
Abstract
To characterize the hydrogen-bonding network in lysozyme, we focused on the residue of Asp48 located at the active site in hen egg-white lysozyme. We constructed a mutant lysozyme (D48A) and analyzed using (GlcNAc)3 and chitin-affinity chromatography. The substrate binding of subsites D–F in D48A and the activity against (GlcNAc)5 were decreased. The parameters of substrate binding and rate constants obtained from computer simulations confirmed these changes. In the crystal structure, (GlcNAc)4 was located at the same position as wildtype. However, the side chains of Arg45 and Thr47 at subsites E–F were moved by the replacement. Further, the loss of the hydrogen bond between Asp48 and Ser50 changed the hydrogen-bonding network, and this resulted in an alteration of the side chain of Asn59. This result suggests that the hydrogen-bonding network plays a crucial in the function of Asp52 and of transglycosylation at subsites E–F.
Graphical Abstract
The changes of the hydrogen-bonding network involving catalytic residue Asp52 of hen egg-white lysozyme by the replacement of Asp48 to Ala.
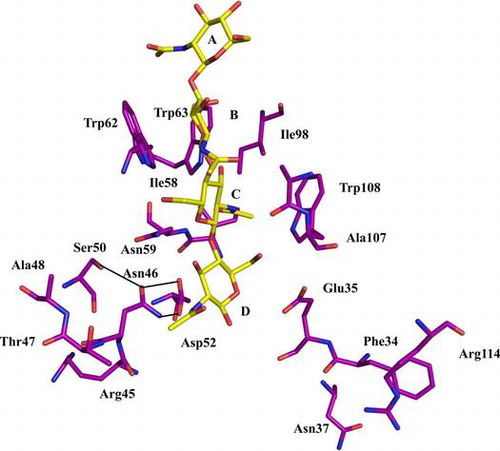
Lysozyme (EC 3.2.1.17) is a widely distributed carbohydrolase that is classified into c (chicken), g (goose), and i (invertebrate)-type lysozymes. Lysozyme catalyzes the hydrolysis of β-1,4-glycosidic bonds of the alternating copolymers of GlcNAc and MurNAc in bacterial cell walls and of chitin, the homopolymer of GlcNAc.Citation1) Crystallographic studies have shown that the substrate-binding site of hen-egg white lysozyme (HEL), a c-type lysozyme, contains six subsites (A–F) that can accommodate six saccharide residues.Citation2,3) A substrate that is bound to the subsites is cleaved at subsites D and E through the cooperative reaction of Glu35 and Asp52.Citation4–7) Catalytic models of the hydrolysis of saccharide chains by HEL were proposed independently by PhillipsCitation8) and Koshland.Citation9) The first step of the hydrolysis is understood in detail and it involves Glu35, which possesses an unusual pKa and protonates the oxygen connecting the saccharide residues at subsites D and E. By contrast, in the next step, the role of the second amino acid Asp52, which was proposed to stabilize the oxocarbonium ionCitation8) or the covalent state of the intermediate in the hydrolysis,Citation9) remains unclear. HEL was reported to not only hydrolyze saccharide chains, but also catalyze transglycosylation, which requires an acceptor oligomer instead of a water molecule during hydrolysis.Citation10–13)
The active site of HEL has been characterized extensively using various methods. The unitary free-energy changes required for the binding of the GlcNAc residue to the subsites have been estimated to be approximately −2.0, −3.0, −5.0, +4.5, −2.5, and −1.5 kcal/mol, respectively.Citation14,15) The X-ray structure of the complex formed between HEL and the GlcNAc trimer (GlcNAc)3 revealed that the structure of active site, subsite C, which provides most of the binding energy for the sugar substrate, was formed by the side chains of Trp62 and Trp63Citation16,17) and the main chains of Asn59 and Ala107.Citation18) Most importantly, a deep hydrophobic hole was formed by the side chains of Ile58, Ile98, and Trp108.Citation19,20) Subsite C is also considered to be crucial for substrate binding, and it plays a key role in determining the cleavage specificity of HEL.Citation19)
As noted in the first paragraph, Asp52, the second carboxyl-containing residue, has been implicated in the hydrolysis of the saccharide chain. The structure of the Asp52 catalytic residue was described in detail by Strynadka et al.Citation21) The side chain of Asp52 protruded from the central strand of a three-stranded antiparallel β-sheet into the active-site cleft. In the native enzyme, one of the oxygen atoms of Asp52’s side chain was solvent exposed and interacted through hydrogen bonding with two solvent molecules that formed a bridge to the carboxyl group of Glu35. The other oxygen atom was involved in a hydrogen-bonding network that contained the side chains of Asn46, Asp48, Ser50, and Asn59, and the side chains of all of these residues were on the same side of the β-sheet as the side chain of Asp52. Therefore, the residues that comprise this hydrogen-bonding network are all highly conserved in the sequences of c-type lysozyme.Citation1) Consequently, the replacement of Asp52 was previously shown, as expected, to reduce the hydrolytic activity of lysozyme to <5% of the activity of the wild-type enzyme.Citation22) In the hydrogen-bonding network, the role of Asn46, Ser50, and Asn59 have been examined in detail by performing mutation analysis.Citation18,23,24) However, no structural or functional studies have been conducted on the role of one member of this network—Asp48. Consequently, the complete mechanism through which the hydrogen-bonding network functions remains unclear. Here, we report biochemical and crystallographic studies on Asp48-mutated lysozyme (D48A).
Materials and methods
Materials
Restriction enzymes and DNA-modifying enzymes were purchased from Takara (Kyoto, Japan) and Toyobo (Tokyo, Japan). Oligonucleotide DNA primers were synthesized by Hokkaido System Science (Sapporo, Japan). Mutagenesis was performed using HA94-1, which contains the wild-type HEL gene in an mp18-derivative of the cloning vector M13. Escherichia coli strain JM109 was used in routine transformations and plasmid preparations. Multi-Copy Pichia expression kits, which included the expression plasmid pPIC9 K and the host strain GS115, were obtained from Invitrogen (La Jolla, CA, USA). Glycol chitin was prepared using the method of Yamada and Imoto.Citation25) (GlcNAc)n polymers were prepared according to the method of Rupley.Citation26) HPLC was used to confirm the purity of each oligosaccharide. All reagents were of analytical or biochemical grade.
Mutagenesis, expression, and purification of mutant lysozyme
Site-directed mutagenesis was performed using the PCR-based megaprimer method, and HA94-1 served as the template.Citation27) The primer used for mutagenesis was 5′-CGTAACACCGCTGGGAGTACC-3′ (D48A). DNA sequencing was used to confirm the mutation in the lysozyme gene. Wild-type lysozyme and D48A were produced and purified as described previously.Citation28) Protein concentrations were measured using the method of Pace et al.Citation29)
CD spectra
CD spectra in the far ultraviolet (UV) range, 200–250 nm, were recorded at 25 °C on a JASCO J-600 Spectropolarimeter (JASCO CO., Tokyo, Japan). Proteins were dissolved in 10 mM sodium acetate buffer (pH 5.0) to obtain a final concentration of 0.15 mg/mL. The data were expressed in terms of mean residue ellipticity.
Substrate binding
Substrate binding was estimated by measuring fluorescence intensities, which were standardized using N-acetyltryptophan; we used a Hitachi F-4500 fluorescence photometer (Hitachi Co., Tokyo, Japan). We incubated (GlcNAc)3 (0.02–0.2 mM) with lysozyme (0.02 mM) in 10 mM sodium acetate buffer (pH 5.0) for 5 min at 30 °C. The fluorescence intensity of the reaction mixture was measured at excitation and emission wavelengths of 291 and 360 nm, respectively. The dissociation constant (Kd) was calculated in terms of the amounts of enzyme and substrate complex (reduction of the fluorescence intensity of the reaction mixture relative to that of lysozyme) by performing Scatchard plotting with the equation ΔF = -Kd × ΔF/[S] + ΔFMAX (ΔF, reduction of the fluorescence intensity of the reaction mixture relative to that of lysozyme; [S], concentration of substrate).Citation30) We performed affinity HPLC of the lysozymes in order to determine their substrate-binding abilities using was a chitin-coated celite column (7.5 × 75 mm), which was eluted using a gradient of 10 mM sodium phosphate buffer (pH 7.0) and same buffer containing 1 M NaCl at a flow rate of 0.5 mL/min at 0 °C.
Enzymatic activities of lysozyme
The activity of the enzyme against glycol chitin was measured according to the method of Yamada and ImotoCitation25) by measuring the amount of released reducing groups and expressing these values as activity relative to the activity of the wild-type enzyme. Glycol chitin (0.05% w/v) was incubated with or without lysozyme (1 mM) at 50 °C for 30 min in 50 mM sodium acetate buffer (pH 5.0).
The enzymatic activity toward (GlcNAc)5 was measured using the method of Masaki et al.,Citation14) with slight modifications. The reaction mixture, which contained 0.1 mM lysozyme and 1 mM (GlcNAc)5, was incubated in 10 mM sodium acetate buffer (pH 5.0) at 50 °C. After a given reaction time, 200 μL of the reaction mixture was withdrawn and rapidly chilled in a KOOL KUP (Towa Co., Tokyo, Japan). The reaction mixture was centrifuged using Ultrafree-MC filter units (Millipore Co., Billerica, MA, USA), and the filtrate was lyophilized. The dried sample was dissolved in 50 μL of ice-cold water, and then 10 μL of the solution was applied on a TSK-gel G-Oligo-PW column (7.5 × 600 mm; Tosoh Co., Tokyo, Japan) mounted in a JASCO 800 series HPLC system (JASCO Co.). Elution was performed using distilled water at room temperature and the flow rate was 0.3 mL/min. The concentration of each chitooligosaccharide was calculated from the area of its peak, monitored by measuring UV absorption (220 nm), using a standard curve obtained for saccharide solutions of known concentrations. The relative error of measurements was defined as (y−x) = x × 100, where x is the concentration of the initial substrate and y is the recovered concentration of all chitooligosaccharides in (GlcNAc)1 units.
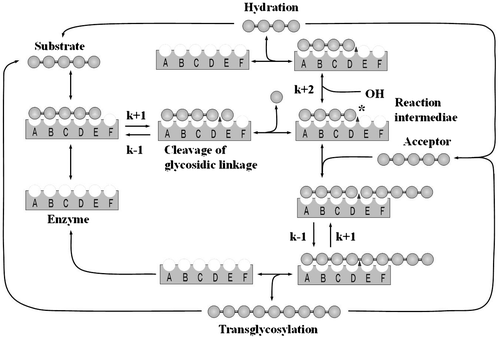
The rate equation used for the lysozyme-catalyzed reaction on the initial substrate, (GlcNAc)5, was solved numerically to obtain the calculated time courses. A kinetic model of the lysozyme-catalyzed reaction of chitooligosaccharides has been reported,Citation13–15,19) and the lysozyme-catalyzed reaction used for the calculation is schematically represented in Fig. . In the calculation, the rate equations were solved repeatedly while changing the values of the binding free energies and the rate constants k+1 (cleavage of the β-1,4-glycosidic linkage), k−1 (transglycosylation), and k+2 (hydration), so that the calculated time courses fit those obtained experimentally. The details of the method of calculation have been described previously.Citation19,31)
Fig. 1. Schematic representation of the lysozyme-catalyzed reaction.
Note: k+1, k−1, and k+2 are the rate constants for cleavage of the glycosidic linkage, transglycosylation, and hydration, respectively.
Equilibrium denaturation
Guanidine hydrochloride (GdnHCl)-induced unfolding of lysozymes was monitored by measuring the intrinsic fluorescence emission at 360 nm at an excitation of wavelength 280 nm. The lysozyme solution (0.015 mg/mL) was incubated in 0.1 M sodium acetate buffer (pH 5.0) containing various concentrations of GdnHCl at 30 °C for 1 h before the measurements in order to equilibrate the proteins with the denaturant. The fluorescence data were analyzed based on a two-state model. From the GdnHCl denaturation profiles, the difference in the free-energy change between the folded and unfolded states (ΔG) was calculated according to the method of Pace.Citation32) The free-energy change in water (ΔGH2O) and the dependence of ΔG on the GdnHCl concentration (m) were determined through least-squares fitting of the data obtained for the transition region; the equation used was ΔG = ΔGH2O−m[GdnHCl]. The GdnHCl concentration at the mid-point of the transition (ΔG = 0) was defined as Cm. The differences in Cm (ΔCm) between the wild-type and mutant enzymes were calculated by subtracting the values of the wild-type enzyme from those of the mutant.
Crystallization
D48A was crystalized in the presence and absence of a substrate analog (GlcNAc)4 at 20 °C using the sitting-drop vapor-diffusion method; we mixed 1 μL of the lysozyme solution (13.8 mg/mL protein in 10 mM sodium phosphate buffer (pH 7.0) containing 200 mM NaCl or 13.8 mg/mL protein in same buffer containing 1.98 mM substrate) with 1 μL of the reservoir solution (1.2 M NaCl, 0.1 M sodium acetate buffer, pH 4.0). We used the coordinates of HEL (protein data bank (PDB) codes 1lza and 1lzcCitation33)) to create an initial model for the structure determination. The atomic parameters were refined using CNSCitation34) and Refmac5Citation35) programs. The mutated residue and the bound substrate saccharide were carefully fitted to the electron densities using COOTCitation36) and MIFitCitation37). Molecular graphics were created using PyMOL (http://www.pymol.org/). The statistics of data collection, phase determination, and refinement are summarized in Table .
Table 1. Statistics of data collection, phase determination and refinement.
Results and discussion
Expression and characterization of mutant lysozyme
To clarify the functional and structural relevance of the hydrogen-bonding network to lysozyme’s catalytic activity, we focused on the residue Asp48. We constructed a mutant lysozyme (D48A) that lacked the carboxyl group provided by Asp48. We prepared D48A as described in Materials and Methods; D48A was expressed in the methylotrophic yeast Pichia pastoris expression system and produced in amounts similar to that of wild-type lysozyme.Citation28) We purified approximately 15 mg of the enzymes from 1 L of culture broth. The purity of the mutant protein was confirmed by means of SDS-PAGE (data not shown). Far UV CD spectra of D48A and the wild-type enzyme were indistinguishable (data not shown), which indicated that lysozyme’s secondary structure was not affected by the D48A mutation.
Substrate binding
In lysozyme, the fluorescence intensity derived from the Trp residues in subsites A–C (Trp62, Trp63, and Trp108) reflects the microenvironments of these subsites.Citation16,Citation17,20) To examine the mutation-induced structural changes around subsites A–C, we analyzed the mutant’s fluorescence spectra that were generated by the Trp residues in the presence and absence of (GlcNAc)3, and we compared these spectra with the spectrum of wild-type lysozyme. Kd of D48A for binding to (GlcNAc)3 was almost the same as that of the wild-type enzyme (Table ). Because the substrate (GlcNAc)3 binds predominantly to subsites A–C at a ratio of 99% in the absence of hydrolysis,Citation38) these results indicate that the structure around subsites A–C of lysozyme is not altered by the D48A mutation.
Table 2. Comparison of two biochemical parameters of wild-type and mutant lysozymes.
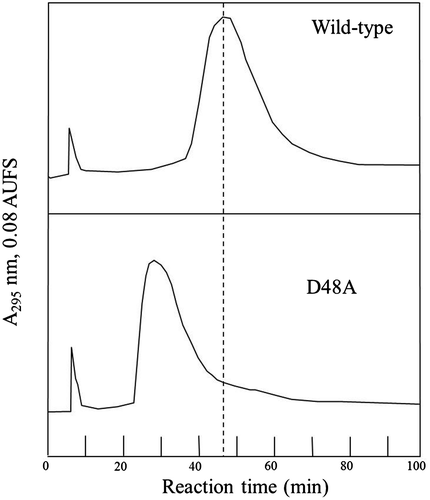
The relative retention times of the lysozyme proteins on an affinity-chromatography column (chitin-coated celite) are also shown in Table . Because chitin, a polymer substrate, occupies all of the subsites (A–F), the relative retention time on the chitin-coated celite column should reflect the substrate-binding ability of the enzyme in equilibrium binding. Our results showed that the relative retention time of D48A on the affinity column was lower than that of the wild-type enzyme (Fig. and Table ), 28.5 min for D48A and 47.0 min for wild-type lysozyme. This reduction in the retention time on the chitin-affinity column reflected the entire binding that includes subsites A–F. However, D48A’s binding affinity for (GlcNAc)3 was the same as that of wild-type lysozyme (Table ). Therefore, the reduction in the binding affinity of the mutant for the polymer substrate can be attributed to changes in the affinity of subsites D–F. Thus, we concluded that the substrate-binding ability of subsites D–F in D48A was weaker than that of the corresponding sites in wild-type lysozyme.
Fig. 2. Comparison of retention times in affinity HPLC performed on a chitin-celite column at 0 °C.
Enzymatic properties of the mutant protein
The enzymatic activity that D48A exhibited the hydrolysis of glycol chitin, a solubilized polymer of the substrate, was measured and expressed as a value relative to the activity of wild-type lysozyme. The enzymatic activity of D48A was 67.5% less than that of the wild-type enzyme.
Next, we analyzed the experimental time courses of (GlcNAc)5 degradation catalyzed by D48A and compared these with the time courses obtained using the wild-type enzyme. When investigating lysozyme-catalyzed reactions in detail, examining the experimental time courses of oligosaccharide-substrate degradation and product formation are critical; this is because the time courses provide substantial amounts of information on lysozyme catalysis, such as information on substrate binding, hydration, and transglycosylation.Citation14,19,39) Time-course analysis performed using the substrate (GlcNAc)5 reflects not only the hydrolysis of the initial substrate but also the transglycosylation produced by the cleavage of the substrate and the product.Citation39) Therefore, we analyzed time courses of the reaction catalyzed by D48A using (GlcNAc)5 as a substrate.
The experimental time courses measured for wild-type lysozyme and D48A using the initial substrate (GlcNAc)5 are shown in Fig. . After 20 min of the reaction catalyzed by the wild-type enzyme, the order of the amounts of the products formed was (GlcNAc)1 > (GlcNAc)2 > (GlcNAc)4 > (GlcNAc)3 (Fig. (A)). The time course obtained for D48A exhibited a profile that was distinct from that of wild-type lysozyme; 240 min were required for the completion of the catalysis, and the order of the amounts of the products was changed to (GlcNAc)1 > (GlcNAc)4 > (GlcNAc)2 > (GlcNAc)3 (Fig. (B)).
Fig. 3. Experimental and calculated time-course plots obtained for (GlcNAc)5 degradation by wild-type and mutant lysozymes. (A) Experimental, wild-type; (B) experimental, D48A; (C) calculated, wild-type; and (D) calculated, D48A.
Notes: Numerals in the figures are the degrees of polymerization of the reaction-product species. (GlcNAc)1, (GlcNAc)2, (GlcNAc)3, (GlcNAc)4, and (GlcNAc)5 are indicated by ○, □, ▲, △, and ●, respectively.
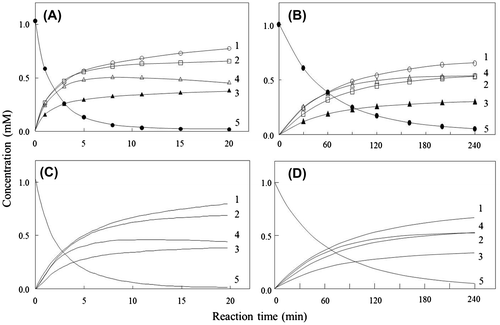
The experimental time courses of D48A and wild-type lysozyme were analyzed using computer simulations in order to obtain the rate constants and binding free-energy values for individual subsites and also for the whole wild-type enzyme. The calculated time courses are shown in Fig. (C) and (D), and the reaction parameters obtained are summarized in Table . The binding free-energy values obtained for subsites A–C were identical in wild-type lysozyme and D48A, which agrees with the finding that lysozyme’s affinity for (GlcNAc)3 was unaffected by the D48A mutation. By contrast, the D48A mutation changed the binding free-energy values measured for subsites D–F; the binding free energies for subsites D and E were decreased to (4.7 and −2.3 kcal/mol, respectively), whereas the binding free energy for subsite F was increased (to −1.6 kcal/mol), as compared with the values measured for the wild-type enzyme. Furthermore, the rate constants of transglycosylation, k−1, and the cleavage of the glycosidic linkage, k+1, were decreased to 19 and 0.065 s−1, respectively, in the case of D48A (Table ). These results suggest that Asp48 key roles in lysozyme catalysis, contributing to substrate binding at subsites D–F and to the transglycosylation activity of the enzyme. Furthermore, the reduction in the catalytic activity of D48A was attributed to diminished substrate binding and cleavage of the glycosidic linkage. Subsites E–F were critical for transglycosylation, because the binding site for the oligosaccharide acceptor is located at subsites E–F.Citation31) Our results suggest that the rate constant of the cleavage of the glycosidic linkage, k+1, and transglycosylation, k−-1, might be decreased because of the change in the binding free energies of subsites E–F as a result of the D48A mutation.
Table 3. Estimated values of the reaction parameters of wild-type and mutant lysozymes with the substrate (GlcNAc)5.
Effect of D48A mutation on lysozyme stability in GdnHCl
The contribution of Asp48 to the structural stability of HEL was assessed by means of unfolding experiments performed using GdnHCl as the denaturant. The GdnHCl-induced unfolding curves of wild-type lysozyme and D48A were obtained through fluorescence measurements. The thermodynamic parameter Cm used for evaluating denaturation was calculated from the unfolding curves. The Cm values of D48A and wild-type lysozyme were 3.15 and 3.99 M, respectively. Thus, the ΔCm is −0.84 M, which indicates that D48A was less stable than the wild-type enzyme (Table ). This result suggests that the side chain of Asp48 is directly involved in the enzyme’s structural stability because CD spectra revealed that the wild-type and mutant enzymes carried equal main chains. Furthermore, previous mutation analysis of HEL demonstrated that Asn46 and Ser50 contribute to the enzyme’s structural stability because both residues are members of the hydrogen-bonding network.Citation23,24) The mutation of Asp48 causes the loss of the charge on acidic residue. Therefore, the alteration of the charge may contribute to the stability in addition to the effect for hydrogen-bonding network. This was supported by the previous report by Abe et al. for D48 N mutant.Citation40) Collectively, these results support the conclusion that the hydrogen-bonding network that includes the catalytic residue Asp52 plays a critical role in the structural stability of lysozyme.
Table 4. Parameters characterizing Gdn-HCl denaturation at pH 5.0 at 30 °C.
Structural analysis of D48A performed using X-ray crystallography
In order to elucidate the structural effects of replacing Asp48 with Ala, we used X-ray crystallography to analyze the structures of D48A–with and without the substrate analog (GlcNAc)4.
The crystals of D48A (PDB Code 3wvx) and its complexes were isomorphous with those of the wild-type enzyme (PDB Code 1lza). The resolution limits of the diffraction for the free and complexed forms were 1.6 Å. The model of the D48A-(GlcNAc)4 complex (PDB code 3wvy) was similar to that of the wild-type (GlcNAc)4 complex (PDB code 1lzc). The results in Fig. (A) show that the bound (GlcNAc)4 was located at the binding subsites A–B–C–D, as observed in the wild-type enzyme. Moreover, the amino acids around subsites A–D that participate in substrate binding were not changed. The results of crystal-structure analysis agree with the results obtained from the CD spectra and (GlcNAc)3 binding. Taken together these results suggest that the backbone structure and the substrate binding of subsites A–D are not affected by the replacement of Asp48.
Fig. 4. Comparison of the structures of wild-type (GlcNAc)4 and D48A-(GlcNAc)4.
Notes: Wild-type (GlcNAc)4 (PDB code 1lzc), green; D48A-(GlcNAc)4 (PDB code 3wvy), purple. The electron density of Arg45, Thr47, and (GlcNAc)4 is contoured in gray at 1σ. (A) Superposition of the interaction of (GlcNAc)4 with binding sites. (GlcNAc)4 of wild type and D48A is indicated in cyan and yellow. (B) Superposition of the structures of left-sided and right-sided binding sites of wild type and D48A. (C) The hydrogen-bonding network involving the catalytic residue Asp52 of wild type. (D) The hydrogen-bonding network involving the catalytic residue Asp52 of D48A.
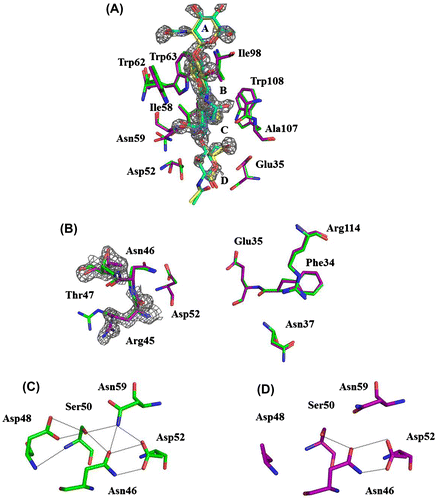
As described in the subsection Substrate binding in Results and Discussion, the substrate-binding ability of subsites D–F in D48A was shown to be weaker than that of the corresponding sites in wild-type lysozyme using (GlcNAc)3 and affinity chromatography. To clarify how substrate binding at subsites D–F was decreased, we focused on the structure of subsites D–F. A catalytic study of several avian lysozymes containing amino acid substitutions suggested that subsites E and F participate in the transglycosylation catalyzed by lysozyme.Citation41) Experiments involving a calculation of conformational energy and monoclonal antibody binding indicated that two binding modes are used in the interaction of (GlcNAc)6 with subsites E and F, the left-sided and right-sided binding modes.Citation10,42) In HEL, Arg45, Asn46, and Thr47 are responsible for the left-sided binding, whereas Phe34, Asn37, and Arg114 mediate the right-sided binding (Fig. (B)). The left-site structure of subsites E and F in D48A was changed relative to that in HEL, as observed at the side chains of Arg45 and Thr47 (Fig. (B)). This result confirmed the change in the binding energy observed at subsites E and F in the computer simulation of (GlcNAc)5 binding. Thus, we conclude that these two amino acids at subsites E and F were affected by the replacement of Asp48.
HEL features a hydrogen-bonding network that involves the catalytic residue Asp52. We focused on the structure of the hydrogen-bonding network in the D48A mutant and compared it with that of the wild-type enzyme. In the structure of D48A, the hydrogen-bonding network was changed because of the loss of a hydrogen bond between Asp48 and Ser50, and this resulted in an alteration of the side chain of Asn59 (Fig. (D). The water molecules in a hydrogen-bonding network of D48A were not changed comparing with wild type. Therefore, the change of the side chain of Ser50 and Asn59 was ascribed to the replacement of Asp48. Asn59 was reported to be critical for lysozyme catalytic activity.Citation18) Thus, the reduction in the catalytic activity of D48A was caused by changes in the affinity and the modes of substrate binding and the hydrogen bonding network structure, which was ascribed to the loss of the hydrogen bonds between Asp48 and Ser50 and Ser50 and Asn59. Asp48 functions in fixing the side chain of Ser50, which forms hydrogen bonds with Asn46 and Asn59.
Several previous studies have been conducted on Asn46, Ser50, and Asn59 mutants. Asn46 was reported to function in the maintenance of environment around Asp52,Citation23) Ser50 was reported to be involved in substrate binding and catalytic activity,Citation24) and Asn59 was reported to retain the orientation of the side chain of Asp52.Citation18) In this study, we obtained new information on the hydrogen-bonding network. Our results suggest that the role of the hydrogen-bonding network involving Asp52 is critical for the expression of the function of Asp52 and for the transglycosylation function at the substrate-binding subsites E–F. In conclusion, we suggest that the environment around the catalytic residue Asp52 is crucial for the catalytic mechanism of HEL. However, the residue Asp52 of c-type lysozyme does not exist in g-type lysozyme.Citation43) Recently, we reported the structure of an i-type lysozyme isolated from Tapes japonica and Meretrix lusoria and compared it with the structure of HEL: the position and the orientation of the Asp residue in i-type lysozyme were distinct from those in c-type lysozyme.Citation44,45) In this study, we revealed that a second catalytic residue, Asp52, in c-type lysozyme is critical for the catalytic activity. Therefore, the function of the catalytic residue Asp52 in HEL must be analyzed further in comparison with g- and i-type lysozymes.
Notes
Abbreviations: HEL, hen egg-white lysozyme; GlcNAc, N-acetylglucosamine; MurNAc, N-acetylmuramic acid; CD, circular dichroism; GdnHCl, guanidine hydrochloride; (GlcNAc)n, β-1,4-linked oligosaccharide of GlcNAc featuring a polymerization degree of n.
Related Research Data
References
- Jollès J, Jollès P. What’s new in lysozyme research? Always a model system, today as yesterday. Mol. Cell. Biochem. 1984;63:165–189.
- Blake CC, Koenig DF, Mair GA, North ACT, Phillips DC, Sarma VR. Structure of hen egg-white lysozyme: a three-dimensional Fourier synthesis at 2 Å resolution. Nature. 1965;206:757–761.10.1038/206757a0
- Blake CC, Johnson LN, Mair GA, North AC, Phillips DC, Sarma VR. Crystallographic studies of the activity of hen egg-white lysozyme. Proc. R. Soc. Lond., Ser. B, Biol. Sci. 1967;167:378–388.10.1098/rspb.1967.0035
- Kuroki R, Yamada H, Moriyama T, Imoto T. Chemical mutations of the catalytic carboxyl groups in lysozyme to the corresponding amides. J. Biol. Chem. 1986;261:13571–13574.
- Malcolm BA, Rosenberg S, Corey MJ, Allen JS, de Baetselier AD, Kirsch JF. Site-directed mutagenesis of the catalytic residues Asp-52 and Glu-35 of chicken egg white lysozyme. Proc. Natl. Acad. Sci. USA. 1989;86:133–137.10.1073/pnas.86.1.133
- Hashimoto Y, Yamada K, Motoshima H, Omura T, Yamada H, Yasukochi T, Miki T, Ueda T, Imoto T. A mutation study of catalytic residue Asp52 in hen egg lysozyme. J. Biochem. 1996;119:145–150.10.1093/oxfordjournals.jbchem.a021199
- Vocadlo DJ, Davies GJ, Laine R, Withers SG. Catalysis by hen egg-white lysozyme proceeds via a covalent intermediate. Nature. 2001;412:835–838.10.1038/35090602
- Phillips DC. The three-dimensional structure of an enzyme molecule. Sci. Am. 1966;215:78–90.10.1038/scientificamerican1166-78
- Koshland DE Jr, Neet KE. The catalytic and regulatory properties of enzymes. Annu. Rev. Biochem. 1968;37:359–411.10.1146/annurev.bi.37.070168.002043
- Smith-Gill SJ, Rupley JA, Pincus MR, Carty RP, Scheraga HA. Experimental identification of a theoretically predicted “left-sided” binding mode for (GlcNAc)6 in the active site of lysozyme. Biochemistry. 1984;23:993–997.10.1021/bi00300a030
- Pollock JJ, Sharon N. Acceptor specificity of the lysozyme-catalyzed transglycosylation reaction. Biochemistry. 1970;9:3913–3925.10.1021/bi00822a009
- Inoue M, Yamada H, Yasukochi T, Miki T, Horiuchi T, Imoto T. Left-sided substrate binding of lysozyme: evidence for the involvement of asparagine-46 in the initial binding of substrate to chicken lysozyme. Biochemistry. 1992;31:10322–10330.10.1021/bi00157a021
- Pincus MR, Burgess AW, Scheraga HA. Conformational energy calculations of enzyme-substrate complexes of lysozyme. I. Energy minimization of monosaccharide and oligosaccharide inhibitors and substrates of lysozyme. Biopolymers. 1976;15:2485–2521.10.1002/(ISSN)1097-0282
- Masaki A, Fukamizo T, Otakara A, Torikata T, Hayashi K, Imoto T. Lysozyme-catalyzed reaction of chitooligosaccharides. J. Biochem. 1981;90:527–533.
- Masaki A, Fukamizo T, Otakara A, Torikata T, Hayashi K, Imoto T. Estimation of rate constants in lysozyme-catalyzed reaction of chitooligosaccharides. J. Biochem. 1981;90:1167–1175.
- Teshima K, Kuramitsu S, Hamaguchi K, Sakiyama F, Mizuno K, Yamasaki N. Binding of substrate analogs to hen lysozyme in which Trp62 is modified to Kynurenine. J. Biochem. 1980;87:1015–1027.
- Ueda T, Nakashima A, Hashimoto Y, Miki T, Yamada H, Imoto T. Formation of alpha-helix 88-98 is essential in the establishment of higher-order structure from reduced lysozyme. J. Mol. Biol. 1994;235:1312–1317.10.1006/jmbi.1994.1084
- Ose T, Kuroki K, Matsushima M, Maenaka K, Kumagai I. Importance of the hydrogen bonding network including Asp52 for catalysis, as revealed by Asn59 mutant hen egg-white lysozymes. J. Biochem. 2009;146:651–657.10.1093/jb/mvp110
- Kawamura S, Chijiiwa Y, Torikata T, Araki T. The mutation effect of Ile58 at subsite C in hen egg-white lysozyme on substrate binding, enzymatic activity, and protein stability. Biosci. Biotechnol. Biochem. 2013;77:560–565.10.1271/bbb.120811
- Inoue M, Yamada H, Yasukochi T, Kuroki R, Miki T, Horiuchi T, Imoto T. Multiple role of hydrophobicity of tryptophan-108 in chicken lysozyme: structural stability, saccharide binding ability, and abnormal pKa of glutamic acid-35. Biochemistry. 1992;31:5545–5553.10.1021/bi00139a017
- Strynadka NC, James MN. Lysozyme revisited: crystallographic evidence for distortion of N-acetylmuramic acid residue bound in site D. J. Mol. Biol. 1991;220:401–424.10.1016/0022-2836(91)90021-W
- Matsumura I, Kirsch JF. Synergistic contributions of asparagine 46 and aspartate 52 to the catalytic mechanism of chicken egg white lysozyme. Biochemistry. 1996;35:1890–1896.10.1021/bi951672i
- Inoue M, Yamada H, Yasukochi T, Miki T, Horiuchi T, Imoto T. Left-sided substrate binding of lysozyme: evidence for the involvement of asparagine-46 in the initial binding of substrate to chicken lysozyme. Biochemistry. 1992;31:10322–10330.10.1021/bi00157a021
- Kawaguchi Y, Kawaura S, Yoneda K, Totikata T, Araki T. The role of Ser50 in the catalytic site of hen egg-white lysozyme for substrate binding and activity toward oligomer of chitin. Chitin and Chitosan Research. 2012;18:242–251. Japanese.
- Yamada H, Imoto T. A convenient synthesis of glycolchitin, a substrate of lysozyme. Carbohydr. Res. 1981;92:160–162.10.1016/S0008-6215(00)85993-5
- Rupley JA. The hydrolysis of chitin by concentrated hydrochloric acid, and the preparation of low-molecular-weight substrates for lysozyme. Biochim. Biophys. Acta. 1964;83:245–255.
- Sarkar G, Sommer SS. The “megaprimer” method of site-directed mutagenesis. Biotechniques. 1990;8:404–407.
- Kawamura S, Toshima G, Imoto T, Araki T, Torikata T. Amino acid residues in subsites E and F responsible for the characteristic enzymatic activity of duck egg-white lysozyme. J. Biochem. 2002;131:663–670.10.1093/oxfordjournals.jbchem.a003149
- Pace CN, Vajdos F, Fee L, Grimsley G, Gray T. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 1995;4:2411–2423.10.1002/pro.v4:11
- Scatchard G. The attractions of proteins for small molecules and ions. Ann. N. Y. Acad. Sci. 1949;51:660–672.10.1111/nyas.1949.51.issue-4
- Kawamura S, Chijiiwa Y, Minematsu T, Fukamizo T, Varum KM, Torikata T. The role of Arg114 at subsites E and F in reactions catalyzed by hen egg-white lysozyme. Biosci. Biotechnol. Biochem. 2008;72:823–832.10.1271/bbb.70694
- Pace CN. Measuring and increasing protein stability. Trends Biotechnol. 1990;8:93–98.10.1016/0167-7799(90)90146-O
- Maenaka K, Matsushima M, Song H, Sunada F, Watanabe K, Kumagai I. Dissection of protein–carbohydrate interactions in mutant hen egg-white lysozyme complexes and their hydrolytic activity. J. Mol. Biol. 1995;247:281–293.10.1006/jmbi.1994.0139
- Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta. Crystallogr. Sec. D. Biol. Crystallogr. 1998;54:905–921.10.1107/S0907444998003254
- Murshudov GN, Skubák P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA. REFMAC 5 for the refinement of macromolecular crystal structures. Acta Crystallogr. Sec. D. Biol. Crystallogr. 2011;67:355–367.10.1107/S0907444911001314
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. Sec. D. Biol. Crystallogr. 2004;60:2126–2132.10.1107/S0907444904019158
- McRee DE. Differential evolution for protein crystallographic optimizations. Acta. Crystallogr. Sec. D. Biol. Crystallogr. 2004;60:2276–2279.10.1107/S0907444904025491
- Kuhara S, Ezaki E, Fukamizo T, Hayashi K. Estimation of the free energy change of substrate binding in lysozyme-catalyzed reactions. J. Biochem. 1982;92:121–127.
- Fukamizo T, Minematsu T, Yanase Y, Hayashi K, Goto S. Substrate size dependence of lysozyme-catalyzed reaction. Arch. Biochem. Biophys. 1986;250:312–321.10.1016/0003-9861(86)90732-0
- Abe Y, Ueda T, Iwashita H, Hashimoto Y, Motoshima H, Tanaka Y, Imoto T. Effect of salt concentration on the pKa of acidic residues in lysozyme. J. Biochem. 1995;118:946–952.10.1093/jb/118.5.946
- Fukamizo T, Torikata T, Nagayama T, Minematsu T, Hayashi K. Enzymatic activity of avian egg-white lysozymes. J. Biochem. 1983;94:115–122.
- Pincus MR, Zimmerman SS, Scheraga HA. Structures of enzyme-substrate complexes of lysozyme. Proc. Natl. Acad. Sci. 1977;74:2629–2633.10.1073/pnas.74.7.2629
- Weaver LH, Grütter MG, Matthews BW. The refined structures of goose lysozyme and its complex with a bound trisaccharide show that the “Goose-type” ly'sozymes lack a catalytic aspartate residue. J. Mol. Biol. 1995;245:54–68.10.1016/S0022-2836(95)80038-7
- Goto T, Abe Y, Kakuta Y, Takeshita K, Imoto T, Ueda T. Crystal structure of tapes japonica lysozyme with substrate analogue: structural basis of the catalytic mechanism and manifestation of its chitinase activity accompanied by quaternary structural change. J. Biol. Chem. 2007;282:27459–27467.10.1074/jbc.M704555200
- Kuwano Y, Yoneda K, Kawaguchi Y, Araki T. The tertiary structure of an i-type lysozyme isolated from the common orient clam (Meretrix lusoria). Acta. Crystallogr. Sec. F. 2013;69:1202–1206.10.1107/S1744309113028170