
Abstract
For the production of enantiopure β-amino acids, enantioselective resolution of N-acyl β-amino acids using acylases, especially those recognizing N-acetyl-β-amino acids, is one of the most attractive methods. Burkholderia sp. AJ110349 had been reported to exhibit either (R)- or (S)-enantiomer selective N-acetyl-β-Phe amidohydrolyzing activity, and in this study, both (R)- and (S)-enantioselective N-acetyl-β-Phe acylases were purified to be electrophoretically pure and determined the sequences, respectively. They were quite different in terms of enantioselectivities and in their amino acids sequences and molecular weights. Although both the purified acylases were confirmed to catalyze N-acetyl hydrolyzing activities, neither of them show sequence similarities to the N-acetyl-α-amino acid acylases reported thus far. Both (R)- and (S)-enantioselective N-acetyl-β-Phe acylase were expressed in Escherichia coli. Using these recombinant strains, enantiomerically pure (R)-β-Phe (>99% ee) and (S)-β-Phe (>99% ee) were obtained from the racemic substrate.
Graphical abstract
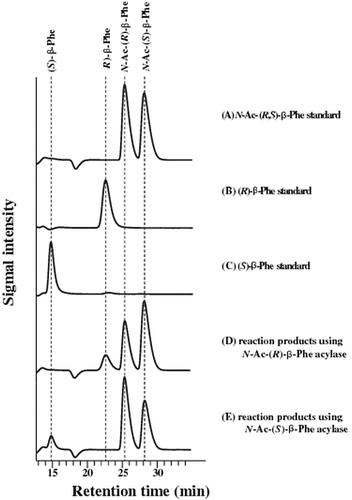
Enantioselectivity of purified acylases. N-Ac-(R,S)-β-Phe was incubated with either purified N-Ac-(R)- or (S)-β-Phe acylase. The reaction products were analyzed by HPLC.
Enantiopure β-amino acids reportedly possess biological properties that make them useful as the building blocks of pharmaceutically important compounds. Therefore, the synthetic methods of enantiopure β-amino acids are widely developed.Citation1,2)
Various methods for synthesizing β-amino acids have been developed, but enzymatic methods seem to be superior to chemical methods from the viewpoint of enantioselectivities, which are easily achieved by way of the enzymes’ properties. Numerous enzymatic methodologies have been devised and extensively reviewed.Citation3)
Among the enzymatic methods, enantioselective resolution using enzymes has some specific merits, i.e. easy preparation of the starting material, from racemic β-amino acids, high enantioselectivity, high conversion yield, generally reported in several examples. In particular, the enantioselective resolution of N-phenylacetyl-β-amino acid by Penicillin G amidase has been reported to be highly useful.Citation4–8) With this method, however, bulky and expensive acylation reagents are required, if the acids are to be recognized by known N-acyl-β-amino acid acylases. One of the most cost-effective acylation methods is acetylation, and enzymatic resolution of N-acetyl-β-amino acids was successful in the industrial production of β-amino acids.Citation9,10) So enantioselective acylases recognizing N-acetyl-β-amino acids are attractive from a cost viewpoint.
In our previous report,Citation11) we newly discovered two microorganisms capable of enantioselectively amidohydrolyzing (R,S)-N-acetyl-3-amino-3-phenylpropionic acid [(R,S)-N-acetyl β-Phe] and identified them to be Burkholderia sp. AJ110349 and Variovorax sp. AJ110348, respectively. β-Phe is one of the most valuable β-amino acids; it is a useful ingredient for the synthesis of pharmaceuticals such as paclitaxel, a complex diterpene isolated from the bark of Taxus brevifolia that possesses strong anticancer properties.Citation12) The enzyme activities observed in these microorganisms were confirmed to recognize the N-acetyl group as their substrate; the enzymes catalyzing these activities are therefore considered useful, for the aforementioned cost reasons.
One microorganism we discovered, Variovorax sp. AJ110348, was shown to possess (R)-enantioselective N-acetyl-β-Phe acylase. The other microorganism, Burkholderia sp. AJ110349, was demonstrated to possess two different enantioselective N-acetyl-β-Phe acylases. The presence of these two kinds of acylases was supported by the different optimal reaction temperatures of these two enantioselective activities. Therefore, the latter microorganism, Burkholderia sp. AJ110349, is considered an attractive enzyme source of both (R)- and (S)-enantioselective acylases. However, it is difficult to control strictly the enantioselective production of β-amino acids by wild-type Burkholderia sp. AJ110349—which co-produces two different enantioselective enzymes—even though their optimal reaction temperatures differ. In addition, the enzyme activity needs to be strengthened, if it is to be used in efficient conversion in industrial production.
To solve these problems, we sought here to purify, clone, and characterize both N-acetyl-(R)- and (S)-β-Phe acylases from Burkholderia sp. AJ110349. In addition, both (R)- and (S)-enantioselective N-acetyl-β-Phe acylases were expressed in Escherichia coli. Using these recombinant strains in the resting cell reaction, enantiomerically pure (R)- and (S)-β-Phe were obtained from the racemic substrate.
Materials and methods
Materials
(R,S)-3-amino-3-phenylpropionic acid[(R,S)-β-Phe], dl-β-leucine, dl-β-homophenylalanine, l-β-leucine hydrochloride, and l-β-homophenylalanine hydrochloride were purchased from Sigma-Aldrich (St. Louis, MO, USA). (R)-3-amino-3-phenylpropionic acid and (S)-3-amino-3-phenylpropionic acid were purchased from Watanabe Chemical Industries (Hiroshima, Japan). (R,S)-3-amino-5-methyl-hexanoic acid (dl-β-homoleucine) and (R)-3-amino-5-methyl-hexanoic acid (d-β-homoleucine) were purchased from AstaTech (Bristol, PA, USA). (R,S)-3-amino-3-(4-hydroxyphenyl)-propanoic acid (dl-β-tyrosine) and (R,S)-3-amino-3-(4-fluorophenyl)-propanoic acid (dl-4-fluoro-β-phenylalanine) were purchased from Bionet (Cornwall, UK). β-valine was purchased from Apollo Scientific (Cheshire, UK). (R)-3-amino-3-(4-hydroxyphenyl)-propanoic acid (l-β-tyrosine) and (R)-3-amino-3-(4-fluorophenyl)-propanoic acid (l-4-fluoro-β-phenylalanine) were purchased from PepTech (Bedford, MA, USA).
N-acetyl-β-Ala was purchased from Watanabe Chemical Industries. N-acetyl-dl-α-phenylalanine was purchased from Bachem AG (Bubendorf, Switzerland). N-acetyl-(R,S)-β-Phe, N-acetyl-dl-4-fluoro-β-phenylalanine, N-acetyl-dl-β-tyrosine, N-acetyl-dl-β-homoleucine, N-acetyl-dl-β-homophenylalanine, N-acetyl-dl-β-leucine, and N-acetyl-β-valine were acetylated with acetic anhydride from each corresponding racemic amino acid, as previously described.Citation13)
Bacterial strain, culture media, and plasmid
The isolation and characterization of Burkholderia sp. AJ110349 has been reported previously.Citation11) For the purification of acylases, AJ110349 was grown in enzyme-producing medium comprising 10 g/L d-glucose, 10 g/L (NH4)2SO4, 10 g/L (R,S)-β-Phe, 2 g/L casamino acid, 1 g/L KH2PO4, 0.4 g/L MgSO4·7H2O, 1 g/L NaCl, 19.5 g/L 2-(N-morpholino)ethanesulfonic acid (MES), 5 mg/L nicotinamide, 0.2 mg/L thiamin, 10 mg/L FeSO4·7H2O, and 10 mg/L MnSO4·4–5H2O. Cultivation was carried out with shaking at 120 rpm in a 500-mL Sakaguchi flask, for 66 h at 30 °C.
E. coli JM109 was selected as the host strain for the recombinant DNA studies. For the cloning experiments, E. coli strains were routinely cultured at 37 °C in LB medium supplemented with ampicillin (100 µg/mL). For the purification and expression experiments, E. coli JM109 was cultured at 37 °C in LB medium or terrific broth (TB) medium composed of 4 g/L glycerol, 12 g/L peptone, 24 g/L yeast extract, 2.3 g/L KH2PO4, and 12.5 g/L K2HPO4 with 100 µg/mL ampicillin.
pUC118 (TaKaRa Bio Inc., Shiga, Japan) was used for most of the cloning experiments. The plasmid ptrp4,Citation14) an expression plasmid that contain the tryptophan promoter and the rrnB terminator of E. coli, was used for the expression of (R)-enantioselective N-acetyl-β-Phe acylase. The plasmid pSFN_Sm_Aet,Citation15) an expression plasmid that contain the mutated acid-phosphatase promoter of Enterobactor aerogenes, was used for the expression of (S)-enantioselective N-acetyl-β-Phe acylase.
Preparation of cell-free extract
For enzyme purification, the cells of Burkholderia sp. AJ110349 were collected through a 6800 g, 10-min centrifugation operation, from 2000 mL of culture broth. Since precipitation of the bacterial cells was not adequately observed after centrifugation, about 1600 mL of supernatant was removed and the remainder was rendered uniform by pipetting. The concentrated culture broth thus prepared, without being washed with a buffer, and was then disrupted by ultrasonication for 20 min at 200 W. The sonicate was centrifuged at 200,000 g for 30 min, and the obtained supernatant was used as a cell-free extract.
The extract from recombinant E. coli JM109/pSFN_BS was similarly prepared. The cells obtained from 250 mL of culture broth were collected via 10 min of 8000 g centrifugation and re-suspended of the buffer containing Tris–HCl (pH 7.6). The cell suspension was ultrasonicated; the supernatant obtained by centrifugation at 15,000 g for 10 min was used as a cell-free extract.
Enzyme purification from Burkhordelia sp AJ110349
Protein purification was conducted at temperatures <4 °C. The cell-free extract obtained above was a resultant supernatant applied to ammonium sulfate fractionation. (NH4)2SO4 was added to the cell-free extract, to a final concentration of 40% saturation. The mixture was stirred on ice for 1 h and centrifuged for 15 min at 9200 g. The precipitate obtained was dissolved in a small quantity of 25 mM Tris–HCl (pH 7.6) and dialyzed against 25 mM Tris–HCl (pH 7.6). After dialysis, the protein solution was used as a sample in chromatography, as further described below.
| (1) | Phenyl Sepharose 26/10 (GE Healthcare UK Ltd., Buckinghamshire, England). | ||||
The ammonium sulfate fractions obtained as described above were dialyzed against a buffer solution comprising 25 mM Tris–HCl (pH 7.6) and 0.6 M (NH4)2SO4; they were then placed on Phenyl Sepharose 26/10 that had been equilibrated with the same buffer solution. Following non-adsorptive protein elution, the adsorptive protein was eluted by linearly varying the (NH4)2SO4 concentration in the buffer solution, from 0.6 to 0 M. This operation resulted in the N-acetyl-(R)-β-Phe acylase activity being detected when the (NH4)2SO4 concentration was about 0.2 M, and the N-acetyl-(S)-β-Phe acylase activity being detected when the (NH4)2SO4 concentration was about 0.1 M. The fractions exhibiting activity were divided into N-acetyl-(R)-β-Phe acylase activity elution fractions and N-acetyl-(S)-β-Phe acylase activity elution fractions, and they were subsequently recovered. About all purification process, each fraction of their high stereoselectivity was also recovered separately and purified, respectively.
| (2) | Q-Sepharose 16/10 (GE Healthcare UK Ltd.). | ||||
The Phenyl Sepharose fractions obtained were concentrated and dialyzed against 25 mM Tris–HCl (pH 7.6); they were then applied to Q-Sepharose 16/10 equilibrated with the same buffer solution. Following non-adsorptive protein elution, the adsorptive protein was eluted by linearly varying the NaCl concentration in the buffer solution, from 0 to 0.5 M. This operation resulted in N-acetyl-(R)-β-Phe acylase activity being detected when the NaCl concentration was about 0.22 M, and the N-acetyl-(S)-β-Phe acylase activity being detected when the NaCl concentration was about 0.3 M. The fractions exhibiting activity were collected.
| (3) | Superdex 200 16/60 (GE Healthcare UK Ltd.). | ||||
The Q-Sepharose fractions obtained were concentrated and placed on a Superdex 200 16/60 equilibrated with 25 mM Tris–HCl (pH 7.6). This operation resulted in the detection of N-acetyl-(R)-β-Phe acylase activity at an elution position estimated to correspond to a molecular weight of 206 kDa; the detection of N-acetyl-(S)-β-Phe acylase activity was at an elution position estimated to correspond to a molecular weight of 101 kDa. Estimations of the molecular weights of the enzymes were calculated from the elution positions of standard proteins (Bio-Rad Laboratories, Carlsbad, CA, USA) containing thyroglobulin, 670 kDa; IgG, 158 kDa; ovalbumin, 44 kDa; myoglobin, 17 kDa; and vitamin Β-12, 1.35 kDa. The fractions exhibiting activity were recovered.
| (4) | Resource phenyl (GE Healthcare UK Ltd.). | ||||
The Superdex fractions obtained were concentrated, dialyzed against a buffer solution comprising 25 mM Tris–HCl (pH 7.6) and 0.6 M (NH4)2SO4, and placed on Resource phenyl equilibrated with the same buffer solution. Following non-adsorptive protein elution, the adsorptive protein was eluted by linearly varying the (NH4)2SO4 concentration in the buffer solution, from 0.6 to 0 M. This operation resulted in N-acetyl-(R)-β-Phe acylase activity being detected when the (NH4)2SO4 concentration was about 0.35 M, and N-acetyl-(S)-β-Phe acylase activity being detected when the (NH4)2SO4 concentration was about 0.45 M. The fractions exhibiting activity were recovered.
| (5) | Mono Q 5/5 (GE Healthcare UK Ltd.). | ||||
The Resource phenyl fractions obtained were concentrated, dialyzed against 25 mM Tris–HCl (pH 7.6), and placed on Mono Q 5/5 equilibrated with the same buffer solution. Following non-adsorptive protein elution, the adsorptive protein was eluted by linearly varying the NaCl concentration in the buffer solution, from 0 to 0.5 M. This operation resulted in the N-acetyl-(R)-β-Phe acylase activity being detected when the NaCl concentration was about 0.2 M, and the N-acetyl-(S)-β-Phe acylase activity being detected when the NaCl concentration was about 0.28 M. The fractions exhibiting activity were recovered and adopted as purified enzyme solutions of N-acetyl-(R)-β-Phe acylase and N-acetyl-(S)-β-Phe acylase, respectively.
N-terminal amino acid sequence analysis
The purified enzymes were applied to SDS-PAGE and transferred to a polyvinylidene difluoride membrane (Trans-Blot; Bio-Rad Laboratories). The proteins were stained with Coomassie blue; the positions at which bands appeared were cut and placed on a protein sequencer (Model 476A; Applied Biosystems, Carlsbad, CA, USA).
Gene-cloning and nucleotide sequencing
Chromosomal DNA was prepared from Burkholderia sp. AJ110349, using a Genomic-Tip and Genomic Buffer set (QIAGEN, Hilden, Germany). The N-acetyl-(R)-β-Phe acylase gene and N-acetyl-(S)-β-Phe acylase gene were partially cloned using an LA PCR in vitro cloning kit (TaKaRa Bio Inc.), as described in the manufacturer’s manual. The principle of the kit is as follows: (1) Target DNA is completely digested by an appropriate restriction enzyme, (2) Ligate the cassette (double-strand synthetic oligonucleotide with the restriction site at one end) with the restriction site, (3) Perform the first PCR using cassette primer C1 and primer S1 of the known region, and (4) Perform the second PCR using primers designed for inner sequences, cassette primer C2 and primer S2 of the known region. The primers for N-acetyl-(R)-β-Phe acylase gene-cloning were 5′-AGNACRTCSSWRTANCCNGT-3′ (first PCR) and 5′-RTANCCNGTDATNGTDATCAT-3′ (second PCR). The primers for N-acetyl-(S)-β-Phe acylase gene-cloning were 5′-CGNAARGGNCGNATHCARAC-3′, 5′-ATSARNCCSAGNACNGTYTG-3′ (first PCR) and 5′-CARACNGTNCTSGGNYTSAT-3′, 5′-GTYTGDATNCGNCCYTTNCG-3′ (second PCR) These primers were designed from the N-terminal amino acid sequence of each protein determined in this study. The second PCR products were sequenced with an ABI 3100 (Applied Biosystems) and labeled with a DIG Nucleotide Detection kit (Roche Diagnostics, Basel, Switzerland). The DIG labeling DNA was then used as a probe for Southern hybridization. Southern hybridization and colony hybridization were performed on positively charged nylon membranes (Roche Diagnostics). For southern hybridization, the genomic DNA from Burkholderia sp. AJ110349 was restricted by the use of some kinds of restriction enzymes. The digested DNA was ligated into pUC118. The ligation products were used to transform E. coli JM109 for the colony hybridization.
Sequence analysis was done with the ABI PRISM SeqSpace software package, version 2.1. (Applied Biosystems) The database searches and comparisons were performed using Scifinder2007 (American Chemical Society, Washington, DC, USA).
Expression of acylase genes in E. coli
For the expression of N-acetyl-(R)-β-Phe acylase, PCR was performed using the chromosomal DNA of Burkholderia sp. AJ110349 as the template and the primers R_7F 5′-CGAGGATCCGCAGCAGGTTCAGGTCGATATC-3′ and R_R_HindIII 5′-CCCAAGCTTTCAGTCGACCTCGGTGTGAG-3′. The 2.3-kb amplified fragment was treated with BamH I and HindIII and inserted into the BamH I/HindIII site of ptrp4 to create ptrp4_3BR. E. coli JM109 was transformed with this plasmid, and the resultant transformant was named JM109/ptrp4_3BR.
For the expression of N-acetyl-(S)-β-Phe acylase, PCR was performed using the chromosomal DNA of Burkholderia sp. AJ110349 as the template and the primers S_F_NdeI 5′-AACGACCCCATATGGGTTTCTGCCAGATGAA-3′ and S_R_HindIII 5′-CCCAAGCTTCGCTAGACGAAAGTCAGAAG-3′. The 1.1-kb amplified fragment was treated with NdeI/HindIII. Separately, pSFN_Sm_Aet was cleaved with NdeI/HindIII, and approximately, 3 kb of DNA was excised and purified. A 1.1-kb PCR product that had been treated with restriction nucleases was inserted at the NdeI/HindIII sites of pSFN to create pSFN_BS. E. coli JM109 was transformed with this plasmid, and the resultant transformant was named JM109/pSFN_BS.
Recombinant N-acetyl-(S)-β-Phe acylase purification
Recombinant N-acetyl-(S)-β-Phe acylase was purified using the same methods as with the Burkhordelia sp AJ110349. The enzyme was purified from a crude extract of E. coli JM109 pSFN_BS, using Phenyl Sepharose 26/10, Q-Sepharose 16/10, Superdex 200 16/60, and Resource phenyl column.
Protein assay
Protein concentrations were measured by the Bradford method, with bovine serum albumin as a standard.
SDS-PAGE
SDS-PAGE was performed on a 10–20% polyacrylamide gel (Daiichi Pure Chemicals, Tokyo, Japan) with a protein marker (Precision Protein Marker; Bio-Rad Laboratories).
Enzyme assay
About the purification of enzyme, the reaction solution—containing 10 mM N-acetyl-(R,S)-β-Phe, 50 mM Tris–HCl (pH 7.6), and a suitable amount of enzyme—was left standing from 10 min to 2 h at 30 °C; it was then processed by boiling for 5 min or by adding 1% phosphoric acid, to terminate the reaction. The reaction solution was then centrifuged, and the supernatant was analyzed by HPLC. For the analysis of specific activity, a column Inertsil Ph-3 (0.46 × 25 cm; GL Science) was adopted. The other conditions were as below: mobile phase 10% (v/v) acetonitrile and 90% (v/v) water (pH 3.0, adjusted by phosphoric acid), flow rate 1.0 mL/min, column temperature 40 °C, and UV detection at 210 nm.
Acylase activity equivalent to 1 µmol of (R)-β-Phe or (S)-β-Phe produced per min was defined as 1 unit (U).
Characterization of amidohydrolyzing activity
Unless otherwise noted, the experiments were carried out as following standard assay condition. The substrate was 5 mM N-acetyl-racemic-β-Phe in 50 mM Tris–HCl buffer. For characterization of N-acetyl-(R)-β-Phe acylase, the reaction was carried out at 50 °C, pH 7.6, with 25 mU/mL purified enzyme. For characterization of the N-acetyl-(S)-β-Phe acylase, the reaction was carried out at 37 °C, pH 7.6, with 25 mU/mL purified recombinant enzyme.
Kinetic assays were performed with each enantiopure substrate. N-acetyl-(R)-β-Phe (0.019–0.93 mM) was used for N-acetyl-(R)-β-Phe acylase and N-acetyl-(S)-β-Phe (0.019–0.93 mM) was used for N-acetyl-(S)-β-Phe acylase. The Km values and Vmax values were determined from secondary Lineweaver–Burk plots.
The optimum pH was determined by measuring the activity in buffers at various pH values. The buffers used were 100 mM acetate (pH 3.5–5.5), 100 mM MES (pH 5.5–7.0), Tris–HCl (pH 7.0–9.0), and borate (pH 9.0–11.0). Temperature dependence was determined at a temperature range of 25–70 °C. Temperature stability was determined after an incubation of 30 min at temperature range of 30–70 °C. The effects of various compounds (CaCl2, CoCl2, CuCl2, FeSO4, FeCl3, MgSO4, MnCl2, ZnCl2, and EDTA) on enzyme activity were evaluated. After pre-incubation for 60 min (pH 7.6, 4 °C) with 10 mM of each compound listed above, the enzyme activities were determined under standard assay conditions.
To determine the substrate specificities, various N-acetyl-amino acids were used as the substrates, and enzyme reactions were carried out using 12.5 mU/mL purified enzyme under the above aforementioned standard assay conditions. The enzyme activities were determined by the amount of acetic acid released, which was measured using an acetic assay kit (Roche Diagnostics) following the instructions provided in the manual. The enantioselectivities were confirmed beforehand by chiral analysis using HPLC.
Enantioselectivity of purified native enzyme
Enantioselectivity was measured using the purified native enzymes as enzyme sources. Reactions were conducted under the aforementioned assay conditions for a period of 30 min, and purified enzymes were added in the following volumes: 4.6 μg/mL (N-acetyl-(R)-β-Phe acylase) and 3.1 μg/mL (N-acetyl-(S)-β-Phe acylase).
Chiral analysis
For chiral analysis of N-acetyl-(R)-β-Phe, N-acetyl-(S)-β-Phe, (R)-β-Phe, and (S)-β-Phe, a Chiralpak WH column (0.46 × 25 cm; Daicel Chemical Industries, Osaka, Japan) was utilized. Additionally, to improve the analytical resolution, another column (Inertsil ODS3, 0.46 × 5 cm; GL Science, Tokyo, Japan) was tandem-added to the flow line prior to the Chiralpak WH. The other conditions were as below: mobile phase 0.25 mM CuSO4 and 2% (v/v) acetonitrile, flow rate 1.5 mL/min, column temperature 50 °C, and UV detection at 210 nm. For the analysis of the resolution of dl-β-Tyr and dl-β-4-fluoro-Phe resolution, the same 2 columns were adapted. The other conditions were as below: 1 mM CuSO4 and 10% (v/v) methanol, 1.0 mL/min, 50 °C, and UV detection at 210 nm. For chiral analysis of dl-β-homoLeu, an Astec Chirobiotic T column (0.46 × 25 cm, Sigma-Aldrich) was adopted. The other conditions were as below: 90% (v/v) methanol, 0.4 mL/min, 40 °C, and UV 205 nm.Citation16) Under these conditions, optical selectivity was determined by comparison with standard rac-β-amino acids and d- or l-β-amino acids.
Production of enantioselective β-Phe using whole-cell biocatalyst
The reactions using E.coli were performed as follows. E. coli JM109/ptrp4_3BR and E. coli JM109/pSFN_BS cells were cultured in TB medium with ampicillin (100 µg/mL) at 37 °C for 16 h. For reactions with whole cells, after washing the cells with 5 mL of each culture, the mixtures (final volume 2.5 mL) contained 242 mM N-acetyl-(R,S)-β-Phe and 100 mM Tris–HCl (pH 7.6). The mixtures were incubated at 37 °C for 24 h. The consumption of substrates and the production of β-Phe were analyzed by HPLC. The final products were analyzed by chiral analysis using HPLC.
Results
Purification, molecular mass, and subunit structure of the enantioselective acylase
As a result of the first chromatography procedure using Phenyl Sepharose, N-acetyl-β-Phe amidohydrolyzing activity was detected in some of the fractions, shaping two peaks (Fig. ). The first peak mainly consisted of (R)-enantioselective activity, while the second peak mainly consisted of (S)-enantioselective activity, indicating that either (R)- or (S)-enantioselective N-acetyl-β-Phe amidohydrolyzing enzyme had independently occurred in Burkholderia sp. AJ 110349. The specific activities of both (R)- and (S)-enantioselective N-acetyl-β-Phe acylase were increased through the use of the following purification procedures using Q-Sepharose and Superdex; the enantioselectivities of both enzymes were also increased by more than 95% ee (data not shown). However, the purity of both the resulting proteins, as assessed by SDS-PAGE, remained low. Because of the difficulties encountered in the aforementioned purification procedure, an additional two chromatographic purification steps using Resource phenyl and Mono Q were adopted; as a result of these steps, either of the acylases consequently generated a single band on SDS-PAGE (Fig. ).
Fig. 1. Separation of N-Ac-(R)- and (S)-β-Phe acylase activities using a Phenyl Sepharose column.
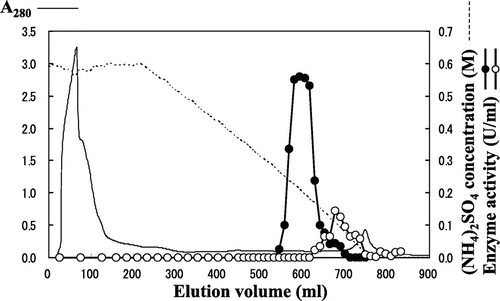
Fig. 2. SDS-PAGE analysis of the purified (A) N-Ac-(R)-β-Phe acylase and (B) N-Ac-(S)-β-Phe acylase.
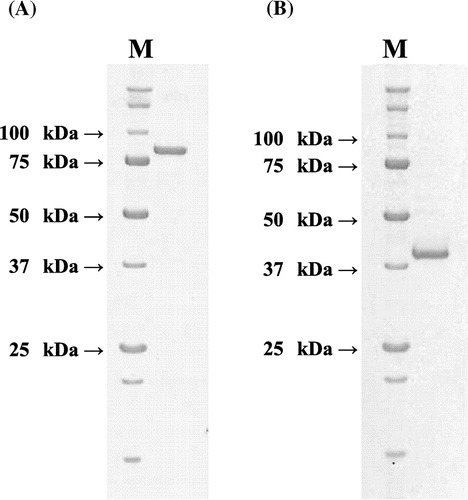
By SDS-PAGE analysis, the purified N-acetyl-(R)-β-Phe acylase was found to have a molecular mass of ~81 kDa, while that of the N-acetyl-(S)-β-Phe acylase was ~39 kDa. With gel-filtration chromatography (data not shown), both the enzyme activities were eluted at positions corresponding to the estimated molecular masses of 206 kDa (N-acetyl-(R)-β-Phe acylase) and 101 kDa (N-acetyl-(S)-β-Phe acylase), so both the enzymes were thought to have homodimer or homotrimer structures.
After all purification steps had been executed, the specific activity of the purified N-acetyl-(R)-β-Phe acylase was found to have increased to 12 U/mg—a 262-fold increase compared to the extract. The N-acetyl-(S)-β-Phe acylase had been purified about 809-fold, providing a specific activity of 13 U/mg. The overall purification results are summarized in Table .
Table 1. Purification of N-Ac-(R)-β-Phe acylase and N-Ac-(S)-β-Phe acylase.
Enantioselectivity of the purified acylase from Burkholderia sp. AJ110349
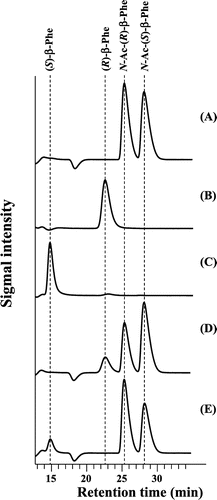
The enantioselectivities of the purified acylases were determined. As a result, only the production of (R)-β-Phe was observed when N-acetyl-(R)-β-Phe acylase was used as an enzyme source, and only the production of (S)-β-Phe was observed when N-acetyl-(S)-β-Phe acylase was so used (Fig. ). Judging from the products after 30 min of reaction, the ee value was estimated to be >95%, using either purified enzyme.
Fig. 3. Enantioselectivity of purified acylases.
Determination of the N-terminal amino acid sequences
The N-terminal amino acid sequence of the purified enzyme was determined by automated Edman degradation. As a result, the 20 residues of the N-terminal amino acid sequence of N-acetyl-(R)-β-Phe acylase were determined to be MITITGYSDVLSAGPGETVE, and the 26 residues of N-acetyl-(S)-β-Phe acylase were determined to be NDLASRKGRIQTVLGLIDPHELGPAL. As the sequence of N-acetyl-(S)-β-Phe acylase was initiated from Asn and not from Met, the occurrence of some sort of post-translational modification is suspected.
Neither sequence showed any significant sequence similarity to other acylase sequences reported thus far.
Cloning and sequencing of acylase genes
Based on the N-terminal amino acid sequences of the purified proteins, a 8.0-kb fragment containing the gene encoding N-acetyl-(R)-β-Phe acylase and a 6.1-kb fragment containing the gene encoding N-acetyl-(S)-β-Phe acylase were cloned and sequenced.
N-acetyl-(R)-β-Phe acylase, as the deduced protein of the cloned gene (Genbank accession number FB701603), consisted of 760 amino acid residues (Genbank accession number CAS03316). The initial 20 amino acid residues of the protein completely coincided with those determined from the purified authentic N-acetyl-(R)-β-Phe acylase. The molecular mass of the N-acetyl-(R)-β-Phe acylase deduced from the sequence was calculated to be 81 kDa. As a result of a homology search using the overall amino acid sequence of N-acetyl-(R)-β-Phe acylase, a sequence similarity (i.e., 32% identical) was observed with N,N-dimethylformamidase from Alcaligenes sp. KUFA-1 (Genbank accession number BAA90664).Citation17)
On the other hand, N-acetyl-(S)-β-Phe acylase, as the deduced protein of the cloned gene(Genbank accession number FB701605), consisted of 352 amino acid residues(Genbank accession number CAS03317). The N-terminal amino acid sequence of the deduced protein was initiated from MDFCQMNDLASRKGRIQTVLGLIDPHELGPAL, and the N-terminal amino acid sequence determined from the authentic enzyme was observed in the region from seventh residues. The molecular mass of the deduced protein assumed to be initiated from authentic N-terminal was calculated to be 39 kDa. The protein found to be most similar to N-acetyl-(S)-β-Phe acylase, as found with a BLAST search, was a phosphotriesterase-related protein from Homo sapiens (Genbank accession number AAH03793),Citation18) with a 40% sequence identity.
Some kinds of putative open reading frames (ORFs) were found in both the cloned fragments. Around the gene encoding N-acetyl-(R)-β-Phe acylase, there existed the genes encoding transcriptional regulators (ORF 1 and 2), amino transferase (ORF 4), and major facilitator superfamily metabolite (ORF 5). On the other hand, the genes encoding aldehyde dehydrogenase (ORF 6), histidinol-phosphate aminotransferase (ORF 7), and transcriptional regulator (ORF 9) were observed around the gene encoding N-acetyl-(S)-β-Phe acylase. The structures of the genes are illustrated in Fig. , and their properties are summarized in Table .
Fig. 4. Genetic organizations around the gene encoding (A) N-Ac-(R)-β-Phe acylase and (B) N-Ac-(S)-β-Phe acylase.
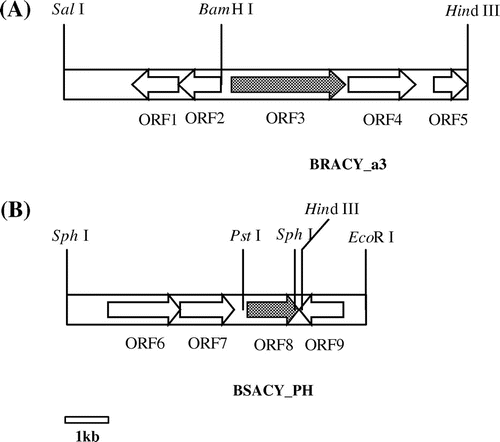
Table 2. Location and properties of sequenced genes and proteins predicted to be encoded by those genes.
The gene expression plasmid ptrp4_3BR having N-acetyl-(R)-β-Phe acylase gene and pSFN_BS having N-acetyl-(S)-β-Phe acylase gene were introduced into E. coli. A level of enzyme activity of 0.094 and 0.88 U/mg was detected in a cell extract prepared from the cell cultured in TB medium with ampicillin (100 µg/mL). No activity was detected in E. coli JM109 cells.
Characterization of N-acetyl-(R)-β-Phe acylase and N-acetyl-(S)-β-Phe acylase
It was not sufficient to characterize the expression level of N-acetyl-(R)-β-Phe acylase using E. coli JM109/ptrp4_3BR. Therefore, we used N-acetyl-(R)-β-Phe acylase from the wild-type strain. As the volume of purified N-acetyl-(S)-β-Phe acylase from the wild-type strain was insufficient, N-acetyl-(S)-β-Phe acylase from the recombinant strain was used for the characterization. After the purification was complete, the specific activity of the purified recombinant N-acetyl-(S)-β-Phe acylase was found to have increased to 13 U/mg, was 70-fold higher than that in the extract of the recombinant E. coli, and was as high as that obtained from wild-type enzyme (13 U/mg, Table ).
With purified N-acetyl-(R)-β-Phe acylase, the Km and Vmax values for N-acetyl-(R)-β-Phe were 0.35 mM and 245 U/mg (pH 7.6, 50 °C). With purified recombinant N-acetyl-(S)-β-Phe acylase, the Km and Vmax values for N-acetyl-(S)-β-Phe were 0.14 mM and 14 U/mg (pH 7.6, 37 °C). The optimal amidohydrolyzing activities of N-acetyl-(R)-β-Phe acylase and recombinant N-acetyl-(S)-β-Phe acylase were observed at pH 8.0, 60 °C, and pH 7.6–8.5, 50 °C, respectively (Fig. ). These enzymes showed no loss of activity after treatment at 50 and 40 °C for 30 min at pH 7.6. For N-acetyl-(R)-β-Phe acylase activity, Cu2+ and Zn2+ addition caused an inhibition of 61 and 6.0%, respectively. In contrast, Fe2+ and Fe3+ addition caused an activation of 187 and 169%, respectively. Similarly, for N-acetyl-(S)-β-Phe acylase activity, Fe3+ and Cu2+ addition caused an inhibition of 63 and 39%, respectively, while Zn2+ addition caused an activation of 139% (Table ). N-acetyl-(R)-β-Phe acylase showed relative activities on N-acetyl-(R,S)-β-Phe, N-acetyl-dl-β-4-fluoro-phenylalanine, N-acetyl-dl-β-tyrosine, and N-acetyl-dl-β-homoleucine were 100, 52, 36, and 11% with N-acetyl-(R,S)-β-Phe (N-acetyl-dl-β-Phe). By chiral analysis of HPLC, it was confirmed that each l-form was used preferentially. N-acetyl-(S)-β-Phe acylase showed relative activities on N-acetyl-(R,S)-β-Phe, N-acetyl-dl-β-4-fluoro-phenylalanine, N-acetyl-dl-β-tyrosine, and N-acetyl-dl-β-homoleucine. Each relative activity was 100, 185, 76, and 4% with N-acetyl-(R,S)-β-Phe (N-acetyl-dl-β-Phe). By chiral analysis, each d-form was confirmed (Table ). It was confirmed that N-acetyl-(R)-β-Phe acylase amidohydrolyzed N-acetyl-l-β-amino acid and N-acetyl-(S)-β-Phe acylase amidohydrolyzed N-acetyl-d-β-amino acid, and both enzymes were identical in the d,l enantiospecificity for N-acetyl-β-Phe. In the R,S notation of β-amino acids, it is determined by the order of –NH3, –CCOOH, –R (side chain), –H. In the case of β-amino acids, it is change of the order of –CCOOH and –R by the amino acid. d,l notation is fixed the position of functional group (–NH3, –CCOOH). So it is suitable of the notation of the enantioselectivity of β-amino acid.
Fig. 5. Optimum pH (A) and temperature (B) of N-Ac-(R)-β-Phe acylase and N-Ac-(S)-β-Phe acylase.
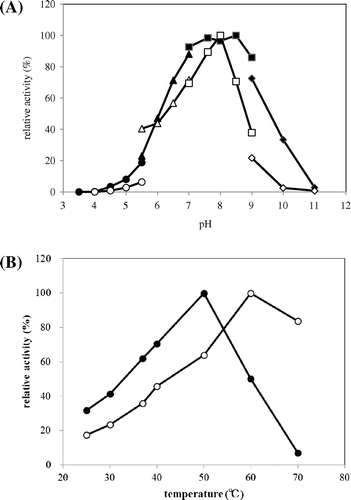
Table 3. Influences of different metal ions on acylase activity.
Table 4. Substrate specificities of the purified acylase (12.5 mU/mL).
Production of enantioselective β-Phe using the whole-cell biocatalyst
Using JM109/ptrp4_3BR, 242 mM N-acetyl-racemic-β-Phe was converted to (R)-β-Phe with a 25% yield (58 mM) and 99% ee (enantiomer excess) for 24 h. Using JM109/pSFN_BS, N-acetyl-racemic-β-Phe was converted to (S)-β-Phe with a 45% yield (108 mM) and 99% ee for 24 h.
Discussion
In our previous report,Citation11) we discovered two microorganisms that enantioselectively amidohydrolyze N-acetyl-(R,S)-β-Phe. In one of the microorganisms, Burkholderia sp. AJ110349, we found either enantioselective N-acetyl-(R)- or (S)-β-Phe acylase activity. In aiming to achieve effective β-amino acid production, it seemed necessary to clone the genes encoding the N-acetyl-(R) and (S)-β-Phe acylases. For this purpose, we sought to purify the authentic enzymes from wild-type Burkholderia sp. AJ110349 and consequently obtained electrophoretically pure N-acetyl-(R)- and (S)-β-Phe acylases (Fig. ). The separation of both the acylases from one another proceeded well, for example, on a Phenyl Sepharose column (as described in Fig. ), but the achievement of electrophoretical purity required five kinds of chromatography (Table ). Difficulties in achieving this purity were thought to be rooted in the contamination of the culture broth elements to the crude extract. The contamination was inevitable, due to the poor sedimentation performance of the cells of Burkholderia sp. AJ110349; multiple washings of the cells had to be abandoned, to secure a sufficient amount of cells for study.
Although N-acetyl-(R)- and (S)-β-Phe acylase catalyzed similar reactions, the obtained properties of both the enzymes were all quite different. The elution positions of the enzymes on hydrophobic interaction chromatographies (Phenyl Sepharose and Resource phenyl), anion-exchange chromatographies (Q-Sepharose and Mono Q), and gel filtration were all different, indicating their differences in terms of hydrophobicity, electrostaticity, and native molecular masses. The differences were confirmed by sequence analysis. The amino acid sequences of both the acylases were so different, they could not be aligned at all.
N-acetyl-(R)-β-Phe acylase showed sequence homology with a N,N-dimethylformamidase large subunit from Alcaligenes sp. KUFA-1.Citation17) In the strain, N,N-dimethylformamidase reportedly required not only a large subunit but also a small subunit for the expression of enzyme activity.Citation17,19) However, there was no sequence encoding the N,N-dimethylformamidase small subunit-like protein in the region upstream of the gene encoding N-acetyl-(R)-β-Phe acylase. In addition, heterologously produced N-acetyl-(R)-β-Phe acylase in E. coli JM109/ ptrp4_3BR surely showed enzyme activity. Therefore, N-acetyl-(R)-β-Phe acylase had different quaternary structure from the N,N-dimethylformamidase.
The overall amino acid sequence of N-acetyl-(S)-β-Phe acylase was also determined as a result of gene-cloning. The N-terminal amino acid sequence of the predicted gene product had an additional six residues compared to that determined with authentic purified protein. In the extra sequence MDFCQM, there are two possible translation initiation positions. A native initiation position will be determined via further studies into gene expression, but at the present time, it is unknown. The difference between authentic and predicted N-terminal amino acid sequences suggests the occurrence of some sort of post-translational modification. On the other hand, it cannot be said that there is no possibility of the artificial digestion during the purification processes. The sequence was found to have similarities with that of a phosphotriesterase-related protein from Homo sapiens. A well-known phosphotriesterase from Pseudomonas has been reported to be an enzyme releasing phosphate from paraoxon, dursban, and parathion.Citation20)
The characterization data for N-acetyl-(R)-β-Phe acylase were compared with data for the corresponding wild-type strain-derived activity.Citation11) The pH and thermal profiles showed an almost identical trend; there was no activity under pH 5 and high activity at 60 °C. About the effects of metal ions, the addition of Fe ions increased the enzyme activities but EDTA showed no effect. About N,N-dimethylformamidase which is high similarity, it was reported an iron-containing amidohydrolase and the addition of 1 mM EDTA showed no effect but the addition of 20 mM EDTA showed inhibition.Citation19) In the process of 10 mM EDTA, Fe ions might not have been completely removed. N-acetyl-(R)-β-Phe acylase recognized N-acetyl-β-amino acids with bulky side chains, but not short alkyl side chains and recognized N-acetyl-β-amino acids of l-form, the corresponding positions of hydrogen, carboxyl groups, amino groups, and side chains. N-acetyl-(R)-β-Phe acylase did not recognize N-acetyl-dl-α-phenylalanine.
The characterization data for N-acetyl-(S)-β-Phe acylase were compared with data for the corresponding wild-type strain-derived activity.Citation11) The pH profile and thermal stability showed the same trend, but the thermal profile was slightly different. This could be attributed to the reaction conditions or the enzyme source with not purified. N-acetyl-(S)-β-Phe acylase recognized N-acetyl-β-amino acids with bulky side chains and d-form but not with short alkyl side chains. Additionally, N-acetyl-(S)-β-Phe acylase did not recognize N-acetyl-dl-α-phenylalanine.
The results of the characterization showed that both acylases were similar in terms of substrate specificity, although the amino acid sequences were very different. The structural analysis of the active site produced interesting results. Two acylases did not show activity toward N-acetyl-α-phenylalanine. This result could be due to the fact that the activity of microorganism 130F (Burkhordelia sp. AJ110349) is induced by N-acetyl-(R,S)-β-Phe and (R,S)-β-Phe, but not induced by N-acetyl-(R)-α-phenylalanine and N-acetyl-(S)-α-phenylalanine.Citation11)
Productions of enantiopure β-Phe from JM109/ptrp4_3BR and JM109/pSFN_BS showed high preferential enantioselectivity. These findings may be potentially useful with respect to industrial application. The production of (S)-β-Phe by JM109/pSFN_BS was a higher molar conversion. However, the production of (R)-β-Phe by JM109/ptrp4_3BR was less than the production of (S)-β-Phe, because the expression level of N-acetyl-(R)-β-Phe acylase in JM109/ptrp4_3BR was lower. Although some plasmids were constructed for the expression of N-acetyl-(R)-β-Phe acylase, the expression levels were too low (data not shown). In our previous report,Citation11) N-acetyl-(R)-β-Phe acylase was discovered from Variovorax sp. AJ110348. With further study of N-acetyl-(R)-β-Phe acylase from Variovorax sp., the efficiency of the (R)-β-Phe production is expected to improve.
While the potential for their use in industrial applications is clearly anticipated due to their high enantioselectivity, the physiological roles of both N-acetyl-(R)- and (S)-β-Phe acylases remain to be clarified. A more detailed analysis to resolve the structures should provide a better understanding of the reaction mechanisms. Further investigations may help to determine the physiological role(s) of these enzymes.
Author contributions
Y. I. and S. S. planned and carried out the experiments. Y. I. and S. S. wrote the manuscript. All authors discussed the results and commented on the manuscript. Final approval of the article was given by Y. I.
Disclosure statement
No potential conflict of interest was reported by the authors.
Acknowledgments
We thank Dr. Kunihiko Watanabe, Dr. Ryo Natsume, and Dr. Yasuhiro Mihara for their encouragement and useful suggestions. We also thank Dr. Shinji Kuroda for kind advice regarding substrate synthesis, and Mayuko Yoda, and Shinsuke Natsui for providing technical support.
References
- Liu M, Sibi MP. Recent advances in the stereoselective synthesis of β-amino acids. Tetrahedron. 2002;58:7991–8035.10.1016/S0040-4020(02)00991-2
- Juaristi E, Soloshonok VA. Enantioselective synthesis of β-amino acids. 2nd ed. Hoboken (NJ): Wiley; 1997.
- Liljeblad A, Kanerva LT. Biocatalysis as a profound tool in the preparation of highly enantiopure β-amino acids. Tetrahedron. 2006;62:5831–5854.10.1016/j.tet.2006.03.109
- Soloshonok VA, Svedas VK, Kukhar VP, et al. An enzymatic entry to enantiopure β-amino acids. Synlett. 1993;5:339–341.10.1055/s-1993-22447
- Soloshonok VA, Kirilenko AG, Fokina NA, et al. Biocatalytic resolution of β-fluoroalkyl-β-amino acids. Tetrahedron: Asymmetry. 1994;5:1119–1126.10.1016/0957-4166(94)80063-4
- Soloshonok VA, Fokina NA, Rybakova AV, et al. Biocatalytic approach to enantiomerically pure β-amino acids. Tetrahedron: Asymmetry. 1995;6:1601–1610.10.1016/0957-4166(95)00204-3
- Topgi RS, Ng JS, Landis B, Wang P, Behling JR. Use of enzyme penicillin acylase in selective amidation/amide hydrolysis to resolve ethyl 3-amino-4-pentynoate isomers. Bioorg. Med. Chem. 1999;7:2221–2229.10.1016/S0968-0896(99)00155-8
- Cardillo G, Gentilucci L, Tolomelli A, Tomasini C. A stereoselective synthesis of (2R,3S)-N-benzoylphenylisoserine methyl ester. J. Org. Chem. 1998;63:2351–2353.10.1021/jo9714066
- Chenault HK, Dahmer J, Whitesides GM. Kinetic resolution of unnatural and rarely occurring amino acids: enantioselective hydrolysis of N-acyl amino acids catalyzed by acylase I. J. Am. Chem. Soc. 1989;111:6354–6364.10.1021/ja00198a055
- Liese A, Seelbach K, Wandrey C. Industrial Biotransformations. Weinheim: Wiley-VCH; 2000. p. 300–303.10.1002/9783527614165
- Kawasaki H, Koyama K, Kurokawa S, et al. Production of (R)-3-amino-3-phenylpropionic acid and (S)-3-amino-3-phenylpropionic acid from (R, S)-N-acetyl-3-amino-3-phenylpropionic acid using microorganisms having enantiomer-specific amidohydrolyzing activity. Biosci. Biotechnol. Biochem. 2006;70:99–106.10.1271/bbb.70.99
- Wani MC, Taylor HL, Wall ME, Coggon P, McPhail AT. Plant antitumor agents. VI. Isolation and structure of taxol, a novel antileukemic and antitumor agent from Taxus brevifolia. J. Am. Chem. Soc. 1971;93:2325–2327.10.1021/ja00738a045
- Suzuki S, Imabayashi Y, Watanabe K, Kawasaki H, Nakamatsu T. United Status patent US20080241895. 2008 Oct 2.
- Nozaki H, Kira I, Watanabe K, Yokozeki K. Purification and properties of d-hydantoin hydrolase and N-carbamoyl-d-amino acid amidohydrolase from Flavobacterium sp. AJ11199 and Pasteurella sp. AJ 11221. J. Mol. Catal. B Enzym. 2005;32:205–211.10.1016/j.molcatb.2004.12.003
- Abe I, Hara S, Kai Y, et al. PCT WO2006075486. 2006 Jul 20.
- Antal P, Anita A, Ericka V, et al. Direct and indirect high-performance liquid chromatographic enantioseparation of beta-amino acids. J. Chromatogr. 2004;1031:171–178.
- Hasegawa Y, Tokuyama T, Iwaki H. Cloning and expression of the N,N-dimethylformamidase gene from Alcaligenes sp. strain KUFA-1. Biosci. Biotechnol. Biochem. 1999;63:2091–2096.10.1271/bbb.63.2091
- Gerhard DS, Wagner L, Feingold EA, et al. The status, quality, and expansion of the NIH full-length cDNA project: the mammalian gene collection (MGC). Genome Res. 2004;14:2121–2127.
- Schar HP, Holzmann W, Ramos Tombo GM, Ghisalba O. Purification and characterization of N,N-dimethylformamidase from DMF 3/3. Eur. J. Biochem. 1986;158:469–475.10.1111/ejb.1986.158.issue-3
- Dumas DP, Caldwell SR, Wild JR, Raushel FM. Purification and properties of the phosphotriesterase from Pseudomonas diminuta. J. Biol. Chem. 1989;64:19659–19665.