
Abstract
Cryptocarya diacetate and each of its stereoisomers were stereoselectively synthesized in 8–16 steps. One of the three chiral carbons was converted from the chiral center of a yeast-reduction product. The other two chiral carbons were constructed by employing stereoselective allylation and syn-and anti-1,3-reductions. The enantiomeric excesses of the synthesized cryptocarya diacetate and its stereoisomers were determined to be more than 99%ee using a chiral column.
Graphical Abstract
Cryptocarya diacetate and its all stereoisomers were stereoselectively synthesized through 8-16 steps. The enantiomeric excess of all stereoisomers were determined as more than 99%ee.
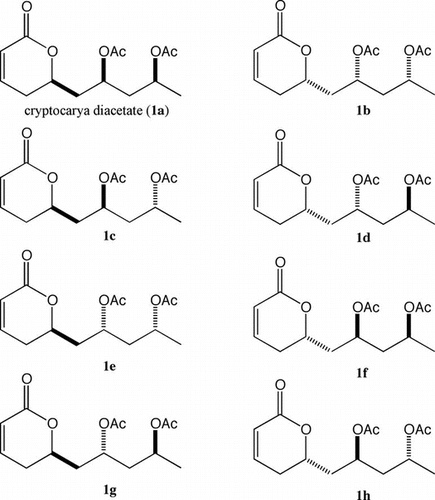
The 6-substituted 5,6-dihydropyran-2-one structures have a variety of useful properties.Citation1) Although each biological activity is interesting, the effect of stereochemistry on the activity is unknown. Cryptocarya diacetate (1a) has been isolated from the African medicinal plant Cryptocarya latifoliaCitation2) and its absolute configuration is determinedCitation3) (Fig. ). The stereochemistry–bioactivity relationship is an interesting area of research: hence, in this project, we focused on the stereoselective syntheses of each of the stereoisomers of cryptocarya diacetate. Cryptocarya diacetate is an important synthetic targetCitation4–9) and the synthesis of all the stereoisomers of cryptocarya diacetate has been reported;Citation10) however, the enantiomeric excess of each stereoisomer has not been determined. The development of new synthetic routes to get an optically pure natural compound and optically pure stereoisomers is necessary in order to pursue research on the structure–activity relationship.
Fig. 1. Cryptocarya diacetate (1a) and its stereoisomers 1b–1h.
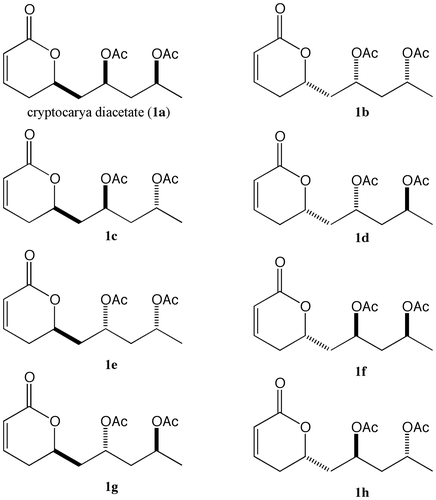
The retrosynthetic analysis is shown in Scheme . In this research, the synthesis of each of the eight optically pure stereoisomers (1a–1h) was started from the yeast-reduction products, 8 and 9.Citation11) The stereoselective allylation to aldehyde, obtained from the yeast-reduction product 8, gives alcohols 6 and 7. After the conversion of allyl alcohols 6 and 7 to β-hydroxy ketones by Wacker oxidation, syn- and anti-stereoselective reductions give the syn-diols 2 and 4 and the anti-diols 3 and 5, respectively. These compounds can be transformed to a 6-membered lactone by employing Baeyer–Villiger oxidation as the key reaction, giving cryptocarya diacetate (1a) and its stereoisomers 1c, 1e, and 1g. The remaining enantiomers 1b, 1d, 1f, and 1h can be obtained from the yeast-reduction product 9 using similar processes. This is the new stereoselective synthetic strategy for obtaining optically pure stereoisomers of cryptocarya diacetate.
Scheme 1. Retrosynthetic analysis of cryptocarya diacetate (1a) and its stereoisomers.
Results and discussion
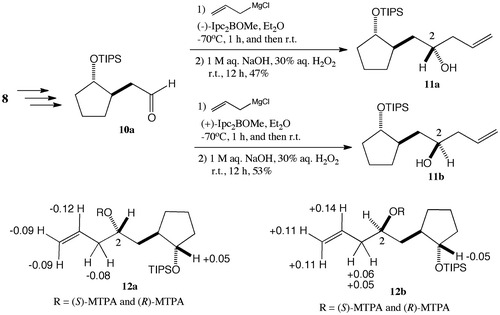
Our synthesis started with stereoselective allylation to aldehyde 10a,Citation12) which was prepared from the yeast-reduction product 8, using (+)-Ipc2BOMe and (-)-Ipc2BOMeCitation13) with allylmagnesium chloride to give secondary alcohols 11a (47% yield, along with 8% of 11b) and 11b (53% yield, along with 8% of 11a), respectively. The stereochemistries of 11a and 11b were determined by the modified Mosher’s methodCitation14) (Scheme ). The secondary alcohols 11a and 11b were converted to (S)- and (R)-α-methoxy-α-(trifluoromethyl)phenylacetate (MTPA) 12a and 12b by employing MTPACl and pyridine, respectively. The ΔδS−R values on the left side of the C2 position of 12a were negative, and those on the right side were positive, showing that the C2 position of 11a was 2S. On the other hand, the ΔδS−R values on the left side of the C2 position of 12b were positive, and those on the right side were negative. With these results, the C2 position of 11b was assigned to be 2R. Stereoselectivity was not observed in the allylation to aldehyde 10a without (+)- and (−)-Ipc2BOMe, which gave 11a (46%) and 11b (47%), respectively.
Scheme 2. Determination of stereochemistry of allyl alcohols 11a and 11b. Chemical shift differences (ΔδS−R) of (S)- and (R)-MTPA 12a and 12b.
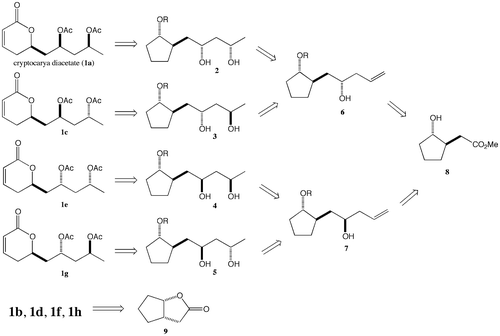
To construct the chiral carbons at the seventh and ninth positions, syn and anti- reductions of β-hydroxy ketone were planned (Scheme ). To achieve this plan, allyl alcohol 11a was subjected to Wacker oxidation using PdCl2 and Cu(OAc),Citation2,15) giving β-hydroxy ketone 13a (48% yield). Subjecting the β-hydroxy carbonyl compound 13a to 1,3-syn-reduction— employing the Et2BOMe-NaBH4 system—Citation16) provided syn-diol 14a (90% yield) as a single isomer. Diacetylation of the resulting diol using acetic anhydride in pyridine (95% yield), followed by the removal of the triisopropylsilyl group by treatment with tetra-n-butylammonium fluoride, gave the secondary alcohol 16a (67% yield), which was subjected to pyridinium chlorochromate oxidation to give ketone 17a (76% yield). By employing m-chloroperoxybenzoic acid, Baeyer–Villiger oxidation was performed to give lactone 18a (70% yield), which was converted to (+)-cryptocarya diacetate (1a) (84% yield) by treating it with benzeneseleninic acid anhydride.Citation17) The NMR spectroscopic data of the synthesized compound 1a were identical to those previously reported for this compound.Citation9) To obtain stereoisomer 1c, the application of 1,3-anti-reduction, using Me4NHB(OAc)3,Citation18) to β-hydroxy ketone 13a was attempted as the first step. The obtained anti-diol 14c (76% yield, along with 11% of syn-diol) was converted to diacetate 15c (92% yield). The cleavage of the silyl ether group from 15c, led to the formation of the secondary alcohol 16c (77% yield), which was then subjected to pyridinium chlorochromate oxidation (75% yield) followed by Baeyer–Villiger oxidation, to give lactone 18c (74% yield). Finally, conversion to α,β-unsaturated lactone gave the stereoisomer 1c (80% yield). Stereoisomers 1e and 1g were obtained from 11b, using the same synthetic method described for the syntheses of (+)-cryptocarya diacetate (1a) and stereoisomer 1c, respectively. The syn-reduction of 13b exclusively proceeded to give 14e as a single isomer with 88% yield. In the case of anti-reduction of 13b, the desired anti-diol 14g was stereoselectively obtained (79% yield) along with a syn-product (0.9% yield). The NMR data of all the stereoisomers agreed with those in the literature.Citation4,10) The syntheses of enantiomers 1b, 1d, 1f, 1h were accomplished from the yeast-reduction product 9 via aldehyde 10b by employing the same method described for the syntheses of 1a, 1c, 1e, and 1g (Scheme ). The enantiomeric excesses of 1a–1h were determined to be more than 99%ee by HPLC analysis using a chiral column.
Scheme 3. Syntheses of cryptocarya diacetate (1a) and its stereoisomers 1c, 1e, and 1g. (a) PdCl2, Cu(OAc)2 · H2O, O2, DMF, r.t., 48 h (13a: 48%, 13b: 51%). (b) Et2BOMe, NaBH4, THF-MeOH, −70 °C, 3 h, warmed to 0 °C (14a: 90%, 14e: 88%). (c) Ac2O, DMAP, pyridine, r.t., 12 h (15a: 95%, 15c: 92%, 15e: 98%, 15g: 94%). (d) n-Bu4NF, THF, r.t., 12 h (16a: 67%, 16c: 77%, 16e: 73%, 16g: 78%). (e) PCC, MS 4A, CH2Cl2, r.t., 12 h (17a: 76%, 17c: 75%, 17e: 80%, 17g: 79%). (f) MCPBA, Na2HPO4–NaH2PO4 buffer (pH 8.0), CHCl3, r.t, 12 h (18a: 70%, 18c: 74%, 18e: 69%, 18g: 69%). (g) (C6H5Se = O)2O, ClC6H5, 130 °C, 72 h (1a: 84%, 1c: 80%, 1e: 81%, 1g: 85%). (h) Me4NHB(OAc)3, MeCN, AcOH, −40–0 °C, 1 h (14c: 76%, 14g: 79%).
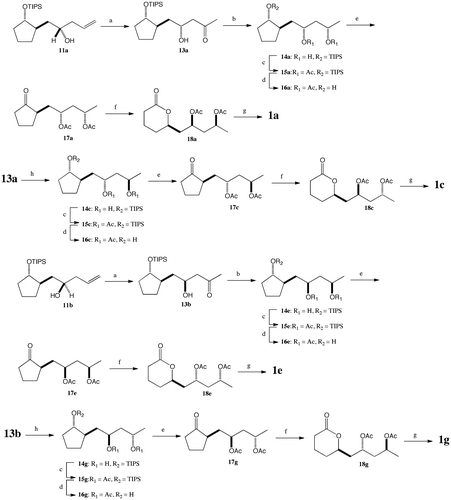
Scheme 4. Syntheses of stereoisomers of cryptocarya diacetate 1b, 1d, 1f, and 1h. (a) (1) 2 M aq. NaOH, EtOH, 60 °C, 16 h; (2) TIPSOTf, 2,6-lutidine, CH2Cl2, r.t., 1 h (84%, 2 steps); (3) HCO2H, ether, r.t., 1 h (86%); (4) PCC, MS 4A, CH2Cl2, r.t., 16 h (84%).
A stereoselective synthetic method for producing cryptocarya diacetate (1a) and all its stereoisomers was exploited by employing the yeast-reduction products 8 and 9 as starting materials. The enantiomeric excesses of all the stereoisomers 1a–1h were determined to be more than 99%ee. The three chiral carbons were constructed by yeast-reduction, asymmetric allylation, and 1,3-syn- and anti-reductions. This is the new report on the syntheses of all the optically pure stereoisomers of cryptocarya diacetate. The success of this work provides a significant contribution to biological research.
Experimental
Optical rotations were measured on a Jasco P-2100 polarimeter. IR data were obtained using a Horiba FT-720 instrument. NMR data were obtained using a JNM-EX400 spectrometer. EIMS data were recorded with a JMS-MS700 V spectrometer. The silica gel used was Wakogel C-300 (Wako, 200–300 mesh). The purity of each compound containing enantiomers was confirmed by 1H NMR. HPLC analysis was performed with Shimadzu LC-6AD and SPD-6AV instruments. The chiral column used for HPLC analysis of enantiomeric excess was CHIRALCEL AD-H (DAICEL Chemical Industries Ltd, Tokyo, Japan).
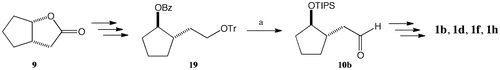
[(1S,2R)-2-(Triisopropylsilyloxy)cyclopent-1-yl]acetaldehyde (10b)
A reaction solution of benzoate 19Citation19) (4.56 g, 9.57 mmol) in 2 M aq. NaOH solution (84 mL) and EtOH (240 mL) was stirred at 60 °C for 16 h before the addition of CHCl3 and H2O. The organic solution was separated and then dried (Na2SO4). Concentration of the solvent gave crude secondary alcohol. To an ice-cooled solution of the crude secondary alcohol and 2,6-lutidine (2.47 mL, 21.2 mmol) in CH2Cl2 (10 mL) was added TIPSOTf (3.44 mL, 12.8 mmol), and then the reaction solution was stirred at room temperature for 1 h before the addition of sat. aq. CuSO4 solution. The organic solution was separated, washed with sat. aq. NaHCO3 solution, and then dried (Na2SO4). Concentration of the solvent followed by silica gel column chromatography (2%EtOAc/hexane), gave silyl ether (4.67 g, 8.83 mmol, 92%) as a colorless oil. To a solution of silyl ether (4.67 g, 8.83 mmol) in ether (260 mL) was added formic acid (230 mL) at −10 °C. After stirring at room temperature for 1 h, CHCl3 and H2O were added. The organic solution was separated, washed with sat. aq. NaHCO3 solution, and then dried (Na2SO4). Concentration of the solvent followed by silica gel column chromatography (5% EtOAc/hexane) gave primary alcohol (2.18 g, 7.61 mmol, 86%) as a colorless oil. A reaction mixture of the primary alcohol (2.18 g, 7.61 mmol), PCC (1.97 g, 9.14 mmol), and MS 4A (0.5 g) in CH2Cl2 (20 mL) was stirred at room temperature for 16 h before the addition of ether. After the mixture was filtered, the filtrate was concentrated. The residue was applied to silica gel column chromatography (5% EtOAc/hexane) to give aldehyde 10b (1.82 g, 6.40 mmol, 84%) as a colorless oil, [α]20D −45 (c 1.3, CHCl3). NMR data agreed with those in the literature.Citation12)
(2S)-1-[(1R,2S)-2-(Triisopropylsilyloxy)cyclopent-1-yl]-4-penten-2-ol (11a)
To a solution of (-)-Ipc2BOMe (1.04 g, 3.19 mmol) in ether (30 mL) was added allylmagnesium chloride (1.64 mL, 2 M in THF, 3.28 mmol) at −75 °C, and then the mixture was stirred at −70 °C for 15 min before warming to room temperature. After stirring at room temperature for 1 h, the mixture was cooled to −70 °C, and then aldehyde 10a (0.47 g, 1.65 mmol) in ether (15 mL) was added. The resulting reaction solution was stirred at −70 °C for 1 h, and then warmed to room temperature. The reaction was quenched by the addition of 1 M aq. NaOH solution and 30% aq. H2O2 solution. After the resulting mixture was stirred at room temperature for 12 h, the organic solution was separated and then dried (Na2SO4). Concentration of the solvent, followed by silica gel column chromatography (5% EtOAc/hexane), gave (2S)-1-[(1R,2S)]-11a (0.25 g, 0.77 mmol, 47%) as a colorless oil, Rf = 0.40 (EtOAc/hexane = 1/9 × 2), [α]20D +47 (c 0.7, CHCl3); 1H NMR (400 MHz, CDCl3) δ:1.06 (21H, s), 1.18 (1H, m), 1.36 (1H, m), 1.56–1.62 (3H, m), 1.72 (1H, m), 1.85–1.92 (3H, m), 2.18–2.29 (2H, m), 3.13 (1H, s), 3.70 (1H, m), 3.90 (1H, ddd, J = 5.4, 5.4, 5.4 Hz), 5.10 (1H, d, J = 9.8 Hz), 5.11 (1H, d, J = 17.3 Hz), 5.83 (1H, m); 13C NMR (100 MHz, CDCl3) δ: 12.6, 18.10,18.14, 21.2, 29.3, 34.5, 41.0, 42.4, 45.5, 70.1, 80.3, 117.7, 135.0; IR νmax (CHCl3): 3410, 2947, 1643, and 1103 cm−1. HRFABMS m/z (M + H)+: calcd for C19H39O2Si: 327.2720, found: 327.2739. (2R)-1-[(1S,2R)]-(ent-11a). By employing (+)-Ipc2BOMe and allylmagnesium chloride against aldehyde 10b, (2R)-1-[(1S,2R)]-(ent-11a) was obtained in 56% yield, [α]20D −50 (c 0.3, CHCl3).
(2R)-1-[(1R,2S)-2-(Triisopropylsilyloxy)cyclopent-1-yl]-4-penten-2-ol (11b)
By employing (+)-Ipc2BOMe and allylmagnesium chloride against aldehyde 10a, (2R)-1-[(1R,2S)]-11b was obtained in 53% yield as a colorless oil, Rf = 0.34 (EtOAc/hexane = 1/9 × 2), [α]20D +39 (c 0.8, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 1.07 (21H, s), 1.20 (1H, m), 1.42 (1H, m), 1.53–1.66 (3H, m), 1.70 (1H, m), 1.84–1.95 (3H, m), 2.00 (1H, br. s), 2.15 (1H, m), 2.30 (1H, m), 3.77 (1H, dddd, J = 7.3, 7.3, 7.3, 7.3 Hz), 3.90 (1H, ddd, J = 5.9, 5.9, 5.9 Hz), 5.12 (1H, d, J = 9.9 Hz), 5.13 (1H, d, J = 17.1 Hz), 5.84 (1H, m); 13C NMR (100 MHz, CDCl3) δ: 12.6, 18.13, 18.17, 21.5, 29.6, 34.6, 41.0, 41.6, 45.1, 69.7, 80.0, 117.9, 135.0; IR νmax (CHCl3): 3409, 2946, 1643, and 1103 cm−1. HRFABMS m/z (M + H)+: calcd for C19H39O2Si: 327.2720, found: 327.2718. (2S)-1-[(1S,2R)]-(ent-11b). By employing (-)-Ipc2BOMe and allylmagnesium chloride against aldehyde 10b, (2S)-1-[(1S,2R)]-(ent-11b) was obtained in 60% yield, [α]20D −39 (c 0.4, CHCl3).
(4R)-4-Hydroxy-5-[(1R,2S)-2-(triisopropylsilyloxy)cyclopent-1-yl]-2-pentanone (13a)
A reaction mixture of allyl alcohol 11a (0.22 g, 0.67 mmol), PdCl2 (12.0 mg, 67.7 μmol), and Cu (OAc)2·H2O (27.0 mg, 0.14 mmol) in DMF (7 ml) was stirred under O2 gas at ambient temperature for 48 h before filtration with ethyl acetate. The filtrate was washed with H2O and brine, and then dried (Na2SO4). After concentration of the solvent, the residue was applied to silica gel column chromatography (EtOAc/hexane = 1/5) to give β-hydroxy ketone 13a (0.11 g, 0.32 mmol, 48%) as a colorless oil, [α]20D +48 (c 1.3, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 1.06 (21H, s), 1.10–1.26 (2H, m), 1.51–1.61 (2H, m), 1.67 (1H, m), 1.73 (1H, m), 1.83 (1H, m), 1.90–1.98 (2H, m), 2.17 (3H, s), 2.58–2.60 (2H, m), 3.17 (1H, br. s), 3.90 (1H, ddd, J = 5.6, 5.6, 5.6 Hz), 4.10 (1H, m); 13C NMR (100 MHz, CDCl3) δ: 12.4, 18.0, 18.1, 21.2, 29.1, 30.7, 34.5, 40.7, 44.9, 50.6, 66.6, 79.9, 209.5; IR νmax (CHCl3): 3745, 2945, 2362, and 1705 cm−1. HRFABMS m/z (M + H)+: calcd for C19H39O3Si: 343.2669, found: 343.2667. (4S)-5-[(1S,2R)]-(ent-13a). 59% yield, [α]20D −48 (c 0.8, CHCl3).
(4S)-4-Hydroxy-5-[(1R,2S)-2-(triisopropylsilyloxy)cyclopent-1-yl]-2-pentanone (13b)
51% yield, [α]20D +54 (c 0.8, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 1.06 (21H, m), 1.19 (1H, m), 1.43 (1H, m), 1.51–1.64 (3H, m), 1.70 (1H, m), 1.73–1.94 (3H, m), 2.19 (3H, s), 2.52 (1H, dd, J = 17.4, 9.2 Hz), 2.65 (1H, dd, J = 17.4, 2.7 Hz), 3.13 (1H, d, J = 2.7 Hz), 3.90 (1H, ddd, J = 5.9, 5.9, 5.9 H), 4.13 (1H, m); 13C NMR (100 MHz, CDCl3) δ: 12.5, 18.10, 18.14, 21.4, 29.4, 30.8, 34.5, 40.6, 45.0, 49.7, 66.8, 79.8, 209.7. IR νmax (CHCl3): 3745, 2945, 2362, and 1705 cm−1. HRFABMS m/z (M + H)+: calcd for C19H39O3Si: 343.2669, found: 343.2650. (4R)-5-[(1S,2R)]-(ent-13b). 51% yield, [α]20D −53 (c 0.8, CHCl3).
(2R,4S)-1-[(1R,2S)-2-(Triisopropylsilyloxy)cyclopent-1-yl]pentane-2,4-diol (14a)
To a solution of β-hydroxy ketone 13a (0.60 g, 1.75 mmol) in THF-MeOH (4/1, 13 mL) was added Et2BOMe (1.91 mL, 1 M in THF, 1.91 mmol) at −70 °C. After the mixture was stirred at −70 °C for 15 min, NaBH4 (72.0 mg, 1.90 mmol) was added. The reaction mixture was stirred at −70 °C for 3 h before warming to 0 °C. The reaction was quenched by the addition of 3 M aq. NaOH (2.5 mL) and 30% aq. H2O2 solution (1 mL). The mixture was then extracted with ether. The organic solution was separated, washed with brine, and then dried (Na2SO4). Concentration of the solvent, followed by silica gel column chromatography (EtOAc/hexane = 1/3), gave 1,3-syn-diol 14a (0.54 g, 1.57 mmol, 90%) as a colorless oil, [α]20D +75 (c 0.7, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 1.05 (3H, s), 1.07 (18H, s), 1.18 (3H, d, J = 6.0 Hz), 1.21 (1H, m), 1.41 (1H, ddd, J = 11.0, 6.4, 3.2 Hz), 1.50–1.64 (5H, m), 1.71 (1H, m), 1.83–1.94 (3H, m), 3.80–3.87 (2H, m), 3.88–3.93 (2H, m), 4.05 (1H, dddd, J = 15.3, 5.7, 5.7, 3.2 Hz); 13C NMR (100 MHz, CDCl3) δ: 12.7, 18.0, 18.1, 21.0, 23.8, 29.5, 34.4, 42.7, 45.2, 45.7, 68.8, 72.6, 80.5; IR νmax (CHCl3): 3500, 2945, 1458, 1092, and 1061 cm−1. HRFABMS m/z (M + H)+: calcd for C19H41O3Si: 345.2825, found: 345.2817. (2S,4R)-1-[(1S,2R)]-(ent-14a). 92% yield, [α]20D −77 (c 0.4, CHCl3).
(2R,4R)-1-[(1R,2S)-2-(Triisopropylsilyloxy)cyclopent-1-yl]pentane-2,4-diol (14c)
To a solution of Me4NHB(OAc)3 (0.89 g, 3.38 mmol) in MeCN (2.5 mL) and AcOH (1.5 mL), a solution of β-hydroxy ketone 13a (0.26 g, 0.76 mmol) in MeCN (1.5 mL) was added at −40 °C. The reaction mixture was stirred at 0 °C for 1 h before the addition of sat. aq. NH4Cl solution and EtOAc. The organic solution was separated, washed with sat. aq. NaHCO3 solution, and brine and then dried (Na2SO4). Concentration of the solvent, followed by silica gel column chromatography (EtOAc/hexane = 1/2), gave 1,3-anti-diol 14c (0.20 g, 0.58 mmol, 76%) as a colorless oil, [α]20D +43 (c 0.8, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 1.05 (3H, s), 1.07 (18H, s), 1.14–1.24 (2H, m), 1.22 (3H, d, J = 6.0 Hz), 1.28 (1H, m), 1.39 (1H, ddd, J = 13.7, 7.6, 2.7 Hz), 1.54–1.64 (3H, m), 1.67–1.75 (2H, m), 1.86–1.93 (2H, m), 3.12 (1H, br. s), 3.44 (1H, br. s), 3.91 (1H, ddd, J = 6.4, 6.4, 6.4 Hz), 3.99 (1H, m), 4.15 (1H, m); 13C NMR (100 MHz, CDCl3) δ: 12.6, 18.0, 18.1, 21.0, 23.4, 29.5, 34.4, 41.6, 44.3, 46.1, 65.4, 69.1, 80.5; IR νmax (CHCl3): 3500, 2945, 1457, 1093, and 1061 cm−1. HRFABMS m/z (M + H)+: calcd for C19H41O3Si: 345.2825, found: 345.2819. (2S,4S)-1-[(1S,2R)]-(ent-14c). 81% yield, [α]20D−40 (c 4.3, CHCl3).
(2S,4R)-1-[(1R,2S)-2-(Triisopropylsilyloxy)cyclopent-1-yl]pentane-2,4-diol (14e)
88% yield, [α]20D +32 (c 0.8, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 1.08 (21H, s), 1.19 (3H, d, J = 6.0 Hz), 1.19–1.27 (1H, overlapped), 1.46–1.62 (6H, m), 1.72 (1H, m), 1.85–1.91 (3H, m), 3.39 (1H, br. s), 3.69 (1H, br. s), 3.92 (1H, ddd, J = 6.4, 6.4, 6.4 Hz), 3.99–4.08 (2H, m); 13C NMR (100 MHz, CDCl3) δ: 12.7, 18.08, 18.13, 21.3, 23.9, 29.6, 34.6, 42.3, 44.28, 44.34, 69.0, 71.8, 80.1. IR νmax (CHCl3): 3500, 2946, 1458, 1092, and 1060 cm−1. HRFABMS m/z (M + H)+: calcd for C19H41O3Si: 345.2825, found: 345.2823. (2R,4S)-1-[(1S,2R)]-(ent-14e). 85% yield, [α]20D −33 (c 0.4, CHCl3).
(2S,4S)-1-[(1R,2S)-2-(Triisopropylsilyloxy)cyclopent-1-yl]pentane-2,4-diol (14g)
79% yield, [α]20D +43 (c 0.6, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 1.05 (3H, s), 1.07 (18H, s), 1.21 (1H, m), 1.23 (3H, d, J = 6.0 Hz), 1.50–1.59 (2H, m), 1.59–1.64 (3H, m), 1.68–1.78 (2H, m), 1.81–1.93 (3H, m), 2.73 (1H, br. s), 2.88 (1H, br. s), 3.92 (1H, ddd, J = 6.5, 6.5, 6.5 Hz), 4.10 (1H, m), 4.16 (1H, m); 13C NMR (100 MHz, CDCl3) δ: 12.6, 18.08, 18.12, 21.4, 23.4, 29.5, 34.6, 41.5, 43.4, 44.8, 65.4, 68.0, 80.0; IR νmax (CHCl3): 3500, 2945, 1458, 1091, and 1060 cm−1. HRFABMS m/z (M + H)+: calcd for C19H41O3Si: 345.2825, found: 345.2818. (2R,4R)-1-[(1S,2R)]-(ent-14g). 83% yield, [α]20D −40 (c 0.2, CHCl3).
(1S,3R)-1-Methyl-3-[(1R,2S)-2-(triisopropylsilyloxy)cyclopent-1-yl]methylpropylene diacetate (15a)
A reaction mixture of syn-diol 14a (0.39 g, 1.13 mmol) and DMAP (10.0 mg, 81.9 μmol) in pyridine (3 mL) and Ac2O (3 mL) was stirred at room temperature for 12 h before the addition of ice. After 6 h at room temperature, the mixture was extracted with EtOAc. The organic solution was washed with 1 M aq. HCl solution, sat. aq. NaHCO3 solution, and brine and then dried (Na2SO4). Concentration of the solvent, followed by silica gel column chromatography (EtOAc/hexane = 1/9), gave diacetate 15a (0.46 g, 1.07 mmol, 95%) as a colorless oil, [α]20D +24 (c 0.8, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 1.05 (21H, s), 1.05–1.15 (1H, m), 1.24 (3H, d, J = 6.4 Hz), 1.28 (1H, m), 1.51–1.58 (2H, m), 1.65 (1H, ddd, J = 14.2, 6.0, 6.0 Hz), 1.68–1.84 (4H, m), 1.86–2.05 (2H, m), 2.03 (3H, s), 2.04 (3H, s), 3.83 (1H, ddd, J = 5.5, 5.5, 5.5 Hz), 4.94 (1H, m), 4.97 (1H, m); 13C NMR (100 MHz, CDCl3) δ: 12.3, 18.1, 20.1, 21.2, 21.3, 21.4, 28.9, 34.6, 38.4, 41.0, 44.5, 68.0, 69.9, 79.6, 170.4, 170.6; IR νmax (CHCl3): 2945, 2360, 1734, and 1250 cm−1. HRFABMS m/z (M-H)+: calcd for C23H43O5Si: 427.2880, found: 427.2866. (1R,3S)-3-[(1S,2R)]-(ent-15a). 95% yield, [α]20D −22 (c 1.3, CHCl3).
(1R,3R)-1-Methyl-3-[(1R,2S)-2-(triisopropylsilyloxy)cyclopent-1-yl]methylpropylene diacetate (15c)
92% yield, [α]20D +20 (c 0.9, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 1.05 (21H, s), 1.14 (1H, m), 1.21–1.28 (1H, overlapped), 1.22 (3H, d, J = 6.0 Hz), 1.50–1.59 (2H, m), 1.68–1.84 (6H, m), 1.96 (1H, m), 2.01 (3H, s), 2.02 (3H, s), 3.85 (1H, ddd, J = 5.5, 5.5, 5.5 Hz), 4.92 (1H, m), 5.05 (1H, dddd, J = 13.3, 4.6, 3.6, 3.6 Hz); 13C NMR (100 MHz, CDCl3) δ: 12.2, 18.01, 18.03, 20.4, 20.9, 21.1, 21.3, 29.1, 34.5, 38.9, 41.1, 44.5, 66.9, 68.9, 79.5, 170.4, 170.5; IR νmax (CHCl3): 2944, 2360, 1732, and 1251 cm−1. Found: C, 64.50; H, 10.41. Calcd for C23H44O5Si: C, 64.44; H, 10.35. (1S,3S)-3-[(1S,2R)]-(ent-15c). 96% yield, [α]20D −20 (c 1.3, CHCl3).
(1R,3S)-1-Methyl-3-[(1R,2S)-2-(triisopropylsilyloxy)cyclopent-1-yl]methylpropylene diacetate (15e)
98% yield, [α]20D +40 (c 0.3, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 1.05 (21H, s), 1.23 (3H, d, J = 6.0 Hz), 1.38 (1H, m), 1.50–1.60 (3H, m), 1.67–1.82 (5H, m), 1.83–1.94 (2H, m), 2.02 (3H, s), 2.03 (3H, s), 3.83 (1H, ddd, J = 5.5, 5.5, 5.5 Hz), 4.89–4.98 (2H, m); 13C NMR (100 MHz, CDCl3) δ: 12.3, 18.08, 18.11, 19.8, 21.3, 21.4, 28.8, 34.2, 38.3, 39.8, 45.3, 68.2, 71.1, 79.5, 170.4, 170.5; IR νmax (CHCl3): 2945, 2360, 1733, and 1249 cm−1. Found: C, 64.30; H, 10.62. Calcd for C23H44O5Si: C, 64.44; H, 10.35. (1S,3R)-3-[(1S,2R)]-(ent-15e). 94% yield, [α]20D −42 (c 1.0, CHCl3).
(1S,3S)-1-Methyl-3-[(1R,2S)-2-(triisopropylsilyloxy)cyclopent-1-yl]methylpropylene diacetate (15g)
94% yield, [α]20D +52 (c 0.7, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 1.05 (21H, m), 1.23 (3H, d, J = 6.4 Hz), 1.25 (1H, m), 1.39 (1H, ddd, J = 13.3, 10.3, 5.2 Hz), 1.48–1.61 (2H, m), 1.62–1.93 (7H, m), 2.01 (3H, s), 2.02 (3H, s), 3.82 (1H, ddd, J = 5.5, 5.5, 5.5 Hz), 4.89–4.98 (2H, m); 13C NMR (100 MHz, CDCl3) δ: 12.3, 18.07, 18.10, 20.5, 21.19, 21.23, 21.3, 28.6, 34.3, 38.5, 40.0, 45.2, 67.0, 70.0, 79.7, 170.5, 170.6; IR νmax (CHCl3): 2945, 2360, 1733, and 1250 cm−1. Found: C, 64.47; H, 10.65. Calcd for C23H44O5Si: C, 64.44; H, 10.35. (1R,3R)-3-[(1S,2R)]-(ent-15g). 92% yield, [α]20D −50 (c 0.6, CHCl3).
(1R,3S)-1-[(1R,2S)-2-Hydroxycyclopent-1-yl]methyl-3-methylpropylene diacetate (16a)
To a solution of silyl ether 15a (0.45 g, 1.05 mmol) in THF (10 mL) was added n-Bu4NF (1.16 mL, 1.0 M in THF, 1.16 mmol). After the reaction solution was stirred at room temperature for 12 h, sat. aq. NH4Cl solution and EtOAc were added. The organic solution was separated, washed with sat. aq. CuSO4 solution, sat. aq. NaHCO3 solution, and brine and then dried (Na2SO4). Concentration of the solvent, followed by silica gel column chromatography (EtOAc/hexane = 1/3), gave alcohol 16a (0.19 g, 0.70 mmol, 67%) as a colorless oil, [α]20D +27 (c 0.6, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 1.14 (1H, m), 1.23 (1H, m), 1.24 (3H, d, J = 6.4 Hz), 1.42 (1H, ddd, J = 13.7, 9.1, 4.6 Hz), 1.50–1.62 (2H, m), 1.65–1.73 (2H, m), 1.71 (1H, ddd, J = 14.2, 6.0, 6.0 Hz), 1.81 (1H, ddd, J = 13.7, 8.2, 5.5 Hz), 1.86–1.99 (2H, m), 2.03 (3H, s), 2.05 (3H, s), 2.17 (1H, s), 3.80 (1H, ddd, J = 6.4, 6.4, 6.4 Hz), 4.96 (1H, m), 5.02 (1H, m); 13C NMR (100 MHz, CDCl3) δ: 20.0, 21.17, 21.21, 21.3, 29.8, 34.3, 38.2, 40.5, 44.1, 68.0, 70.1, 78.9, 170.5, 170.7; IR νmax (CHCl3): 3745, 2962, 2360, 1734, 1774, and 1252 cm−1. HRFABMS m/z (M + H)+: calcd for C14H25O5: 273.1702, found: 273.1709. (1S,3R)-1-[(1S,2R)]-(ent-16a). 65% yield, [α]20D −27 (c 2.9, CHCl3).
(1R,3R)-1-[(1R,2S)-2-Hydroxycyclopent-1-yl]methyl-3-methylpropylene diacetate (16c)
77% yield, [α]20D +17 (c 0.8, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 1.16 (1H, m), 1.23 (3H, d, J = 6.4 Hz), 1.38 (1H, ddd, J = 13.7, 8.9, 5.0 Hz), 1.49–1.62 (2H, m), 1.63–2.00 (7H, m), 1.93 (1H, br. s), 2.02 (3H, s), 2.03 (3H, s), 3.80 (1H, ddd, J = 6.4, 6.4, 6.4 Hz), 4.93 (1H, m), 5.06 (1H, m); 13C NMR (100 MHz, CDCl3) δ: 20.4, 21.1, 21.2, 21.4, 30.2, 34.3, 38.8, 40.9, 44.3, 67.1, 69.3, 79.1, 170.65, 170.74; IR νmax (CHCl3): 3745, 2962, 2360, 1734, 1774, 1694, and 1252 cm−1. HRFABMS m/z (M + H)+: calcd for C14H25O5: 273.1702, found: 273.1707. (1S,3S)-1-[(1S,2R)]-(ent-16c). 82% yield, [α]20D −19 (c 1.9, CHCl3).
(1S,3R)-1-[(1R,2S)-2-Hydroxycyclopent-1-yl]methyl-3-methylpropylene diacetate (16e)
73% yield, [α]20D +13 (c 0.9, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 1.19 (1H, m), 1.24 (3H, d, J = 6.0 Hz), 1.52–1.64 (3H, m), 1.65–1.75 (4H, m), 1.83–2.00 (3H, m), 2.04 (3H, s), 2.07 (3H, s), 2.35 (1H, br. s), 3.81 (1H, ddd, J = 6.4, 6.4, 6.4 Hz), 4.96 (1H, m), 5.09 (1H, m); 13C NMR (100 MHz, CDCl3) δ: 20.0, 21.26, 21.29, 22.2, 31.2, 34.5, 38.8, 40.4, 44.6, 67.9, 70.6, 78.6, 170.5, 171.2; IR νmax (CHCl3): 3745, 2962, 2360, 1734, 1694, and 1252 cm−1. HRFABMS m/z (M + H)+: calcd for C14H25O5: 273.1702, found: 273.1710. (1R,3S)-1-[(1S,2R)]-(ent-16e). 72% yield, [α]20D −12 (c 3.1, CHCl3).
(1S,3S)-1-[(1R,2S)-2-Hydroxycyclopent-1-yl]methyl-3-methylpropylene diacetate (16g)
78% yield, [α]20D +18 (c 0.7, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 1.20 (1H, m), 1.23 (3H, d, J = 6.4 Hz), 1.53–1.63 (4H, m), 1.64–1.72 (2H, m), 1.72–1.82 (2H, m), 1.83–1.96 (2H, m), 2.02 (3H, s), 2.05 (3H, s), 2.48 (1H, br. s), 3.84 (1H, ddd, J = 6.5, 6.5, 6.5 Hz), 4.96 (1H, m), 5.10 (1H, m); 13C NMR (100 MHz, CDCl3) δ: 20.4, 21.1, 21.2, 22.4, 31.3, 34.6, 39.4, 40.8, 44.7, 66.7, 69.6, 78.6, 170.6, 171.5; IR νmax (CHCl3): 3744, 2962, 2360, 1733, 1692, and 1252 cm−1. HRFABMS m/z (M + H)+: calcd for C14H25O5: 273.1702, found: 273.1703. (1R,3R)-1-[(1S,2R)]-(ent-16g). 80% yield, [α]20D −17 (c 1.3, CHCl3).
(1S,3R)-1-Methyl-3-[(1R)-2-oxocyclopent-1-yl]methylpropylene diacetate (17a)
A reaction mixture of alcohol 16a (0.24 g, 0.88 mmol), PCC (0.23 g, 1.07 mmol), and MS 4A (0.50 g) in CH2Cl2 (10 mL) was stirred at room temperature for 12 h before the addition of ether. After the mixture was filtered, the filtrate was concentrated. The residue was applied to silica gel column chromatography (5% EtOAc/hexane) to give ketone 17a (0.18 g, 0.67 mmol, 76%) as a colorless oil, [α]20D +89 (c 0.4, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 1.24 (3H, d, J = 6.4 Hz), 1.41–1.51 (2H, m), 1.67 (1H, ddd, J = 14.2, 5.5, 5.5 Hz), 1.72–1.83 (2H, m), 1.97 (1H, ddd, J = 14.2, 7.3, 7.3 Hz), 1.98–2.16 (3H, m), 2.04 (3H, s), 2.05 (3H, s), 2.28–2.36 (2H, m), 4.95 (1H, m), 5.02 (1H, m); 13C NMR (100 MHz, CDCl3) δ: 20.2, 20.7, 21.3, 21.4, 30.0, 34.5, 37.9, 40.9, 45.9, 67.9, 69.3, 170.6, 170.7, 220.6; IR νmax (CHCl3): 3026, 2972, 2359, 1732, 1375, 1250, and 1022 cm−1. HRFABMS m/z (M + H)+: calcd for C14H23O5: 271.1545, found: 271.1528. (1R,3S)-3-[(1S)]-(ent-17a). 81% yield, [α]20D −89 (c 2.3, CHCl3).
(1R,3R)-1-Methyl-3-[(1R)-2-oxocyclopent-1-yl]methylpropylene diacetate (17c)
75% yield, [α]20D +90 (c 0.7, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 1.22 (3H, d, J = 6.4 Hz), 1.33–1.52 (2H, m), 1.71–1.82 (3H, m), 1.97–2.07 (2H, m), 2.025 (3H, s), 2.030 (3H, s), 2.07–2.15 (2H, m), 2.29–2.42 (2H, m), 4.92 (1H, dddd, J = 13.3, 6.4, 6.4, 6.4 Hz), 5.10 (1H, m); 13C NMR (100 MHz, CDCl3) δ: 20.4, 20.6, 21.0, 21.2, 30.2, 35.0, 37.8, 41.0, 46.0, 66.9, 68.3, 170.6, 170.7, 220.5; IR νmax (CHCl3): 3025, 2971, 2358, 1731, 1374, 1251, and 1021 cm−1. Found: C, 62.11; H, 8.14. Calcd for C14H22O5: C, 62.20; H, 8.20. (1S,3S)-3-[(1S)]-(ent-17c). 78% yield, [α]20D −89 (c 1.6, CHCl3).
(1R,3S)-1-Methyl-3-[(1R)-2-oxocyclopent-1-yl]methylpropylene diacetate (17e)
80% yield, [α]20D +70 (c 0.9, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 1.24 (3H, d, J = 6.0 Hz), 1.48–1.62 (2H, m), 1.66–1.81 (3H, m), 1.95 (1H, ddd, J = 14.2, 6.4, 6.4 Hz), 2.00–2.12 (3H, m), 2.038 (3H, s), 2.042 (3H, s), 2.21 (1H, m), 2.30 (1H, m), 4.95 (1H, dddd, J = 12.3, 6.4, 6.4, 6.4 Hz), 5.06 (1H, m); 13C NMR (100 MHz, CDCl3) δ: 20.0, 20.7, 21.3, 30.1, 34.2, 37.4, 40.1, 46.5, 67.9, 70.4, 170.5, 170.6, 219.8; IR νmax (CHCl3): 3026, 2972, 2359, 1733, 1376, 1251, and 1021 cm−1. Found: C, 62.09; H, 8.04. Calcd for C14H22O5: C, 62.20; H, 8.20. (1S,3R)-3-[(1S)]-(ent-17e). 85% yield, [α]20D −68 (c 2.9, CHCl3).
(1S,3S)-1-Methyl-3-[(1R)-2-oxocyclopent-1-yl]methylpropylene diacetate (17g)
79% yield, [α]20D +104 (c 0.8, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 1.23 (3H, d, J = 6.4 Hz), 1.48–1.63 (2H, m), 1.68–1.86 (3H, m), 2.00–2.12 (4H, m), 2.02 (6H, s), 2.22 (1H, m), 2.30 (1H, m), 4.93 (1H, dddd, J = 16.0, 9.3, 3.2, 3.2 Hz), 5.07 (1H, m); 13C NMR (100 MHz, CDCl3) δ: 20.4, 20.7, 21.1, 30.1, 34.7, 37.3, 40.5, 46.7, 66.8, 69.6, 170.5, 219.8; IR νmax (CHCl3): 3024, 2971, 2360, 1732, 1375, 1250, and 1021 cm−1. Found: C, 61.99; H, 8.45. Calcd for C14H22O5: C, 62.20; H, 8.20. (1R,3R)-3-[(1S)]-(ent-17g). 83% yield, [α]20D −101 (c 1.1, CHCl3).
(5R,7S,9S)-7,9-Diacetoxy-5-decanolide (18a)
A reaction mixture of ketone 17a (0.20 g, 0.74 mmol) and MCPBA (0.40 g, 70%, 1.62 mmol) in CHCl3 (8 mL) and Na2HPO4-NaH2PO4 buffer (pH 8.0, 8 mL) was stirred at room temperature for 12 h before the addition of sat. aq. Na2S2O3 solution. The organic solution was separated, washed with sat. aq. NaHCO3 solution, and then dried (Na2SO4). Concentration of the solvent, followed by silica gel column chromatography (EtOAc/hexane = 1/1), gave lactone 18a (0.15 g, 0.52 mmol, 70%) as a colorless oil, [α]20D −35 (c 0.5, CHCl3); 1H NMR (400 MHz, CDCl3) δ:1.25 (3H, d, J = 6.4 Hz), 1.51 (1H, m), 1.77 (1H, ddd, J = 14.2, 6.0, 6.0 Hz), 1.82–1.95 (3H, m), 1.95–2.02 (2H, m), 2.04 (3H, s), 2.04–2.09 (1H, overlapped), 2.07 (3H, s), 2.44 (1H, ddd, J = 17.6, 8.7, 7.1 Hz), 2.58 (1H, dddd, J = 17.6, 6.7, 6.7, 1.0 Hz), 4.37 (1H, m), 4.97 (1H, dddd, J = 13.3, 6.4, 6.4, 6.4 Hz), 5.11 (1H, m); 13C NMR (100 MHz, CDCl3) δ: 18.3, 20.1, 21.1, 21.3, 27.7, 29.3, 40.1, 40.3, 67.7, 67.9, 77.3, 170.5, 170.6, 171.1; IR νmax (CHCl3): 3026, 2360, 1734, 1373, 1244, and 1049 cm−1. HRFABMS m/z (M + H)+: calcd for C14H23O6: 287.1495, found: 287.1484. (5S,7R,9R)-(ent-18a). 74% yield, [α]20D +35 (c 0.6, CHCl3).
(5R,7S,9R)-7,9-Diacetoxy-5-decanolide (18c)
74% yield, [α]20D −40 (c 0.7, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 1.23 (3H, d, J = 6.4 Hz), 1.51 (1H, m), 1.76–1.86 (4H, m), 1.89 (1H, m), 2.01–2.10 (2H, m), 2.02 (3H, s), 2.05 (3H, s), 2.44 (1H, ddd, J = 17.4, 8.7, 7.3 Hz), 2.58 (1H, ddd, J = 17.4, 6.2, 6.2 Hz), 4.35 (1H, m), 4.94 (1H, m), 5.14 (1H, m); 13C NMR (100 MHz, CDCl3) δ: 18.3, 20.4, 21.0, 21.1, 27.8, 29.2, 40.7, 40.8, 66.6, 66.9, 77.3, 170.5, 170.7, 171.2; IR νmax (CHCl3): 3025, 2359, 1734, 1373, 1244, and 1048 cm−1. HRFABMS m/z (M + H)+: calcd for C14H23O6: 287.1495, found: 287.1490. (5S,7R,9S)-(ent-18c). 75% yield, [α]20D +39 (c 1.3, CHCl3).
(5R,7R,9R)-7,9-Diacetoxy-5-decanolide (18e)
69% yield, [α]20D −57 (c 0.9, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 1.25 (3H, d, J = 6.4 Hz), 1.58 (1H, m), 1.75 (1H, ddd, J = 14.2, 6.0, 6.0 Hz), 1.80–1.96 (5H, m), 2.01 (1H, ddd, J = 14.2, 7.3, 7.3 Hz), 2.04 (3H, s), 2.06 (3H, s), 2.44 (1H, ddd, J = 17.9, 7.8, 7.8 Hz), 2.58 (1H, ddd, J = 17.9, 6.9, 6.9 Hz), 4.34 (1H, m), 4.96 (1H, dddd, J = 12.9, 6.4, 6.4, 6.4 Hz), 5.18 (1H, m); 13C NMR (100 MHz, CDCl3) δ: 18.4, 20.1, 21.1, 21.3, 28.0, 29.2, 40.5, 40.6, 67.7, 68.0, 76.8, 170.4, 170.5, 171.0; IR νmax (CHCl3): 3026, 2360, 1734, 1373, 1244, and 1049 cm−1. HRFABMS m/z (M + H)+: calcd for C14H23O6: 287.1495, found: 287.1489. (5S,7S,9S)-(ent-18e). 70% yield, [α]20D −55 (c 0.6, CHCl3).
(5R,7R,9S)-7,9-Diacetoxy-5-decanolide (18g)
69% yield, [α]20D −46 (c 0.6, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 1.24 (3H, d, J = 6.4 Hz), 1.59 (1H, m), 1.76 (1H, ddd, J = 14.2, 9.4, 3.7 Hz), 1.83–1.95 (6H, m), 2.03 (3H, s), 2.04 (3H, s), 2.45 (1H, ddd, J = 17.4, 8.2, 8.2 Hz), 2.58 (1H, m), 4.34 (1H, m), 4.94 (1H, m), 5.19 (1H, m); 13C NMR (100 MHz, CDCl3) δ: 18.3, 20.4, 21.0, 21.2, 28.1, 29.1, 40.9, 41.1, 66.7, 67.3, 77.0, 170.4, 170.6, 171.2; IR νmax (CHCl3): 3025, 2360, 1734, 1373, 1244, and 1049 cm−1. HRFABMS m/z (M + H)+: calcd for C14H23O6: 287.1495, found: 287.1499. (5S,7S,9R)-(ent-18g). 70% yield, [α]20D +45 (c 1.0, CHCl3).
(+)-Cryptocarya diacetate (1a)
A reaction mixture of diacetate 18a (89.0 mg, 0.31 mmol) and benzeneseleninic acid anhydride (0.18 g, 70%, 0.35 mmol) in chlorobenzene (10 mL) was heated at 130 °C for 72 h. After concentration of the solvent, the residue was applied to silica gel column chromatography (EtOAc/hexane = 1/1), to give 1a (74 mg, 0.26 mmol, 84%) as a colorless oil, [α]20D +49 (c 0.5, CHCl3), [α]20D +47.5 (c 0.6, CHCl3) in literature.Citation9) NMR data agreed with those in the literature.Citation9) >99%ee (AD-H, 250 mm × 4.6 mm i.d., 5 μm, 5% iso-PrOH/hexane, 1 mL min−1, 210 nm, tR35 min).
(-)-Cryptocarya diacetate (1b)
[α]20D −49 (c 1.2, CHCl3), [α]20D −44 (c 0.46, CHCl3)Citation10) in literature. NMR data agreed with those in the literature.Citation9) >99%ee (AD-H, 250 mm × 4.6 mm i.d., 5 μm, 5% iso-PrOH/hexane, 1 mL min−1, 210 nm, tR30 min).
(5R,7S,9R)-7,9-Diacetoxy-2-decen-5-olide (1c)
80% yield, NMR data agreed with those in the literature.Citation10) [α]20D +57 (c 0.2, CHCl3), [α]20D +30 (c 0.62, CHCl3) in the literature.Citation10)>99%ee (AD-H, 250 mm × 4.6 mm i.d., 5 μm, 10% EtOH/hexane, 1 mL min−1, 210 nm, tR24 min).
(5S,7R,9S)-7,9-Diacetoxy-2-decen-5-olide (1d)
83% yield, NMR data agreed with those in the literature.Citation10) [α]20D −57 (c 0.1, CHCl3), [α]20D −41.5 (c 1.02, CHCl3) in literature.Citation10)>99%ee (AD-H, 250 mm × 4.6 mm i.d., 5 μm, 10%EtOH/hexane, 1 mL min−1, 210 nm, tR16 min).
(5R,7R,9R)-7,9-Diacetoxy-2-decen-5-olide (1e)
81% yield, NMR data agreed with those in the literature.Citation10) [α]20D +39 (c 0.7, CHCl3), [α]20D +25.6 (c 0.6, CHCl3) in literature.Citation10) >99%ee (AD-H, 250 mm × 4.6 mm i.d., 5 μm, 10% EtOH/hexane, 1 mL min−1, 210 nm, tR43 min).
(5S,7S,9S)-7,9-Diacetoxy-2-decen-5-olide (1f)
80% yield, NMR data agreed with those in the literature.Citation4) [α]20D −39 (c 0.6, CHCl3), [α]20D −20.5 (c 0.6, CHCl3)Citation10) and [α]20D −35.6 (c 0.75, CHCl3)Citation4) in literatures. >99%ee (AD-H, 250 mm × 4.6 mm i.d., 5 μm, 10% EtOH/hexane, 1 mL min−1, 210 nm, tR30 min).
(5R,7R,9S)-7,9-Diacetoxy-2-decen-5-olide (1g)
85% yield, NMR data agreed with those in the literature.Citation10) [α]20D +40 (c 0.8, CHCl3), [α]20D +43.5 (c 0.8, CHCl3) in literature.Citation10) >99%ee (AD-H, 250 mm × 4.6 mm i.d., 5 μm, 10% EtOH/hexane, 1 mL min−1, 210 nm, tR 22 min).
(5S,7S,9R)-7,9-Diacetoxy-2-decen-5-olide (1h)
80% yield, NMR data agreed with those in the literature.Citation10) [α]20D −40 (c 0.5, CHCl3), [α]20D −32 (c 0.17, CHCl3) in literature.Citation10) >99%ee (AD-H, 250 mm × 4.6 mm i.d., 5 μm, 10% EtOH/hexane, 1 mL min−1, 210 nm, tR20 min).
References
- Davies-Coleman MT, Rivett DEA. Naturally occurring 6-substituted 5,6-dihydro-α-pyrones. Fortschr. Chem. Org. Naturst. 1989;55:1–35.
- Drewes SE, Sehlapelo BM, Horn MM, Scott-Shaw R, Sandor P. 5,6-dihydro-α-pyrones and two bicyclic tetrahydro-α-pyrone derivatives from Cryptocarya latifolia. Phytochemistry. 1995;38:1427–1430.10.1016/0031-9422(94)00828-H
- Collett LA, Davies-Coleman MT, Rivett DEA, Drewes SE, Horn MM. Absolute configuration of α-pyrones from Cryptocarya latifolia and Syncolostemon densiflorus. Phytochemistry. 1997;44:935–938.10.1016/S0031-9422(96)00643-7
- Hunter TJ, O’Doherty GA. An enantioselective synthesis of cryptocarya diacetate. Org. Lett. 2001;3:2777–2780.10.1021/ol016399t
- Krishna PR, Reddy VVR. Stereoselective total synthesis of (+)-cryptocarya diacetate by an iterative Jacobsen’s hydrolytic kinetic resolution protocol. Tetrahedron Lett. 2005;46:3905–3907.10.1016/j.tetlet.2005.03.202
- Yadav JS, Rao PP, Reddy MS, Rao NV, Prasad AR. Stereoselective synthesis of (+)-cryptocarya diacetate by an iterative Prins cyclisation and reductive cleavage sequence. Tetrahedron Lett. 2007;48:1469–1471.10.1016/j.tetlet.2006.12.068
- Sabitha G, Reddy NM, Prasad MN, Yadav JS. Stereoselective routes for the total synthesis of (+)-cryptocarya diacetate. Helv. Chim. Acta. 2009;92:967–976.10.1002/(ISSN)1522-2675
- Gurjar MK, Raghupathi N, Chorghade MS. A short total synthesis of (+)-cryptocarya diacetate. Heterocycles. 2009;77:945–952.10.3987/COM-08-S(F)107
- Melillo B, Smith AB III. A unified synthetic strategy to the cryptocarya family of natural products exploiting anion relay chemistry (ARC). Org. Lett. 2013;15:2282–2285.10.1021/ol400857k
- Umarye JD, Leßmann T, García AB, Mamane V, Sommer S, Waldmann H. Biology-oriented synthesis of stereochemically diverse natural-product-derived compound collections by iterative allylations on a solid support. Chem. Eur. J. 2007;13:3305–3319.10.1002/(ISSN)1521-3765
- Yamauchi S, Takeda K, Ganaha M, Kinoshita Y. Synthesis of (R)-6,7-dihydro-5-HETE lactone and (S)-6,7-dihydro-5-HETE lactone by using novel yeast reduction as a key reaction. J. Chem. Soc., Perkin Trans. I. 2002;2156–2160.
- Yoshida T, Yamauchi S, Tago R, Maruyama M, Akiyama K, Sugahara T, Kishida T, Koba Y. Syntheses of all stereoisomers of goniodiol from yeast-reduction products and their antimicrobiological activity. Biosci. Biotechnol. Biochem. 2008;72:2342–2352.10.1271/bbb.80262
- Racherla US, Brown HC. Chiral synthesis via organoboranes. 27. Remarkably rapid and exceptionally enantioselective (approaching 100% ee) allylboration of representative aldehydes at -100 degree under new, salt-free conditions. J. Org. Chem. 1991;56:401–404.10.1021/jo00001a072
- Ohtani I, Kusumi T, Kashman Y, Kakisawa H. High-field FT NMR application of Mosher's method. The absolute configurations of marine terpenoids. J. Am. Chem. Soc. 1991;113:4092–4096.10.1021/ja00011a006
- Smith AB III, Cho YS, Friestad GK. Convenient Wacker oxidations with substoichiometric cupric acetate. Tetrahedron Lett. 1998;39:8765–8768.10.1016/S0040-4039(98)01992-3
- Chen K-M, Hardtmann GE, Prasad K, Repič O, Shapiro MJ. 1,3-diastereoselective reduction of β-hydroxyketones utilizing alkoxydialkylboranes. Tetrahedron Lett. 1987;28:155–158.10.1016/S0040-4039(00)95673-9
- Jefford CW, Moulin M-C. The synthesis of boronolide. Helv. Chim. Acta. 1991;74:336–342.10.1002/(ISSN)1522-2675
- Evans DA, Chapman KT, Carreira EM. Directed reduction of beta-hydroxy ketones employing tetramethylammonium triacetoxyborohydride. J. Am. Chem. Soc. 1988;110:3560–3578.10.1021/ja00219a035
- Yamauchi S, Isozaki Y, Nishimura H, Tsuda T, Nishiwaki H, Shuto Y. Total syntheses of (−)- and (+)-boronolide and their plant growth-inhibitory activity. Biosci. Biotechnol. Biochem. 2012;76:1708–1714.10.1271/bbb.120317