
Abstract
Amilorides, well-known inhibitors of Na+/H+ antiporters, have also shown to inhibit bacterial and mitochondrial NADH-quinone oxidoreductase (complex I). Since the membrane subunits ND2, ND4, and ND5 of bovine mitochondrial complex I are homologous to Na+/H+ antiporters, amilorides have been thought to bind to any or all of the antiporter-like subunits; however, there is no direct experimental evidence in support of this notion. Photoaffinity labeling is a powerful technique to identify the binding site of amilorides in bovine complex I. Commercially available amilorides such as 5-(N-ethyl-N-isopropyl)amiloride are not suitable as design templates to synthesize photoreactive amilorides because of their low binding affinities to bovine complex I. Thereby, we attempted to modify the structures of commercially available amilorides in order to obtain more potent derivatives. We successfully produced two photoreactive amilorides (PRA1 and PRA2) with a photolabile azido group at opposite ends of the molecule.
Graphical Abstract
Amilorides, well-known inhibitors of Na+/H+ antiporters, were reported to inhibit mitochondrial complex I. We attempted to modify the structure of commercially available amilorides and successfully produced two potent photoreactive amilorides.
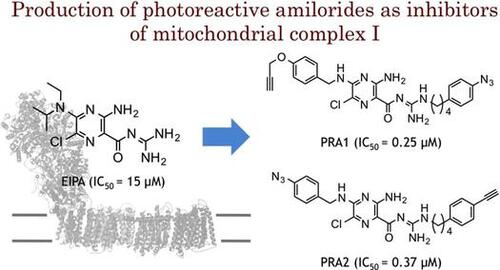
Amilorides are well-known inhibitors of Na+/H+ and Na+/Ca2+ antiporters and Na+ channels.Citation1,2) The structural factors of amilorides that are critical to the inhibitory action greatly vary depending on the different ion transporters and species.Citation1) Amilorides have also shown to inhibit bacterial and mitochondrial NADH-quinone oxidoreductase (complex I),Citation3,4) but their inhibitory potencies, in terms of IC50 values, are much weaker than those of traditional inhibitors such as rotenone, piericidin A, and acetogenins. As the membrane subunits ND2, ND4, and ND5 of bovine mitochondrial complex I are homologous to Na+/H+ antiporters,Citation5–7) amilorides have been thought to bind to any or all of the antiporter-like subunits; however, there is currently no direct experimental evidence in support of this notion. This binding of amilorides to antiporter-like subunit(s) needs to be examined in more detail because if this is the case, they may become useful molecular tools to study the mechanism of proton translocation through the antiporter-like subunits.
Photoaffinity labeling is a powerful technique to identify the binding site of amilorides in bovine complex I.Citation8,9) In the photoaffinity labeling technique, less nonspecific binding occurs at higher binding affinities of photoreactive ligands; therefore, using a ligand with high binding affinity as possible allows for the accurate interpretation of data obtained in photoaffinity labeling experiments. Commercially available amilorides are not suitable as design templates for the synthesis of photoreactive amilorides possessing high binding affinities to complex I because of their low binding affinities to bovine complex I. Therefore, we herein attempted to modify the structures of commercially available amilorides and investigated the structure–activity relationship (SAR) of these synthetic derivatives to obtain more potent derivatives.
Materials and methods
Materials
5-(N-ethyl-N-isopropyl)amiloride (EIPA), 5-(N-methyl-N-isobutyl)amiloride (MIA), and benzamil were purchased from Sigma-Aldrich (St. Louis, MO, USA). Ubiquinone-1 (Q1) was generously provided by Eisai Co., Ltd. (Tokyo, Japan). Other reagents were of analytical grade.
General synthetic methods
1H NMR spectra were recorded at 500 or 400 MHz with Bruker AVANCE III 500 or AVANCE III 400 spectrometers, respectively, using tetramethylsilane (TMS) as the internal standard. 13C NMR spectra were recorded at 125 or 100 MHz. Chemical shifts (δ) are given in ppm relative to TMS with coupling constants (J) in Hz. HPLC purification was carried out with a Shimadzu LC-10 AS using a Wakosil-II 5C18 HG prep column. Elution profiles were detected at 254 nm with a Shimadzu SPD-10A.
General procedures for the preparation of series 1
To a solution of methyl 3-amino-5,6-dichloro-2-pyrazinecarboxylate (1.0 eq.) in 2-propanol (5 mL/mmol), an appropriate primary or secondary amine (3–6 eq.) was added, and the reaction mixture was refluxed for 2 h.Citation10) The solution was cooled to room temperature, concentrated under reduced pressure, and the residue was purified by silica-gel column chromatography (Wako gel® C-200, 30–40% ethyl acetate/n-hexane) to afford the series 1 intermediate (yield: 32–95%). The regioselectivity of the nucleophilic substitution at the 5-position of the pyrazine ring was confirmed by an X-ray structural analysis (Fig. S1).
General procedures for the preparation of series 2
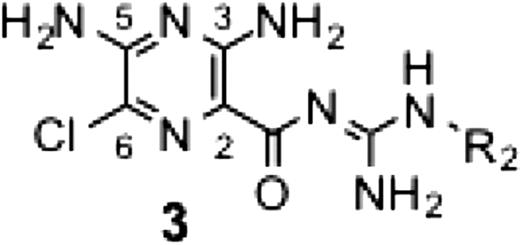
Guanidine hydrochloride (5 eq.) was dissolved in anhydrous methanol (1 mL/mmol) and stirred with sodium methoxide (5.5 eq.) for 30 min at room temperature under N2 atmosphere. The precipitate was removed by filtration, and the resulting methanolic solution was concentrated under reduced pressure to give the guanidine base.Citation10,11) The guanidine base was further dissolved in anhydrous methanol (1 mL/mmol) with an appropriate pyrazine unit 1 (1 eq.), and the mixture was refluxed for 12–18 h under N2 atmosphere. The solution was cooled to room temperature, acidified with 1.0 M aqueous HCl, and concentrated. Excess guanidine was removed from the residue using reversed-phase column chromatography (YMC gel ODS-A-HG, 10–100% methanol/0.1% aqueous HCl). The crude product was further purified by silica-gel column chromatography (Wako gel® C-200, 10% methanol/chloroform) to afford the series 2 derivatives (yield: 29–67%). The spectral data of 2c–2 k were listed in the Supporting Information.
General procedures for the preparation of series 3
To a solution of an appropriate primary amine (2.5 eq.) in a mixture of anhydrous DMF and diisopropylethylamine [3:1 (v/v), 5 mL/mmol], 1H-pyrazol-1-carboxamidine hydrochloride (3.5 eq.) was added under N2 atmosphere, and the mixture was stirred at room temperature for 12–18 h, followed by the addition of diethyl ether (10–20 mL).Citation12) The formed oil containing the guanidine salt was washed twice with ether and dried under the reduced pressure. The resulting oil was further dissolved in anhydrous methanol (2–3 mL/mmol). Then, sodium methoxide (5 eq.) was added and the solution was stirred for 30 min at room temperature, followed by filtration of the precipitate. The filtrate was concentrated under reduced pressure to provide guanidine bases, which were used in the next reaction without further purification.
The guanidine base was dissolved in anhydrous methanol (2–3 mL/mmol) with methyl 3,5-diamino-6-chloropyrazine-2-carboxylate (1 eq.) and refluxed for 2 h under N2 atmosphere. The mixture was cooled to room temperature, concentrated under reduced pressure, and purified by silica-gel column chromatography (Wako gel® C-200, 10% methanol/chloroform) to afford series 3 (yield: 23–28%). The spectral data of 3a–3c were listed in the Supporting Information.
General procedures for the preparation of series 4
To a solution of an appropriate primary amine (1 eq.) in water (0.25 mL/mmol), S-methylisothiourea sulfate (1 eq.) in ethanol (0.25 mL/mmol) was added, and the resulting mixture was refluxed for 2 h.Citation13) The solution was cooled to room temperature, concentrated under reduced pressure, and the residue was then washed with diethyl ether. The guanidine compound was recrystallized from water containing 5–30% ethanol. The crystal was collected by filtration as an alkylguanidine sulfate salt.
Under N2 atmosphere, the guanidine sulfate salt was treated with sodium methoxide (1.1 eq.) in anhydrous methanol (1 mL/mmol). The mixture was stirred for 30 min at room temperature, and the precipitate was removed by filtration. The filtrate was concentrated under reduced pressure, yielding the guanidine base 4 (yield: 59–85%).
General procedure for the preparation of series 5
A solution of an appropriate pyrazine 1 (1 eq. typically ~1 mmol) and guanidine base 4 (2.5–5 equiv) in anhydrous methanol (1.5 mL) was refluxed for 4 h. The reaction mixture was cooled to room temperature, acidified with 1.0 M aqueous HCl, and concentrated under reduced pressure. The residue was purified by reversed-phase column chromatography (YMC gel ODS-A-HG, 10–100% methanol/0.1% aqueous HCl) to remove excess guanidine. The crude product was further purified by silica-gel column chromatography (Wako gel® C-200, 10% methanol/chloroform). Final purification was carried out using preparative HPLC (Wakosil-II 5C18 HG prep, 5 mL/min, 50–90% methanol/water containing 0.1% TFA) to provide 5 (yield: 12–61%). The spectral data of 5a–5s, PRA1, and PRA2 were listed in the Supporting Information.
General procedure for the preparation of 6
To a solution of an appropriate series 1 (1 eq.) in a mixture of THF and methanol [3:1 (v/v), 8 mL/mmol], 1.0 M aqueous LiOH (2 eq.) was added, and the mixture was stirred for 4 h at room temperature. The reaction was quenched with saturated aqueous NH4Cl, extracted with dichloromethane, and the organic layer was dried over MgSO4. The crude product was purified by silica-gel column chromatography (Wako gel® C-200, 10% methanol/chloroform) to give 6 (yield: 75%).
General procedures for the preparation of series 7
To a solution of series 6 (1 eq.) in anhydrous DMF (18 mL/mmol), an appropriate primary amine (1.2 eq.), 4-methylmorphorine (5 eq.), and HATU (1.5 equiv) were added, and the mixture was stirred for 3 h at room temperature.Citation14) The reaction was quenched with 0.1 M aqueous NaOH. The mixture was extracted with dichloromethane, and the organic layer was dried over MgSO4. The crude product was purified by silica-gel column chromatography (Wako gel® C-200, 10–30% ethyl acetate/n-hexane) to give 7 (yield: 64–68%). The spectral data of 7a and 7b were listed in the Supporting Information.
Measurement of complex I activity in bovine heart submitochondrial particles (SMP)
SMP were prepared from isolated bovine heart mitochondria by the method of Matsuno-Yagi and Hatefi.Citation15) and stored in buffer containing 250 mM sucrose and 10 mM Tris-HCl (pH 7.4) at −80 °C before being used. The NADH oxidase activity in SMP was followed spectrometrically with a Shimadzu UV-3000 (340 nm, ε = 6.2 mM−1 cm−1) at 30 °C. The reaction medium (2.5 mL) contained 0.25 M sucrose, 1 mM MgCl2, and 50 mM phosphate buffer (pH 7.5). The final mitochondrial protein concentration was 30 μg of protein/mL. The reaction was started by adding 50 µM NADH after the equilibration of SMP with an inhibitor for 4 min. The NADH-Q1 oxidoreductase activity was also measured under the same experimental conditions, except that the reaction medium contained 50 μM Q1, 0.2 μM antimycin A, and 4 mM KCN.Citation16)
Results and discussion
The structures of commercially available amilorides [EIPA (2a), MIA (2b), and benzamil (3a)], which have been used in the research on complex I, are shown in Fig. . The inhibitory activities of these amilorides were determined by the NADH oxidase assay using bovine heart SMP, and their IC50 values were approximately 10–20 μM (Table ); being comparable to those reported.Citation3) We also confirmed that the IC50 values of these amilorides determined with the NADH-Q1 oxidoreductase assay matched those determined with the NADH oxidase assay, indicating that the inhibition of other respiratory complexes is negligible. Since the IC50 values of the photoreactive inhibitors used in earlier photoaffinity labeling studies with complex I, such as quinazoline,Citation8) fenpyroximate,Citation9) and acetogenins,Citation17) were all at one digit nm levels, the commercially available amilorides are not potent enough to be used as templates for designing photoreactive amilorides. Therefore, we needed to produce more potent amiloride derivatives (Scheme ).
Fig. 1. Structures of EIPA, MIA, benzamil, and photoreactive amilorides (PRA1 and PRA2).
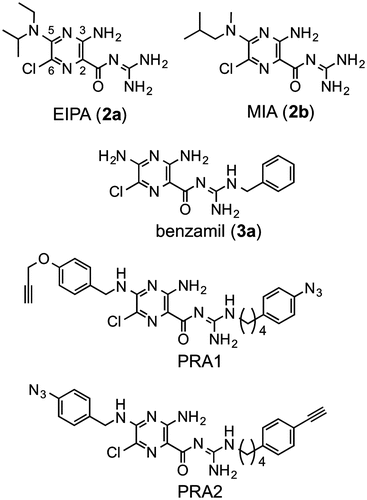
Table 1. Inhibitory activities of series 2 and 3 derivativesTable Footnotea. 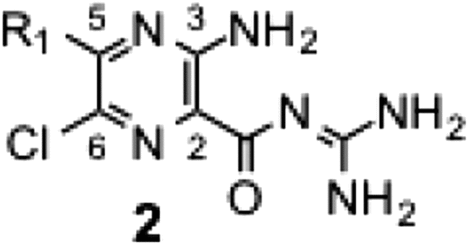
 .
.
Scheme 1. Synthesis of amiloride derivatives (series 2, 3, 5, and 7). Reagents and conditions: (a) primary or secondary amine, 2-propanol, reflux, 2 h; (b) guanidine hydrochloride, NaOMe, MeOH, reflux, 12–18 h; (c) (i) R2–NH2, 1H-pyrazole-1-carboxamidine hydrochloride, DMF/(i-Pr)2EtN (3:1), rt, 12–18 h, (ii) NaOMe, MeOH, rt, 30 min, (iii) R2–guanidine base, reflux, 2 h; (d) (i) R2–NH2, EtOH/H2O (1:1), reflux, 4 h, (ii) NaOMe, MeOH, rt, 30 min; (e) MeOH, reflux, 2 h; (f) aq. LiOH, THF/MeOH (1:1), rt, 4 h; (g) R3–NH2, 4-methylmorphorine, HATU, DMF, rt, 3 h.
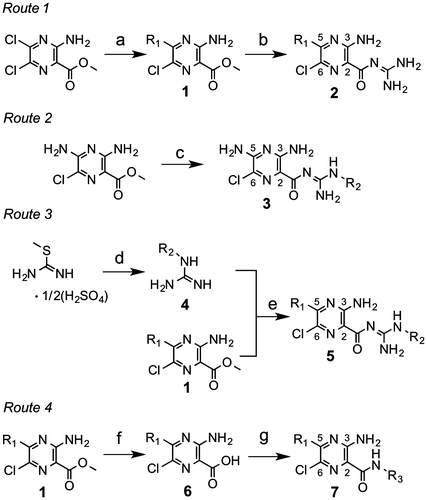
In the series 2 derivatives (Table ), we modified the 5-amino group on the pyrazine ring without modifying the guanidine moiety. In spite of wide modifications, we did not obtain any derivative eliciting more potent activity than those of EIPA and MIA. The hydrogen-bond donating –NH was not critical to the inhibitory activity (2b vs. 2d, 2 h vs. 2 k).
The SAR for series 3 revealed that the activity does not significantly increase with elevation in the hydrophobicity of R2 unless the 5-position is fixed with the primary amino group. However, this was not the case when R1 and R2 were modified simultaneously, as described below.
In the series 5 derivatives (Table ), we modified R1 and R2 simultaneously. A comparison of 5c ~ 5f showed that the mono-benzyl secondary amine at the 5-position is sufficient to elicit potent activity; hence, by taking the synthetic facility into consideration, we fixed this position by nonsubstituted or substituted benzyl secondary amine for this series of derivatives. The SAR of this series indicated that the activity increases with elevation in the hydrophobicity of R2 regardless of R1 (5b vs. 5f, 5j vs. 5k, 5p vs. 5q). However, this increase in the activity was saturated at a certain level in spite of further increase in the hydrophobicity of R1 (5m vs. 5n). The electronic properties of the substituent on the benzyl moiety had no critical effect on the activity (e.g. 5f vs. 5k, 5q vs. 5r). Replacing the pyrazinoyl guanidine skeleton to the amide type resulted in a marked loss in the activity (5m vs. 7a, 5o vs. 7b), indicating the critical role of the guanidine skeleton. This is probably because the positive charge formed at the guanidine moiety (this moiety is protonated at physiological pHs,Citation1) is important for tightly binding to the enzyme. Furthermore, deletion of the chloride atom from the 6-position of 5k resulted in a significant decrease in the activity (IC50 = 1.6 μM), indicating the important role of the 6-chloride group.
Table 2. Inhibitory activities of series 5 and 7 derivatives.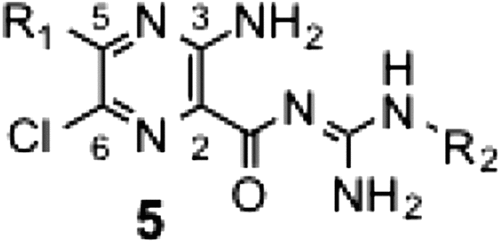
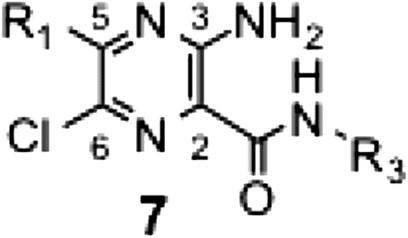
Using potent synthetic amilorides (such as 5m and 5q) as templates, we designed and synthesized two photoreactive amilorides (PRA1 and PRA2, Fig. ). An azido (–N3) group and terminal acetylene (–C≡CH) were incorporated into the two compounds as a photolabile group and “footing” for sequential Cu+1–catalyzed click chemistry, respectively, at opposite ends of the molecules. The use of two amilorides possessing a photolabile azido group in different positions may make characterization of the binding site more reliable. Some molecular tags, such as fluorophore and biotin, can be covalently attached to the terminal acetylene via Cu+1-catalyzed click chemistry following photoaffinity labeling, which enables purification and/or detection of the labeled peptides.Citation18) We confirmed that PRA1 and PRA2 maintain the potent inhibitory activities (Table ), and are the most potent class in this study. Photoaffinity labeling experiments are currently under way in our laboratory using these photoreactive amilorides.
Supporting Information
Fig. S1 (X-ray crystallographic data) and the spectral data of compounds 2c–2k, 3a–3c, 5a–5s, 7a, 7b, PRA1, and PRA2.
Supplemental material
The supplemental material for this paper is available at http://dx.doi.org/10.1080/09168451.2015.1010479.
Conflict of interest
The authors declare that they have no conflict of interest.
Funding
This work was supported by the Grant-in-Aid for Scientific Research [grant 23380064 to H. M.], [grant 26292060 to H. M.]; Grant-in-Aid for Young Scientists [grant 23780116 to M. M.] from the Japan Society for the Promotion of Science.
Supplemental Materials
Download PDF (614.5 KB)Acknowledgments
We thank Dr. Kiyosei Takasu and Dr. Ken-ichi Yamada (Graduate School of Pharmaceutical Sciences, Kyoto University) for the X-ray structural analysis.
References
- Kleyman TR, Cragoe EJ Jr. Amiloride and its analogs as tools in the study of ion transport. J. Membr. Biol. 1988;105:1–21.10.1007/BF01871102
- Putney LK, Denker SP, Barber DL. The changing face of the Na+/H+ exchanger, NHE: structure, regulation, and cellular actions. Annu. Rev. Pharmacol. Toxicol. 2002;42:527–552.10.1146/annurev.pharmtox.42.092001.143801
- Nakamaru-Ogiso E, Seo BB, Yagi T, Matsuno-Yagi A. Amiloride inhibition of the proton-translocating NADH-quinone oxidoreductase of mammals and bacteria. FEBS Lett. 2003;549:43–46.10.1016/S0014-5793(03)00766-X
- Stolpe S, Friedrich T. The Escherichia coli NADH: ubiquinone oxidoreductase (complex I) is a primary proton pump but may be capable of secondary sodium antiport. J. Biol. Chem. 2004;279:18377–18383.10.1074/jbc.M311242200
- Efremov RG, Baradaran R, Sazanov LA. The architecture of respiratory complex I. Nature. 2010;465:441–445.10.1038/nature09066
- Efremov RG, Sazanov LA. Structure of the membrane domain of respiratory complex I. Nature. 2011;476:414–420.10.1038/nature10330
- Baradaran R, Berrisford JM, Minhas GS, Sazanov LA. Crystal structure of the entire respiratory complex I. Nature. 2013;494:443–448.10.1038/nature11871
- Murai M, Mashimo Y, Hirst J, Miyoshi H. Exploring interactions between the 49 kDa and ND1 subunits in mitochondrial NADH-ubiquinone oxidoreductase (complex I) by photoaffinity labeling. Biochemistry. 2011;50:6901–6908.10.1021/bi200883c
- Shiraishi Y, Murai M, Sakiyama N, Ifuku K, Miyoshi H. Fenpyroximate binds to the interface between PSST and 49 kDa subunits in mitochondrial NADH-ubiquinone oxidoreductase. Biochemistry. 2012;51:1953–1963.10.1021/bi300047h
- Cragoe EJ Jr, Woltersdorf OW Jr, Bicking JB, Kwong SF, Jones JH. Pyrazine diuretics II. N-amidino-3-amino-5-substituted 6-halopyrazinecarboxamides. J. Med. Chem. 1967; 10: 66–75.10.1021/jm00313a014
- Matthews H, Ranson M, Tyndall JDA, Kelso MJ. Synthesis and preliminary evaluation of amiloride analogs as inhibitors of the urokinase-type plasminogen activator (uPA). Bioorg. Med. Chem. Lett. 2011;21:6760–6766.10.1016/j.bmcl.2011.09.044
- Hirsh AJ, Molino BF, Zhang J, Astakhova N, Geiss WB, Sargent BJ, Swenson BD, Usyatinsky A, Wyle MJ, Boucher RC, Smith RT, Zamurs A, Johnson MR. Design, synthesis, and structure−activity relationships of novel 2-substituted pyrazinoylguanidine epithelial sodium channel blockers: drugs for cystic fibrosis and chronic bronchitis. J. Med. Chem. 2006;49:4098–4115.10.1021/jm051134w
- Secondo A, Pannaccione A, Molinaro P, Ambrosino P, Lippiello P, Esposito A, Cantile M, Khatri PR, Melisi D, Di Renzo GD, Annunziato L. Molecular pharmacology of the amiloride analog 3-amino-6- chloro-5-[(4-chloro-benzyl)amino]-N-[[(2,4-dimethylbenzyl)-amino]iminomethyl]-pyrazinecarboxamide (CB-DMB) as a pan inhibitor of the Na+-Ca2+ exchanger isoforms NCX1, NCX2, and NCX3 in stably transfected cells. J. Pharmacol. Exp. Ther. 2009;331:212–221.10.1124/jpet.109.152132
- Hunt, T, Atherton-Watson HC, Axford J, Collingwood SP, Coote KJ, Cox B, Czarnecki S, Danahay H, Devereux Na, Howsham C, Hunt P, Paddock V, Paisley D, Young A. Discovery of a novel chemotype of potent human ENaC blockers using a bioisostere approach. Part 1: quaternary amines. Bioorg. Med. Chem. Lett. 2012;22:929–932.10.1016/j.bmcl.2011.12.016
- Matsuno-Yagi A, Hatefi Y. Studies on the mechanism of oxidative phosphorylation. J Biol. Chem. 1985;260:11424–11427.
- Murai M, Ichimaru N, Abe M, Nishioka T, Miyoshi H. Mode of inhibitory action of Δlac-acetogenins, a new class of inhibitors of bovine heart mitochondrial complex I. Biochemistry. 2006;45:9778–9787.10.1021/bi060713f
- Nakanishi S, Abe M, Yamamoto S, Murai M, Miyoshi H. Bis-THF motif of acetogenin binds to the third matrix-side loop of ND1 subunit in mitochondrial NADH-ubiquinone oxidoreductase. Biochim. Biophys. Acta. 2011;1807:1170–1176.10.1016/j.bbabio.2011.05.012
- Masuya T, Murai M, Ifuku K, Morisaka H, Miyoshi H. Site-specific chemical labeling of mitochondrial respiratory complex I through ligand-directed tosylate chemistry. Biochemistry. 2014;53:2307–2317.10.1021/bi500205x