
Abstract
The quinoline antibiotics aurachins C, D, and L, and a structurally simplified analog of aurachin C were synthesized from 1-(2-nitrophenyl)butane-1,3-dione via reductive cyclizations of δ-nitro ketone intermediates, with zinc or iron as key steps. The results of antimicrobial tests indicate that the N-hydroxyquinolone nucleus mimics the electron carrier in the respiratory chain more strongly than the quinoline N-oxide nucleus.
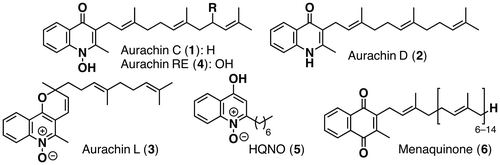
Bacterial respiratory chain inhibitors are promising antibiotics, since there are some differences between the mammalian respiratory chain and the bacterial one. In particular, the quinoline alkaloids have received considerable attention because of their significant activity in the quinol oxidation sites of the bacterial respiratory chain.Citation1,2) Quinoline alkaloids aurachins C (1), D (2), and L (3) were isolated from the culture broth of Stigmatella aurantica by Höfle et al.Citation3,4) Kitagawa et al. isolated the 9′-hydroxyaurachin C from a culture broth of Rhodococcus erythropolis and named it aurachin RE (4).Citation5–7) While 1, 2, and 4 have been reported to exhibit antimicrobial activity against many Gram-positive bacteria, no biological activity of 3 has been reported thus far. Considering the structural similarity among aurachins and 2-heptyl-4-hydroxyquinoline-N-oxide (HQNO) (5), which is an electron transport inhibitor of the respiratory chain,Citation8) the N-hydroxyquinolone nucleus is likely responsible for the activity.Citation9,10) On the other hand, the side chain would not be so important for antibiotic activity, because substrates in the bacterial respiratory chain like menaquinone (6) have different side chain lengths. The unique structures and the potential of aurachins as a new lead compound for practical antibiotics have prompted synthetic efforts toward aurachins and their analogs by organic chemists. Two research groups have succeeded in the total synthesis of 2 and examined the effect of the side chain lengths so far.Citation11,12) Both two groups reported that the geranyl chain analog retained the antibiotic activity but the prenyl chain analog was considerably less active than 2. According to their studies on aurachin D, a structurally simplified analog of aurachin C with a shorter side chain would show as strong of an antimicrobial activity as aurachin C. We describe herein the total synthesis of aurachins C (1), D (2) and L (3), and the structurally simplified analog of aurachin C together with biological activities of 3 and the analog (Fig. ).
Fig. 1. Structures of aurachins C (1), D (2), L (3), RE (4), HQNO (5) & menaquinone (6).
As shown in Scheme , our synthesis of 1 began with the alkylation of known ketone 7Citation13) with farnesyl bromide to give 8 as a 1:1 keto–enol tautomeric mixture. Then the alkylation product 8 was reductively cyclized with zinc dust in the presence of ammonium chloride to afford aurachin C (1) in 46% yield.Citation14) In contrast, the synthesis of aurachin D (2) was accomplished in 63% yield by a reductive cyclization with iron dust in the presence of hydrochloric acid. Although the acids used in these reactions were different, these results are likely due to the smaller first ionization energy of iron atoms (762.5 kJ/mol) than that of zinc atoms (906.4 kJ/mol).Citation15) Stronger reducing power of the iron atom compared to that of the zinc would make it possible to convert the nitro group into the amine. The 1H- and 13C NMR of 1 and 2 were identical to those of natural products.
Scheme 1. Synthesis of aurachins C (1), D (2), L (3) & aurachin C analong 12.
Notes: Reagents and conditions: (a) NaH, farnesyl bromide, HMPA, THF, 0–60 °C (52%, 69% based on recovered 7); (b) Zn dust, NH4Cl, EtOH/H2O (1:1), 80 °C (46%); (c) Fe dust, 6 M HCl, EtOH, 90 °C (63%); (d) farnesal, piperidine, toluene, rt to 60 °C, (39%); (e) Zn dust, NH4Cl, EtOH/H2O (1:1), 80 °C (29%); (f) NaH, allyl bromide, HMPA, THF, 0–50 °C; (g) 1-hexene, Grubbs’ 2nd generation catalyst, CH2Cl2, reflux (11a: 9% from 7, 11b: 12% from 7, 28% based on recovered 7); (h) Zn dust, NH4Cl, EtOH/H2O (1:1), 80 °C (49%).
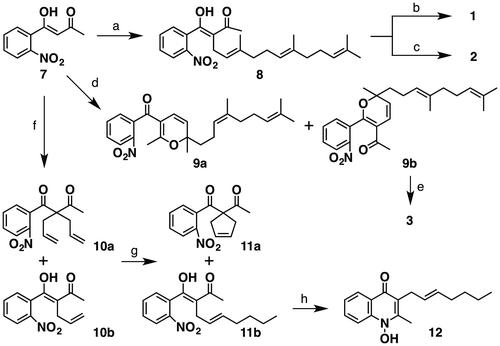
Having completed the synthesis of aurachins C and D, we then turned our attention to the synthesis of aurachin L (3). The plausible biogenesisCitation4) proposed by Höfle et al. suggests that 2H-pyran moiety of 3 is constructed by 6π-electrocyclization from the corresponding dienone precursor. In light of the biogenesis of 3, the moiety would be concisely constructed by a domino Knoevenagel/6π-electrocyclization. Thus we subjected 7 to farnesal under the Knoevenagel conditions to afford an inseparable mixture of desired cycloadduct 9b and presumably its isomer 9a in 39% combined yield.Footnote1 Finally, the mixture was exposed to the reductive cyclization conditions using zinc dust to afford aurachin L (3) in 29% yield together with a separable by-product derived from 9a. The 1H- and 13C NMR of 3 exhibited good agreement with those of natural aurachin L.
Next, we synthesized a structurally simplified analog of 1. According to the SAR study reported by Nay’s and Speicher’s group, the geranyl chain (i.e. 3,7-dimethyl-2,6-octadienyl chain) analog almost conserved the antibiotic activity.Citation11,12) Thus, we set 12 as a target molecule to consider whether the 7-carbon straight side chain analog is able to retain the activity or not. By following the same procedure conducted for the preparation of 8, 7 was alkylated with allyl bromide to give an inseparable 2:3 mixture of 10a and 10b. For side chain elongation of 10b, we employed cross-metathesis with 1-hexene and obtained a separable 4:5 mixture of desired product 11b and RCM product 11a, which was removed by SiO2 chromatography at this stage. The reductive cyclization of 11b with zinc dust proceeded smoothly to afford 12 in 49%. The 1H- and 13C NMR of 12 were analogous to those of 1 except for signals due to the side chain moiety.
The antimicrobial activities of 3 and 12 were tested by an agar diffusion assay. As shown in Table , 3 showed 10–17 mm zones of inhibition when 500 ng/disks of the compounds were applied on paper disks. On the other hand, the analog 12 was found to exhibit strong antimicrobial activity at only 50 ng/disks. These results indicated that the branching in the side chain was not important for the activity and the analog (12) with a shorter side chain than the geranyl group in aurachin C (1) conserved the activity. We are considering that the difference in biological activity between 3 and 12 would be ascribable to the structural difference in their nitrogen-containing heterocyclic rings (quinoline N-oxide nucleus in 3 and N-hydroxyquinolone nucleus in 12). Since the N-hydroxyquinolone nucleus included in 12 seems to be more similar to the naphthoquinone moiety of the electron carrier like 6 than the quinoline N-oxide nucleus in 3, 12 might better mimic the electron carrier than 3.
Table 1. Antimicrobial spectra of 3 & 12.
In conclusion, we successfully achieved two-step syntheses of aurachins C and D from readily available starting material 7 in 24 and 33% overall yields, respectively. It is noteworthy that the two different types of the nucleus, N-hydroxyquinolone and quinolone, could be synthesized by changing the metal employed in the reductive cyclization. The first synthesis of aurachin L was also accomplished in 11% overall yield by utilizing a domino Knoevenagel/6π-electrocyclization for the construction of 2H-pyran moiety. The results of antimicrobial tests for 3 and 12 indicated that N-hydroxyquinolone nucleus better mimics the electron carrier than the quinoline N-oxide nucleus.
Supectral data of aurachins C (1), D (2) & L (3)
Aurachin C (1): IR νmax: 2300–3500 (s), 2919 (s), 1533 (s), 1430 (s), 1530 (s), 756 (m); 1H NMR (270 MHz, CD3OD) δ: 1.53 (3H, s), 1.56 (3H, s), 1.61 (3H, s), 1.81 (3H, s), 1.85–2.15 (8H, m), 2.57 (3H, s), 3.47 (3H, d, J = 6.8 Hz), 4.95–5.17 (2H, m), 7.40 (1H, ddd, J = 8.2, 7.2, 1.35 Hz), 7.71 (1H, ddd, J = 8.5, 7.2, 1.2 Hz), 7.98 (1H, d, J = 8.5 Hz), 8.28 (1H, dd, J = 8.2, 1.2 Hz); 13C NMR (67.5 MHz, CD3OD) δ: 15.8, 16.8, 17.1, 18.4, 26.4, 26.6, 28.2, 28.5, 41.48, 41.52, 116.8, 121.0, 124.5, 125.7, 125.9, 126.0, 126.1, 127.0, 132.7, 133.5, 136.7, 137.0, 141.6, 151.7, 175.3; HRMS (FAB) m/z: calcd. for C25H34O2 N, 380.2590; found, 380.2592 ([M + H]+).
Aurachin D (2): IR νmax: 2916 (s), 1552 (m), 1494 (s); 1H NMR (270 MHz, CDCl3) δ: 1.55 (3H, s), 1.55 (3H, s), 1.65 (3H, s), 1.74 (3H, s), 1.77–2.10 (8H, m), 2.45 (3H, s), 3.41 (2H, d, J = 6.5 Hz), 5.00–5.18 (3H, m), 7.27 (1H, t, J = 8.3 Hz), 7.40 (1H, d, J = 8.3 Hz), 7.50 (1H, d, J = 6.8 Hz), 8.36 (1H, d, J = 7.3 Hz), 9.83 (1H, s); 13C NMR (67.5 MHz, CDCl3) δ: 16.1, 16.4, 17.8, 18.9, 24.2, 25.8, 26.8, 26.9, 39.8, 39.8, 117.3, 120.1, 122.5, 123.2, 124.3, 124.5, 126.3, 131.3, 131.4, 135.0, 135.3, 139.2, 146.3, 177.3; HRMS (FAB) m/z: calcd. for C25H34ON, 364.2638; found, 364.2640 ([M + H]+).
Aurachin L (3): IR νmax: 2924(s), 1325 (m), 1063 (s); 1H NMR (500 MHz, CDCl3) δ: 1.52 (3H, s), 1.53 (3H, s), 1.57 (3H, s), 1.66 (3H, s), 1.78–1.84 (1H, m), 1.85–1.90 (1H, m), 1.90–1.95 (2H, m), 2.00–2.04 (2H, m), 2.14–2.20 (2H, m), 2.75 (3H, s), 5.06 (1H, tt, J = 6.0, 1.0 Hz), 5.11 (1H, td, J = 5.5, 2.0 Hz), 5.76 (1H, d, J = 8.5 Hz), 6.59 (1H, d, J = 8.5 Hz), 7.55 (1H, td, J = 7.0, 2.0 Hz), 7.72 (1H, td, J = 7.0, 0.8 Hz), 8.15 (1H, dd, J = 7.0, 0.8 Hz), 8.72 (1H, d, J = 7.0 Hz); 13C NMR (125 MHz, CDCl3) δ: 14.1, 16.0, 17.7, 22.4, 25.7, 26.5, 26.6, 39.6, 41.2, 80.5, 111.1, 118.4, 119.9, 120.6, 122.1, 123.2, 124.2, 127.1, 130.1, 130.7, 131.4, 135.9, 140.9, 143.6, 147.4; HRMS (FAB) m/z: calcd. for C25H32O2 N, 378.2428; found, 378.2437 ([M + H]+).
Spectral Data for 8, 11b, and 12, and experimental procedures for 1, 2, 3, 8, 11b, and 12 are available on Biosci. Biotechnol. Biochem. Web site.
Supplemental material
The supplemental material for this paper is available at http://dx.doi.org/10.1080/09168451.2014.918494.
bbb-140111-File005_1_.pdf
Download PDF (269.7 KB)Acknowledgement
We thank Ms Yamada (Tohoku University) and Mr Kurashina for measuring NMR, IR, and MS spectra.
Funding
This work was supported, in part, by a grant-in-aid for Young Scientists (B) from the Ministry of Education, Culture, Sports, Science and Technology of Japan [grant number 23780112].
Notes
Abbreviations: HMPA, hexamethylphosphoric triamide; RCM, ring-closing metathesis; MIC, minimum inhibitory concentration; SAR, structure-activity relationship.
1. Comparison of the peak integrations of the double bond proton signals in the resulting pyran rings of 9a ([270 MHz, CDCl3] δ: 6.83 [1H, d, J = 12.2 Hz], 6.39 [1H, d, J = 12.2 Hz]) and 9b ([270 MHz, CDCl3] δ: 5.86 [1H, d, J = 10.3 Hz], 5.06 [1H, d, J = 10.3 Hz]) indicated their ratio to be 1:1.2.
References
- Oettmeier W, Masson K, Soll M, Reil E. Acridones and quinolones as inhibitors of ubiquinone functions in the mitochondrial respiratory chain. Biochem. Soc. Trans. 1994;22:213–216.
- Friedrich T, Ohnishi T, Forche E, Kunze B, Jansen R, Trowitzsch W, Höfle G, Reichenbach H, Weiss H. Two binding sites for naturally occurring inhibitors in mitochondrial and bacterial NADH: ubiquinone oxidoreductase (Complex I). Biochem. Soc. Trans. 1994;22:226–230.
- Kunze B, Höfle G, Reichenbach H. The aurachins, new quinoline antibiotics from myxobacteria: production, physico-chemical and biological properties. J. Antibiot. 1987;40:258–265.10.7164/antibiotics.40.258
- Höfle G, Kunze B. Biosynthesis of aurachins A–L in Stigmatella aurantiaca: a feeding study. J. Nat. Prod. 2008;71:1843–1849.10.1021/np8003084
- Kitagawa W, Tamura T. A quinoline antibiotic from Rhodococcus erythropolis JCM 6824. J. Antibiot. 2008;61:680–682.10.1038/ja.2008.96
- Kitagawa W, Ozaki T, Nishioka T, Yasutake Y, Hata M, Nishiyama M, Kuzuyama T, Tamura T. Cloning and heterologous expression of the aurachin RE biosynthesis gene cluster afford a new cytochrome P450 for quinoline N-hydroxylation. ChemBioChem. 2013;14:1085–1093.10.1002/cbic.201300167
- Yasutake Y, Kitagawa W, Hata M, Nishioka T, Ozaki T, Nishiyama M, Kuzuyama T, Tamura T. Structure of the quinoline N-hydroxylating cytochrome P450 RauA, an essential enzyme that confers antibiotic activity on aurachin alkaloids. FEBS Lett. 2014;588:105–110.10.1016/j.febslet.2013.11.016
- Lightbown JW, Jackson FL. Inhibition of cytochrome systems of heart muscle and certain bacteria by the antagonists of dihydrostreptomycin: 2-alkyl-4-hydroxyquinoline N-oxides. Biochem. J. 1956;63:130–137.
- Miyoshi H, Takegami K, Sakamoto K, Mogi T, Iwamura H. Characterization of the ubiquinol oxidation sites in cytochromes bo and bd from Escherichia coli using aurachin C analogues. J. Biochem. 1999;125:138–142.10.1093/oxfordjournals.jbchem.a022250
- Michael JP. Quinoline, quinazoline and acridone alkaloids. Nat. Prod. Rep. 1997;14:605–618.10.1039/np9971400605
- Li X-W, Herrmann J, Zang Y, Grellier P, Prado S, Müller R, Nay B. Synthesis and biological activities of the respiratory chain inhibitor aurachin D and new ring versus chain analogues. Beilstein J. Org. Chem. 2013;9:1551–1558.10.3762/bjoc.9.176
- Dejon L, Speicher A. Synthesis of aurachin D and isoprenoid analogues from the myxobacterium Stigmatella aurantiaca. Tetrahedron Lett. 2013;54:6700–6702.10.1016/j.tetlet.2013.09.085
- Brie M, Silberg LA. Derivatives of pyrazoles. I. Synthesis and IR-Spectra of some new 3-Methyl-5-aryl-pyrazoles. Revue Roumaine de Chimie [Roumanian J. Chem.]. 1989;34:733–737.
- Kamm O. ß-phenylhydroxylamine. Org. Synth. Colloid. 1941;1:445–447.
- James AM, Lord MP. Macmillan’s Chemical and Physical Data. London: Macmillan; 1992.