, Miyata N. Synthesis and biological activity of optically active NCL-1, a lysine-specific demethylase 1 selective inhibitor. Bioorg. Med. Chem. 2011;19:3702–3708.10.1016/j.bmc.2010.12.024
- Morishita E, Narita H, Nishida M, Kawashima N, Yamagishi K, Masuda S, Nagao M, Hatta H, Sasaki R. Anti-erythropoietin receptor monoclonal antibody: epitope mapping, quantification of the soluble receptor, and detection of the solubilized transmembrane receptor and the receptor-expressing cells. Blood 1996;88:465–471.
- Ribarska T, Ingenwerth M, Goering W, Engers R, Schulz WA. Epigenetic inactivation of the placentally imprinted tumor suppressor gene TFPI2 in prostate carcinoma. Cancer Genomics Proteomics 2010;7:51–60.
- Monk D, Wagschal A, Arnaud P, Muller PS, Parker-Katiraee L, Bourc’his D, Scherer SW, Feil R, Stanier P, Moore GE. Comparative analysis of human chromosome 7q21 and mouse proximal chromosome 6 reveals a placental-specific imprinted gene, TFPI2/Tfpi2, which requires EHMT2 and EED for allelic-silencing. Genome Res. 2008;18:1270–1281.10.1101/gr.077115.108
- Schmidt DM, McCafferty DG. trans-2-Phenylcyclopropylamine is a mechanism-based inactivator of the histone demethylase LSD1. Biochemistry 2007;46:4408–4416.10.1021/bi0618621
- Rao CN, Reddy P, Liu Y, O’toole E, Reeder D, Foster DC, Kisiel W, Woodley DT. Extracellular matrix-associated serine protease inhibitors (Mr 33,000, 31,000, and 27,000) are single-gene products with differential glycosylation: cDNA cloning of the 33-kDa inhibitor reveals its identity to tissue factor pathway inhibitor-2. Arch. Biochem. Biophys. 1996;335:82–92.10.1006/abbi.1996.0484
- Cortez V, Mann M, Tekmal S, Suzuki T, Miyata N, Rodriguez-Aguayo C, Lopez-Berestein G, Sood AK, Vadlamudi RK. Targeting the PELP1-KDM1 axis as a potential therapeutic strategy for breast cancer. Breast Cancer Res. 2012;14:R108.10.1186/bcr3229