
Abstract
Eisenia fetida and Eisenia andrei are vermicomposting species that are used as model animals for testing chemical material toxicology. Eisenia spp. are grown commercially in various fields in Japan. However, these two species have not been classified because it is difficult to distinguish them morphologically; thus, all bred earthworms are called E. fetida. However, it has been proposed that these two species have different expression regulation mechanisms. Here, we classified a sample of earthworms purchased from several farms, confirming that both E. fetida and E. andrei are present in Japanese earthworm breeding programs. We also characterized two highly active endoglucanases (EfEG1 and EfEG2) from the E. fetida Waki strain, which contained strong fibrinolytic enzymes for improving human health. We confirmed that EfEG1 is 1371 bp long and belongs to GHF9. Thus, E. fetida Waki may have commercial application for biomass utilization and as a dietary health supplement.
Graphical abstract
We propose addition of strain name such as ‘Waki’ to species names E. fetida to reduce confusion and differentiate enzyme abilities of these strains.
Introduction
Earthworms are one of the most well-known soil decomposers. Charles Darwin was the famous scientist to study earthworms, leading to the compilation of the “Earthworm Book.” Today, it is widely acknowledged that earthworms are useful animals for forming nutrient-rich soil. Since earthworms are polyphagous animals and the cast contains a large amount of nutrients, the application of earthworms in composting has been extensively studied.Citation1) In particular, Eisenia fetida and Eisenia andrei are useful earthworm species for vermicomposting, and these are used as model animals for chemical material toxicology tests by the Organization for Economic Co-operation and Development (Earthworm, acute toxicity tests, Guideline for Testing Chemicals; 1984 [No. 207]).Citation2) However, the two species have very similar morphologies, making it difficult to distinguish them.Citation2,3) In fact, Dvořák et al. reported that many research groups use these two species indiscriminately.Citation4) The same authors reported that the activity and expression levels of certain enzymes differ between the two species, with these features being affected by the natural habitat occupied by the earthworms.Citation4) Thus, without properly classifying these two species, it is not possible to understand to what extent these features differ. This information gap has only recently been pointed out. Consequently, the Eisenia Barcoding Project focused on addressing this knowledge gap via the Eisenia Barcoding Initiative (EBI in Germany).
Eisenia spp. are common species that are grown commercially in various fields in Japan. However, no reports exist comparing and characterizing these species using genetic information from earthworms that are bred, with the assumption that all earthworms are E. fetida. Since the classification of earthworms remains unclear in Japan, it is not possible to differentiate the two species at present. Furthermore, E. fetida contains strong fibrinolytic enzymes,Citation5,6) with its lyophilized powder being sold as a dietary supplement. In fact, our research group previously developed a new method of producing lyophilized powder with high fibrinolytic activity specifically using the E. fetida Waki strain.Citation7) In addition, E. fetida Waki may contain a high-pressure tolerant fibrinolytic enzyme.Citation8) Therefore, this strain contains essential enzymes for improving human health, and it should be differentiated from other species and strains for appropriate commercial use. Thus, genetic and enzymatic engineering could be used to characterize different earthworm species and strains, in addition to assimilating information about their various functional enzymes such as saccharification enzymes.
For instance, the saccharification enzyme “cellulase” is one of the most important enzymes used in biorefinery.Citation9) This is because cellulose (which is the enzyme substrate of cellulase) is one of the most abundant carbohydrate polymers on earth, and it does not compete against human food requirements, such as starch-based corn.Citation9) Recently, Ueda et al. reported that E. fetida contains endoglucanase (EG), which is one of the cellulolytic enzymes, on a commercial farm in Hokkaido (currently, this earthworm cannot be purchased), Japan.Citation10–12) In addition, earthworm enzymes are beneficial for biomass utilization.Citation10) However, the crude enzymes present in earthworms have not been subjected to the biomass saccharification test.
Here, we classified a sample of earthworms that were purchased from several farms of commercial companies in Japan, with the aim to identify the strains of each species present. In addition, we elucidated the effect of wheat bran reducing sugar production using the cell-free extract of E. fetida Waki. We also characterized the EG of this strain in detail for the potential application of the functional enzymes to biomass utilization, in addition to its current use as a dietary supplement.
Materials and methods
Materials
The earthworm E. fetida Waki and its lyophilized powder were kindly provided by Waki Pharmaceutical Co., Ltd., Nara, Japan. All other earthworms were purchased from Sagami Purification Service Ltd. (Kanagawa, Japan) and Kowa Shoji Co., Ltd. (Hiroshima, Japan). Wheat bran was kindly provided by Nippon Flour Mills Co., Ltd. (Tokyo, Japan). Carboxymethyl cellulose (CMC) was purchased from Sigma-Aldrich Japan K. K. (Tokyo, Japan). All experiments were conducted in accordance with the guidelines of the animal care committee.
Microorganisms and culture media
Two types of media (YPG contained 2.0% peptone, 1.0% Bacto yeast extract, and 1.0% glycerol, and YPM contained 2.0% peptone, 1.0% Bacto yeast extract, 2.0% glucose, and 0.5% methanol) were used for Pichia pastoris strain X-33. Escherichia coli strain DH5α was cultivated in 2 × YT medium containing 1.6% of Bacto tryptone, 1% yeast extract, and 0.5% NaCl (pH 7.0).
Preparation of genomic DNA
The earthworms were starved for 24 h, and then, individuals were separately washed with MilliQ and ground in a mortar with liquid N2. The powdered cells were transferred to a 15-mL conical tube containing 2 mL TE buffer and 2 mL of the lysis buffer (containing 50 mM EDTA and 0.5% SDS). The contents were mixed by gentle inverting. The mixture was shaken gently at 4 °C for 30 min and centrifuged at 6000 × g and 4 °C for 5 min. The supernatant was purified by phenol–chloroform extraction, and the precipitate was dissolved in 400 μL TE buffer. Ribonuclease A (50 μg; Nippon Gene Co., Tokyo, Japan) was added to the DNA solution, and the mixture was incubated at 37 °C for 30 min. After phenol–chloroform extraction, the precipitate was dissolved in the appropriate amount of TE buffer.
Cloning of the gene encoding cytochrome c oxidase I (COI)
This procedure followed the improved methods of Novo et al.Citation13). The genes encoding the COI sequences were amplified by PCR with genomic DNA as a template, using the LCO1490 and HC02198 primer set: 5′-GGTCAACAAATCATAAAGATATTGG-3′ and 5′-TAAACTTCAGGGTGACCAAAAAATCA-3′ as the 5′- and 3′-primer, respectively. PCR was completed under the following conditions: 95 °C for 5 min; 95 °C for 1 min, 40 °C for 1 min, and 72 °C for 1 min (35 cycles); and then 72 °C for 3 min. The purified PCR fragment was ligated with a pMD20-T vector using Mighty TA-cloning Reagent Set for PrimeSTAR (both from Takara Bio Inc., Otsu, Shiga, Japan). E. coli strain DH5α competent cells were transformed with the ligated product by heat shock.
Preparation of cell-free extract of E. fetida Waki
Earthworms were washed and starved for 24 h at 20 °C. Then, they were freeze-dried and pounded in a mortar. The resulting powder (10 g) was suspended in sodium phosphate buffer at pH 7.0. The suspension was gently mixed in a reciprocal shaker for 4 h at 4 °C. Two-percent streptomycin sulfate was added to the suspension to remove nucleic acid, and the solution was centrifuged at 19,000 × g for 2 h to remove cell debris as the precipitates.
Enzyme production of Trichoderma reesei QM9414
T. reesei QM9414 was cultivated at 28 °C on a rotary shaker (220 rpm) for 1 week in 50-mL medium containing 50 mM tartrate buffer with 1.0% Funacel II (Funakoshi Co., Ltd., Tokyo, Japan), 0.3% Bacto peptone, 0.05% Bacto yeast extract, 0.14% (NH4)2SO4, 0.2% KH2PO4, 0.03% CaCl·2H2O, 0.03% MgSO4·7H2O, 0.1% Tween 80, and 0.1% trace elements solution (0.1% FeSO4·7H2O, 0.88% ZnSO4·7H2O, 0.04% CuSO4·5H2O, 0.01% Na2B4O7·10H2O, and 0.005% (NH4)6Mo7O24·4H2O) (pH 4.0). The cultured medium was centrifuged at 7000 × g for 1 h to remove mycelia as the precipitate.
Plate assay
A plate assay was performed by modifying previously described methods.Citation14,15) The plates were prepared with the following substrates that were mixed with 50 mM sodium phosphate buffer (pH 7.0) and 2% agar: 0.1% CMC (Sigma-Aldrich), 0.1% lichenan (Sigma-Aldrich), 0.1% laminarin (Tokyo Chemical Industry Co., Ltd., Osaka, Japan), 0.1% birchwood xylan (Sigma-Aldrich), 0.1% oat spelt xylan (Sigma-Aldrich), and 0.1% xyloglucan (Dainippon Sumitomo Pharma Co., Ltd., Osaka, Japan). Then, each segment of the crude enzyme or purified enzyme was added to the plates. The enzyme reaction on the plates was carried out 40 °C for 2 h. Then, the plates were exposed to 1 M Tris-HCl buffer (pH 7.5). The buffer was then replaced with 0.1% Congo red–0.2% sodium hydrate solution, and the plates were incubated for 30 min. After incubation, the plates were washed with 1 M NaCl and rinsed with 5% acetic acid. The diameter of the clear zone that appeared (which was indicative of enzyme action) was measured.
At the same time, azurine-crosslinked (AZCL) type plates were prepared with the following substrates that were mixed with 50 mM sodium phosphate buffer (pH 7.0) and 2% agar: 0.1% AZCL-β-glucan (Megazyme International Ireland Ltd., Wicklow, Ireland), 0.1% AZCL-β-1,3-glucan (Megazyme), 0.1% AZCL-amylose (Megazyme), 0.1% AZCL-arabinan (Megazyme), 0.1% AZCL-galactan (Megazyme), 0.1% AZCL-mannan (Megazyme), and 0.1% AZCL-xylan (Megazyme). Next, each segment of the crude enzyme or purified enzyme was added to the plates. The enzyme reaction on the plates was carried out 40 °C for 24 h. The diameter of the colored zone that appeared (which was indicative of enzyme activity) was measured. In addition, fluorescence detection type plates were prepared with the following substrates that were mixed with 50 mM sodium phosphate buffer (pH 7.0) and 2% agar: 4-methylumbelliferyl β-d-glucopyranoside (4MU-G) (Sigma-Aldrich) and 4-methylumbelliferyl β-d-cellobioside (4MU-G2) (Sigma-Aldrich). Next, purified enzyme was added to the plates and incubated at 40 °C for 2 h. Subsequently, the plates were exposed to excitation light at 365 nm. The diameter of the fluorescence zone, which was indicative of enzyme activity, was measured.
Enzymatic saccharification of wheat bran
Enzymatic saccharification was carried out on 1.0 g pretreated wheat bran (dry weight) in a 50-mL plastic bottle (15 mL working volume) incubated in a reciprocal shaker (180 rpm) at 30, 40, and 50 °C. The shaker contained 50 mM sodium phosphate buffer at pH 7.0 (E. fetida Waki) or 50 mM sodium acetate buffer at pH 5.0 (T. reesei QM9414), with 3 mM sodium azide and 30 units of CMCase equivalent. The reducing sugar was determined by the Somogyi–Nelson method.Citation16)
Component analysis of wheat bran
Cellulose, hemicellulose, and lignin content were determined by the detergent method.Citation17) Moisture analysis was carried out by drying the sample to a constant weight at 105 °C. Ash content was obtained by heating the moisture-free sample in an electric muffle furnace at 550 °C for 6 h. The protein content of wheat bran was estimated by determining total nitrogen content using the Kjeldahl method.Citation18) Starch content was determined from the amount of glucose using glucose CII-test Wako kit (Wako Pure Chemical Industries, Ltd., Osaka, Japan) from starch. To produce glucose from starch, wheat bran was treated by two-step enzymatic hydrolysis consisting of liquefaction by thermostable α-amylase (Megazyme) and saccharification by amyloglucosidase (Megazyme).Citation9)
Detection of saccharification products
Wheat bran enzymatic saccharification products were identified using the Prominence reducing sugar analysis system (Shimadzu Corp., Kyoto., Japan).
Purification of the two EGs
Purification steps were performed following a modified version of a previously described method.Citation10) All purification steps were completed at 4 °C, and column chromatography was completed with an AKTAprime plus system (GE Healthcare Japan K.K., Tokyo, Japan). E. fetida Waki lyophilized powder (75 g as [dry weight]) was suspended in 750 mL of 50 mM sodium phosphate buffer (containing 1 mM DTT and 2% streptomycin sulfate) at pH 7.0 and was incubated for 24 h. The suspension was centrifuged at 19,000 × g for 1 h to remove cell debris as the precipitates. (NH4)2SO4 powder was added to the supernatant to reach 20% saturation. After gentle stirring for 60 min, the mixture was centrifuged at 19,000 × g for 30 min. The supernatant was adjusted to 60% saturation by (NH4)2SO4 powder. After stirring for another 60 min and centrifugation under the same conditions, the precipitate was dissolved in a minimum volume of buffer A (25 mM sodium phosphate buffer [pH 6.0] containing 1 mM DTT). The enzyme solution was then dialyzed against the same buffer for 24 h and centrifuged at 19,000 × g for 15 min. The supernatant was applied to a DEAE-Toyopearl column (2.2 × 20 cm; Tosoh Co., Tokyo, Japan) equilibrated with buffer A. The column was washed with the same buffer. The adsorbed protein was eluted with a 380-mL linear gradient of 0–0.5 M NaCl in buffer A. The active fractions were suspended in 50 mM sodium acetate buffer (pH 4.5) containing 1 mM DTT and were dialyzed by a VIVASPIN 20 (Sartorius Stedim Japan K.K., Tokyo, Japan) and applied to a HiTrap Capto MMC column (1.6 × 2.5 cm; GE Healthcare) equilibrated with buffer A. After washing the column extensively with the equilibration buffer, elution was completed with a 250-mL linear gradient of 0–1 M NaCl in 50 mM sodium phosphate buffer (pH 7.0). The enzyme-active fractions were collected and dialyzed by a VIVASPIN 6 (Sartorius Stedim) and applied to a Bio-Scale Mini CHT Type I column (1.26 × 4 cm; Bio-Rad Laboratories, Inc., Tokyo, Japan) equilibrated with 1 mM sodium phosphate buffer (pH 6.0) containing 1 mM DTT. The column was washed with the same buffer. The adsorbed protein was eluted with a linear gradient of 0.1 M sodium phosphate buffer (pH 6.0) containing 1 mM DTT. The enzyme-active fractions were collected, dialyzed with a VIVASPIN 500 (Sartorius Stedim) and applied to a Toyopearl HW55-S column (1.6 × 60 cm; Tosoh) equilibrated with buffer A containing 0.1 M NaCl. The active fractions were pooled, dialyzed with a VIVASPIN 500 (Sartorius Stedim) and applied to a Resource Q column (0.64 × 3 cm; GE Healthcare). The column was washed with the same buffer. The adsorbed enzyme was eluted with a linear gradient of 0–0.5 M NaCl in 120 mL of buffer A.
Cloning of the gene encoding EfEG1
Total RNA was prepared from earthworms by homogenizing the bodies with ISOGEN (Nippon Gene) in a glass bead mixer (Mini-Bead Beater; Biospec Products, Bartlesville, OK, USA). First-strand cDNA was prepared with SuperScript III reverse transcriptase using a First-Strand Synthesis System for RT-PCR (Invitrogen Corp., Carlsbad, CA, USA). Partial genes encoding EfEG1 were amplified by PCR with cDNA as a template, using an EG-specific primer set: 5′-CTCTTCTACGAGGCCGAGAG-3′ and 5′-CAACGGCGCCTTGGAATCCG-3′ (positions 102–119 and 1314–1333) as the 5′- and 3′-primers, respectively. The EG-specific primer set was designed from the consensus sequences for earthworm EGs. The nucleotide positions of EG for 5′- and 3′-primer designs were as follows: EG of Lumbricus rubellus at positions 119–138 and 1330–1349, EaEG2 of E. andrei at positions 100–119 and 1314–1333, and phhEG of Metaphire hilgendorfi at positions 91–110 and 1305–1324. PCR was completed under the following conditions: 98 °C for 1 min; 98 °C for 10 s, 55 °C for 5 s, and 72 °C for 2 min (30 cycles); and finally 72 °C for 3 min. The purified PCR fragment was ligated with a pMD20-T vector using Mighty TA-cloning Reagent Set for PrimeSTAR (both from Takara). E. coli strain DH5α competent cells were transformed with the ligated product by heat shock. The 5′-and 3′-flanking regions of the EfEG1 genes were cloned using the 5′ and 3′-Full RACE Core Set (TaKaRa).
Construction of expression systems for the EfEG1 gene
The cDNA fragment attributed to EfEG1 was amplified with the following primers: 5′-CGGAATTCACCAAATATAATTATGATGAAGTTC-3′ and 5′-GCTCTAGAGCCTTGCCGTCTCTCAGCTG-3′. PCR was completed under the following conditions: 95 °C for 5 min; 98 °C for 10 s, 46 °C for 15 s, and 72 °C for 3 min (30 cycles); and then 72 °C for 3 min. The entire cDNA fragment was ligated to pPICZα-A (Invitrogen) and used to transform E. coli strain DH5α competent cells by heat shock. The constructed plasmid was used to transform P. pastoris strain X-33 (Invitrogen).
Purification of His-tag fusion proteins
Five milliliters of an overnight culture of P. pastoris transformant was incubated at 30 °C in 400 mL of 2 × YPG media containing 100 μg/mL zeocin (Invitrogen). The cells were harvested by centrifugation at 6000 × g for 10 min after cultivation for 30 h. The cells were inoculated in 100-mL YPM medium, and endoglucanase activity was induced at 30 °C for 60 h. The supernatant was harvested by centrifugation at 6000 × g for 10 min and was suspended in buffer A (5 mM sodium phosphate buffer (pH 7.4) containing 1 mM DTT, 10 mM imidazole and 500 mM NaCl). The suspension was applied to a HisPrep FF column (1.6 × 10 cm; GE Healthcare) equilibrated with buffer A. The column was washed with the same buffer. The adsorbed protein was eluted with a 100-mL linear gradient of 20 mM sodium phosphate buffer (pH 7.4) containing 1 mM DTT, 500 mM imidazole, and 500 mM NaCl. The active fractions were suspended in 50 mM sodium acetate buffer (pH 4.5) containing 1 mM DTT. The mixture was dialyzed by a VIVASPIN 6 (Sartorius Stedim) and applied to a HiTrap Capto MMC column (1.6 × 2.5 cm; GE Healthcare) equilibrated with the same buffer. After washing the column extensively with the equilibration buffer, elution was completed with a 200-mL linear gradient of 0–1 M NaCl in 50 mM sodium phosphate buffer (pH 7.0). The enzyme-active fractions were collected and dialyzed with VIVASPIN 500 (Sartorius Stedim) and applied to a Toyopearl HW55-S column (Tosoh) equilibrated with 50 mM sodium phosphate buffer (pH 7.0), containing 1 mM DTT and 0.1 M NaCl. The active fractions were pooled and dialyzed with a VIVASPIN 500 (Sartorius Stedim).
Thin-layer chromatography (TLC)
A reaction mixture (200 μL) containing 0.3% (w/v) CMC (Sigma-Aldrich) or 2 mg/mL cellooligosaccharide (cellobiose, cellotriose, cellotetraose, and cellopentaose), enzyme (0.5 units), and 50 mM sodium acetate buffer (pH 7.0) was incubated at 40 °C. The reaction products were identified by TLC with precoated plates (silica gel 60 F254, 0.25 mm thick; E. Merck KGaA, Darmstadt, Germany). The solvent system was 2-propanol–acetic acid–water (3:1:1). Spots on the plates were visually identified by spraying the plates with 20% (v/v) sulfuric acid/ethanol solution, followed by brief heating at 150 °C.
Measurement of enzyme activity
EG and cellobiohydrolase (CBH) activity was measured by determining the amount of reducing sugar related to CMC (Sigma-Aldrich) or Avicel (Funacel II, Funakoshi) using the Somogyi–Nelson method.Citation16) CMC was used as a substrate throughout the purification process and for the examination of substrate specificity at 1.0% (w/v) concentration. Avicel was used as a substrate to examine substrate specificity at a concentration of 0.5% (w/v). A reaction mixture (500 μL) containing 1.0% (w/v) CMC (Sigma-Aldrich) or 0.5% Avicel (Funakoshi), enzyme solution, and 25 mM sodium phosphate buffer (pH 7.0) was incubated at 40 °C for 15 min. Subsequently, the reaction was continued per the Somogyi–Nelson method.Citation16) One unit of the enzyme was defined as EG or CBH activity that produces 1 μmol of glucose per minute.
Protein determination
The amount of protein was determined by the method developed by Bradford Citation19) using a Bio-Rad protein assay kit (Bio-Rad), with bovine serum albumin (BSA; Bio-Rad) as the standard. Gel filtration was carried out with a Toyopearl HW55-S column (Tosoh) in an AKTAprime plus system (GE Healthcare) to determine the molecular mass of proteins. The molecular mass of the subunit of the purified protein was determined by SDS-PAGE on 12.5% gel, as described by Laemmli.Citation20) The protein was stained with Coomassie Brilliant Blue R-250 (Sigma-Aldrich). Internal amino acid sequences were determined by the Edman method, using a PPSQ-33A protein sequencer (Shimadzu Corp., Kyoto, Japan).
Nucleotide sequence accession numbers
The COI sequences that were identified from E. fetida Waki (EfWaCOI), E. andrei Sagami (EaSaCOI), and E. andrei Kowa (EaKoCOI) in this study were submitted to the DDBJ under accession numbers LC006114 (EfWaCOI), LC006115 (EaSaCOI), and LC006116 (EaKoCOI), respectively. The EfEG1 cDNA sequence from E. fetida Waki reported here was submitted to the DDBJ database under the accession number LC006117 (EfEG1). The accession numbers for the additional COI genes used in this study were as follows: HQ534065 for E. andrei Ean1; FJ214228 for E. fetida EF strain; JX531566 for E. fetida; HQ224516 for Eisenia sp. FK-2010; JN870062 for E. andrei; GU206210 for Lumbricus sp. BOLD; U24570 for L. terrestris; DQ092904 for L. rubellus; JQ982486 for Amynthas sp. 14JS-2012; AB542494 for A. hupeiensis; DQ835676 for Metaphire tschiliensis tschiliensis; AY960800 for M. tahanmonta; DQ092890 for Aporrectodea handlirschi; GU013824 for A. caliginosa; GU453370 for Enchytraeus albidus; GU902051 for E. japonensis; EF494507 for Perionyx excavatus; AF064043 for Pirodrilus minutus; AF064042 for Aktedrilus arcticus; and EF457734 for Cooperia oncophora. The accession numbers for the other genes used in this study were as follows: LRC00474_2 (LumbriBASE identifier number) for EG of L. rubellus; EU315749 for EaEG2 of E. andrei; and AB452993 for phhEG of M. hilgendorfi.
Results and discussion
Characterization of earthworms by morphological and phylogenetic analyses
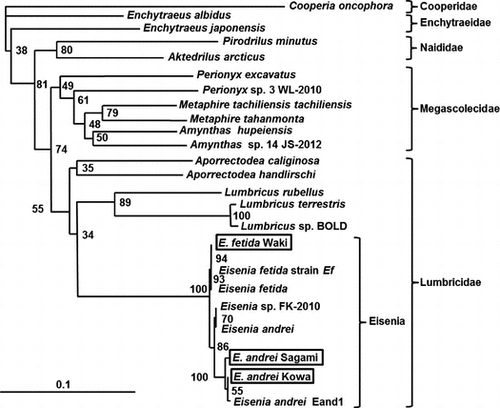
Morphological characterization was carried out following the method developed by Pérez-Losada et al.Citation3). Morphological analysis showed that all analyzed worms had cleaved prostomia and dorsal pores. The setae were paired, and the clitellum was observed after 20 segments. The area around the intersegmental was pale or yellow. These features confirm that all three earthworms were Eisenia spp. Because it is difficult to distinguish E. fetida from E. andrei based on appearance,Citation2) we attempted to distinguish between these species by phylogenetic analysis of COI genes, which are commonly used genes for earthworm phylogenetic analyses,Citation13,21) with the EBI recommending that COI classification be used. Furthermore, it was confirmed that the COI region differs between E. fetida and E. andrei.Citation22) Therefore, we used an internationally recognized method to analyze the COI region.Citation22–24) Phylogenetic analysis showed that all three strains differed to one another (Fig. ). Therefore, we named the three strains of the two species as E. fetida Waki, E. andrei Sagami, and E. andrei Kowa. The fact that all three strains of the two species were present demonstrates the absence of discrimination by breeding companies, with morphological and phylogenetic analyses being necessary for earthworm classification. In addition, Dvořák et al. reported that the activity and expression levels of certain enzymes differ among these species and are affected by the natural habitat occupied by earthworms.Citation4) Thus, if the earthworms are not properly classified, the importance of these features will be misinterpreted. Therefore, we propose the addition of “strains” to species names, such as Waki or Sagami, to reduce confusion and to differentiate the different enzyme abilities of these strains, similar to the procedure followed for microorganisms.
Fig. 1. Phylogenetic analysis of the COI region in earthworms.
Substrate specificity of the cell-free extract and the saccharification of wheat bran
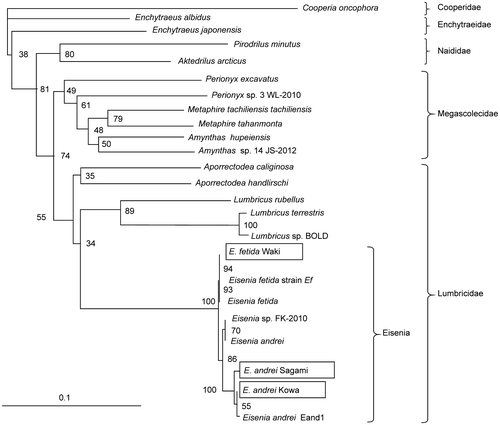
The plate assay showed that the cell-free extract of each segment of E. fetida Waki (Fig. ) was active toward CMC, lichenan, laminarin, birchwood xylan, oat spelt xylan, AZCL-β-glucan, AZCL-1,3-β-glucan, AZCL-amylose, and AZCL-mannan (Table ). However, the extract was not active toward Avicel. These results indicate that the extract exhibited β-1,4-endo-glucanase, β-1,3-endoglucanase, β-1,4-endoxylanase, β-1,4-endomannase, protease, and amylase activity, but did not exhibit cellobiohydrolase activity. Furthermore, the extract exhibited high endoglucanase and amylase activity, but low xylanase activity. In particular, segment 2 appeared to be the most important part of earthworms, because it exhibited extensive enzyme activity, with many vital organs, such as the gut (Table ). In addition, Nozaki et al. identified an endo-β-1,4-glucanase in the gut of Pheretima hilgendorfiCitation25).
Fig. 2. Morphology and cut parts of E. fetida Waki.
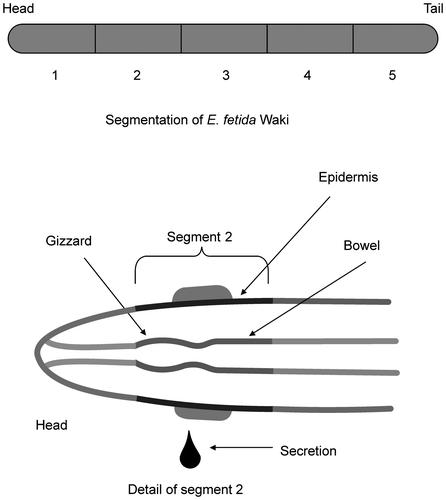
Table 1. Enzyme activity of crude extract from each segment of E. fetida Waki.
The enzymatic saccharification ability of E. fetida Waki extract was compared with that of T. reesei QM9414 cell-free extract. For E. fetida Waki, maximum sugar concentration reached 10,538 μg/mL after 48 h at 50 °C. This value was about 1.5-fold higher than that recorded for T. reesei QM9414 (6992 μg/mL) (Fig. ). Since wheat bran contained starch–cellulose/hemicellulose–other at a ratio of 47:29:24, this result shows that amylase activity is useful for the saccharification of wheat bran because the starch component of wheat bran is relatively higher than the cellulosic biomass of other plant matter, such as rice straw, which has a starch content of about 11%.Citation26) Previous studies have not performed the expression analysis of glycohydrolases from earthworms using various types of feedstuff. In contrast, metallothionein enzymes are induced by the ingestion of heavy metals.Citation27) Therefore, the production of glycohydrolases is also likely to be induced depending on the type of feedstuff. If we select suitable feedstuff to induce the production of these enzymes, enzyme production will increase, resulting in the degradation of soft cellulose, such as wheat bran, by the enzyme.
Fig. 3. Enzymatic saccharification with E. fetida Waki (A) and T. reesei QM9414 (B) cell-free extracts.
Detection of saccharification products
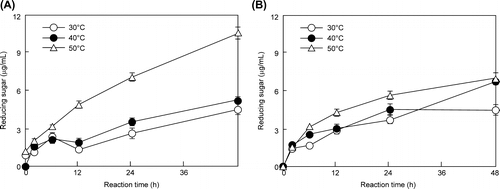
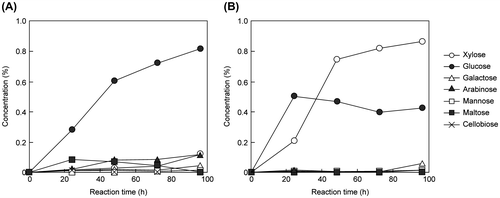
Previous studies have not investigated whether saccharification products, such as glucose from biomass, may be obtained using earthworm crude enzymes. Therefore, we investigated this issue. Glucose production by E. fetida Waki and T. reesei QM9414 cell-free extract was measured by high-performance liquid chromatography. After 96 h, the glucose concentration of E. fetida Waki reached 0.82% (conversion efficiency: 23.8%), which was higher than that of T. reesei QM9414 (0.43%) (conversion efficiency: 12.5%) (Fig. ). In addition, the glucose production of E. fetida Waki increased over time (Fig. (A)). In comparison, xylose was the main component of the T. reesei QM9414 reaction products, with glucose production plateauing after 20 h (Fig. (B)). Plateau formation is common when cellulase is active; however, it did not occur for E. fetida Waki. Thus, amylase may be more active than cellulase in E. fetida Waki. It is relevant that the amylase activity of E. fetida Waki (1.50 units/mg detected from cell-free extract) was stronger than that of T. reesei QM9414 (not detected). This difference affects substrate specificity. Therefore, E. fetida Waki is appropriate for the saccharification of soft cellulose, such as wheat bran.
Fig. 4. Detection of saccharification products in E. fetida Waki (A) and T. reesei QM9414 (B).
Purification and characterization of two endoglucanases from E. fetida Waki
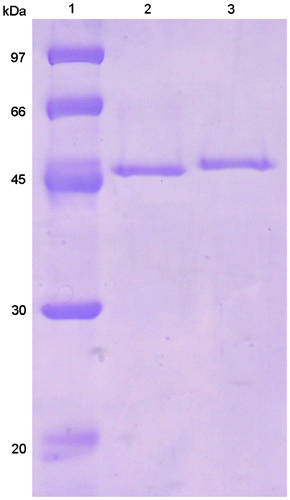
Two endoglucanase-active fractions were found on a DEAE-Toyopearl chromatogram. This enzyme activity was observed in the eluting fractions (EfEG1 and EfEG2) (Fig. ). These fractions were further purified separately. The purification results are shown in Table 2. Each enzyme exhibited a single band on SDS-PAGE (Fig. ). The molecular mass of EfEG1 and EfEG2 was estimated to be 53.3 and 57.5 kDa, respectively. Toyopearl HW55S gel filtration showed that the molecular mass of EfEG1 and EfEG2 was 48.7 and 58.9 kDa, respectively, indicating that both enzymes are monomeric.
Fig. 5. Elution profile of EG isozymes in E. fetida Waki from the DEAE-Toyopearl column.
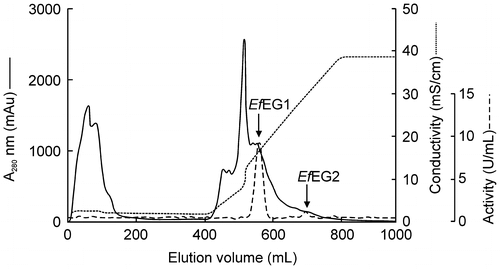
Table 2. Elution profile of EG isozymes in E. fetida Waki from a DEAE-Toyopearl column.
Fig. 6. SDS-PAGE of purified EfEG1 and EfEG2.
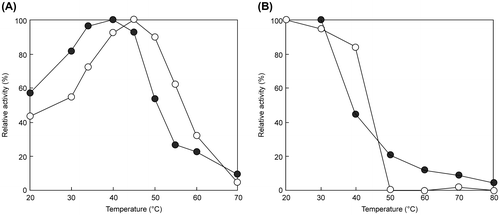
The optimal pH of EfEG1 and EfEG2 was 6.0 and 5.5, respectively (Fig. (A)). The optimal temperature of EfEG1 and EfEG2 was 45 and 40 °C, respectively (Fig. (A)). Enzyme activity was stable over a pH range of 5.0–9.0 (Fig. (B)) and remained stable up to 40 °C for EfEG1 and 30 °C for EfEG2 (Fig. (B)).
Fig. 7. Effect of pH on the activity (A) and stability (B) of EfEG1 and EfEG2.
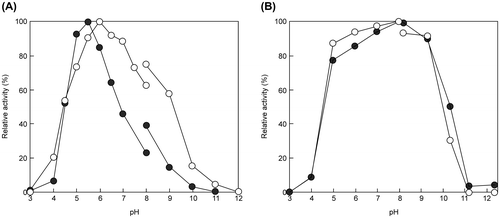
Fig. 8. Effect of temperature on the activity (A) and thermostability (B) of EfEG1 and EfEG2.
The properties of EGs in other animals and T. reesei QM9414 are shown in Table 3.Citation12,28–35) EfEG1 and EfEG2 were not active toward crystalline cellulose (such as Avicel), as reported by Ueda et al.Citation12) for EGs from E. fetida (Nagane Industries Co., Ltd., Sapporo, Japan). However, the specific activity of EfEG2 was half that of EfEG1. In addition, the optimal pH and temperature of EfEG1 and EfEG2 were similar to T. reesei and other animal EGs.
Table 3. Properties of EGs from animal and T. reesei QM9414.
The purified enzymes were active toward CMC, lichenan, and AZCL-β-glucan. Thus, both enzymes had β-1,4-endoglucanase activity. EfEG1 may exhibit β-glucosidase activity because it showed activity against 4MU-G, whereas activity against 4MU-G2 was not detected. In general, β-glucosidase has activity toward 4MU-G2, but this activity was not detected. These results suggest that EfEG1 may have strict substrate specificity. In contrast, EfEG2 did not exert activity against either substrate (Table 4). EfEG2 may be not able to show enzymatic activity against short-chain oligosaccharides. EfEG2 may also exhibit β-1,3-glycosidase activity, but this activity was very weak (Table 4). EfEG1 was active toward oat spelt xylan, but was not detected for AZCL-xylan. Thus, EfEG1 did not exhibit xylanase activity. These results indicate that both enzymes are β-1,4-endoglucanase isozymes.
Table 4. Substrate specificity of purified EfEG1 and EfEG2 from E. fetida Waki.
Cloning of EfEG1 from E. fetida Waki
Since EfEG1 exhibited much higher specific activity toward CMC than EfEG2, we attempted to clone EfEG1 in this study. The ORF of the cloned sequence of EfEG1 was 1371 bp and the deduced amino acid sequence contained 456 amino acid (aa) residues. The internal amino acid sequence (FGTTTNTAWAYD) determined from the purified EfEG1 was also found in the deduced amino acid sequence (258–269 aa). EfEG1 showed 99, 99, and 73% identity with endo-1,4-β-glucanase from E. fetida (AB679653), β-1,4-endoglucanase from E. andrei (AY914054), and β-1,4-endoglucanase from M. hilgendorfi (AB452993), respectively. These results show that EfEG1 encodes the β-1,4-endoglucanase gene.
The purified form of both EGs from E. fetida Waki was not active toward Avicel, which is the model crystalline cellulose compound. Analysis of the sequence indicated that EfEG1 belongs to GHF9 (Table ) and that the active sites (Asp64, Asp77, His378, Asp422, and Glu431) conserved in this sequence were the same as EF-EG2 from E. fetida Nagane.Citation12) Analysis of Signal P showed that the sequence contained a signal peptide (1–21), which is also the same as that in EF-EG2. GHF9 EGs depend on the cellulose-binding domain (CBD) sequence.Citation12) Although EfEG1 does not contain CBD, HdEG66 of H. discus hannai has CBD and weak hydrolytic activity toward crystalline cellulose.Citation32) Thus, animal GHF9 EG activity against crystalline cellulose is weak or too low to be detected. Furthermore, analysis of the partial 5′UTR sequence (about 500 bp) showed that two deduced TATA boxes were found at the nucleotide positions −24 and −278 bp from ATG. In contrast, the CCAAT box and the carbon catabolite repression site CRE in filamentous fungi were not detected.Citation36,37) This interesting result requires further investigation.
Expression of EfEG1 and TLC analysis of the reaction products
The EfEG1 gene was expressed in P. pastoris X-33 as a His-fusion protein that was purified until apparent homogeneity on SDS-PAGE. The control strain did not show endoglucanase activity. The molecular mass of rEfEG1 was estimated to be 53.8 kDa, and the specific activity against CMC was 86.4 units/mg. These results were almost the same as those obtained for purified enzymes from E. fetida Waki.
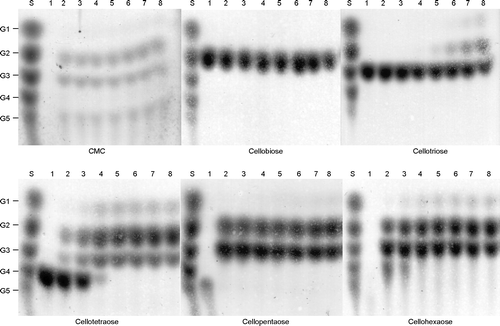
To examine the reaction mechanism of EfEG1, the reaction products were analyzed by TLC (Fig. ). The incubation of CMC, cellobiose, cellotriose, cellotetraose, cellopentaose, and cellohexaose with rEfEG1 produced some spots that may be the reaction product on TLC. TLC analysis showed that rEfEG1 can degrade CMC to cellobiose, cellotriose, and cellopentaose. In addition, this enzyme reacted with the cellotriose substrate, with glucose and cellobiose being detected in the reaction products (Fig. ). These results demonstrate that this enzyme recognizes sugars with a chain structure of more than three, with glucose and cellobiose being produced as the final products of this enzyme reaction. These results indicate that rEfEG1 is β-1,4-endoglucanase. The results of the characterization of EfEG1 were similar to those obtained for EF-EG2 from E. fetida Nagane.Citation12) Although Ueda et al.Citation10) reported the presence of a cold-adapted cellulase complex from the same earthworm, we did not detect any cellulase complex.
Fig. 9. TLC analysis of the reaction products of purified rEfEG1.
It is difficult to classify earthworms belonging to the Eisenia genus, with the wrong species name often being used over many years, as shown for E. fetida in the current study. At present, the name E. fetida is accidentally used for several species.Citation2) For instance, it is quite difficult to distinguish E. fetida from E. andrei by morphological analysis alone. The common name of E. fetida is the tiger earthworm, while that of E. andrei is the red earthworm. However, Lumbricus rubellus is also termed the “red earthworm,” leading to confusion.Citation2,22) In this study, we demonstrated that a mixture of E. fetida and E. andrei is present in commercial breeding farms. Although the characterization of EG in E. fetida Waki was similar to that in the E. fetida Nagane, enzyme activity toward CMC differed between the two strains, facilitating differentiation. Thus, before using earthworms for various types of scientific research, it is essential to carry out morphological and barcoding analyses to prevent species with different enzyme activities being confused. For instance, we found that E. fetida Waki contains many functional enzymes that may be important for medical application and biomass utilization. We are currently attempting to elucidate other functional enzymes in this strain for use in the bioindustry.
Author contributions
S.A. designed the study and wrote the manuscript. H.N. contributed to the development of the manuscript. Y.I., J.Y., K.Y., T.K., and H.N. performed the experiments. W.O. and Y.M. supervised all the enzyme experiments and analyzed the data. All authors have read and approved the final manuscript.
Disclosure statement
The authors declare that they have no conflict of interests to declare.
Funding
This work was supported by Grant-in-Aid for Japan Science and Technology Agency (JST) on Adaptable and Seamless Technology transfer Program (A-STEP) through target-driven R&D [grant number 231Z03721], [grant number AS251Z02531L].
Acknowledgments
We thank Yuki M. for advice on gene cloning, Kaneko N. and Minamiya Y. for advice on phylogenetic analysis, and Shida Y. and Eugene R. for their kind advice. We also thank all the laboratory members for their assistance during the study.
Related Research Data
References
- Aira M, Monroy F, Dominguez J. Earthworms strongly modify microbial biomass and activity triggering enzymatic activities during vermicomposting independently of the application rates of pig slurry. Sci. Total Environ. 2007;385:252–261.10.1016/j.scitotenv.2007.06.031
- Domínguez J, Velando A, Ferreiro A. Are Eisenia fetida (Savigny, 1826) and Eisenia andrei Bouché (1972) (Oligochaeta, Lumbricidae) different biological species? Pedobiologia. 2005;49:81–87.10.1016/j.pedobi.2004.08.005
- Pérez-Losada M, Ricoy M, Marshall JC, Domínguez J. Phylogenetic assessment of the earthworm Aporrectodea caliginosa species complex (Oligochaeta: Lumbricidae) based on mitochondrial and nuclear DNA sequences. Mol. Phylogenet. Evol. 2009;52:293–302.10.1016/j.ympev.2009.04.003
- Dvořák J, Park JY, Mančíková V, et al. Microbial environment affects innate immunity in two closely related earthworm species Eisenia andrei and Eisenia fetida. PLoS One. 2013;8:e79257.
- Yuan X, Cao C, Shan Y, Zhao Z, Chen J, Cong Y. Expression and characterization of earthworm Eisenia foetida Lumbrokinase‐3 in Pichia pastoris. Prep. Biochem. Biotechnol. 2006;36:273–279.10.1080/10826060600716703
- Zhao J, Pan R, He J, Liu Y, Li D-F, He R-Q. Eisenia fetida protease-III-1 functions in both fibrinolysis and fibrogenesis. J. Biomed. Biotechnol. 2007;2007:97654–97664.
- Akazawa S, Wakimoto S, Watanabe T. Japan patent 5548931. 2014 May 30.
- Akazawa S, Wakimoto S, Watanabe T. Japan patent 5505750. 2014 Mar 28.
- Palmarola-Adrados B, Chotěborská P, Galbe M, Zacchi G. Ethanol production from non-starch carbohydrates of wheat bran. Biores. Technol. 2005;96:843–850.10.1016/j.biortech.2004.07.004
- Ueda M, Goto T, Nakazawa M, Miyatake K, Sakaguchi M, Inouye K. A novel cold-adapted cellulase complex from Eisenia foetida: characterization of a multienzyme complex with carboxymethylcellulase, β-glucosidase, β-1,3 glucanase, and β-xylosidase. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2010;157:26–32.10.1016/j.cbpb.2010.04.014
- Arimori T, Ito A, Nakazawa M, Ueda M, Tamada T. Crystal structure of endo-1,4-β-glucanase from Eisenia fetida. J. Synchrotron Radiat. 2013;20:884–889.10.1107/S0909049513021110
- Ueda M, Ito A, Nakazawa M, Miyatake K, Sakaguchi M, Inouye K. Cloning and expression of the cold-adapted endo-1,4-β-glucanase gene from Eisenia fetida. Carbohydr. Polym. 2014;101:511–516.10.1016/j.carbpol.2013.09.057
- Novo M, Almodóvar A, Fernández R, Giribet G, Díaz Cosín DJ. Understanding the biogeography of a group of earthworms in the Mediterranean basin—the phylogenetic puzzle of Hormogastridae (Clitellata: Oligochaeta). Mol. Phylogenet. Evol. 2011;61:125–135.10.1016/j.ympev.2011.05.018
- Farkaš V, Lišková M, Biely P. Novel media for detection of microbial producers of cellulase and xylanase. FEMS Microbiol. Lett. 1985;28:137–140.10.1111/fml.1985.28.issue-2
- Nakazawa H, Okada K, Kobayashi R, et al. Characterization of the catalytic domains of Trichoderma reesei endoglucanase I, II, and III, expressed in Escherichia coli. Appl. Microbiol. Biotechnol. 2008;81:681–689.10.1007/s00253-008-1667-z
- Somogyi M. Notes on sugar determination. J. Biol. Chem. 1952;195:19–23.
- Van Soest P, McQueen R. The chemistry and estimation of fibre. Proc. Nutr. Soc. 1973;32:123–130.10.1079/PNS19730029
- Bremner J. Determination of nitrogen in soil by the Kjeldahl method. J. Agric. Sci. 1960;55:11–33.10.1017/S0021859600021572
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254.10.1016/0003-2697(76)90527-3
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685.10.1038/227680a0
- Minamiya Y, Ohga K, Hayakawa H, Ito K, Shimano S, Fukuda T. Coelomic fluid: a noninvasive source of DNA in earthworms. Mol. Ecol. Resour. 2011;11:645–649.10.1111/men.2011.11.issue-4
- Pérez-Losada M, Eiroa J, Mato S, Domínguez J. Phylogenetic species delimitation of the earthworms Eisenia fetida (Savigny, 1826) and Eisenia andrei Bouché, 1972 (Oligochaeta, Lumbricidae) based on mitochondrial and nuclear DNA sequences. Pedobiologia. 2005;49:317–324.10.1016/j.pedobi.2005.02.004
- Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 1994;3:294–299.
- Minamiya Y, Hayakawa H, Ohga K, Shimano S, Ito MT, Fukuda T. Variability of sexual organ possession rates and phylogenetic analyses of a parthenogenetic Japanese earthworm, Amynthas vittatus (Oligochaeta: Megascolecidae). Genes Genet. Syst. 2011;86:27–35.10.1266/ggs.86.27
- Nozaki M, Miura C, Tozawa Y, Miura T. The contribution of endogenous cellulase to the cellulose digestion in the gut of earthworm (Pheretima hilgendorfi: Megascolecidae). Soil Biol. Biochem. 2009;41:762–769.10.1016/j.soilbio.2009.01.016
- Park JY, Seyama T, Shiroma R, et al. Efficient recovery of glucose and fructose via enzymatic saccharification of rice straw with soft carbohydrates. Biosci. Biotech. Biochem. 2009;73:1072–1077.10.1271/bbb.80840
- Blue F, Mitta G, Leroux R, et al. The strong induction of metallothionein gene following cadmium exposure transiently affects the expression of many genes in Eisenia fetida: a trade-off mechanism? Comp. Biochem. Physiol. Part C. 2007;144:334–341.
- Van Arsdell JN, Kwok S, Schweickart VL, Ladner MB, Gelfand DH, Innis MA. Cloning, characterization, and expression in Saccharomyces cerevisiae of endoglucanase I from Trichoderma reesei. Bio/Technol. 1987;5:60–64.10.1038/nbt0187-60
- Saloheimo M, Lehtovaara P, Penttilä M, et al. EGIII, a new endoglucanase from Trichoderma reesei: the characterization of both gene and enzyme. Gene. 1988;63:11–21.10.1016/0378-1119(88)90541-0
- Nakazawa H, Okada K, Onodera T, Ogasawara W, Okada H, Morikawa Y. Directed evolution of endoglucanase III (Cel12A) from Trichoderma reesei. Appl. Microbiol. Biotechnol. 2009;83:649–657.10.1007/s00253-009-1901-3
- Watanabe H, Nakamura M, Tokuda G, Yamaoka I, Scrivener AM, Noda H. Site of secretion and properties of endogenous endo-β-1,4-glucanase components from Reticulitermes speratus (Kolbe), a Japanese subterranean termite. Insect Biochem. Mol. Biol. 1997;27:305–313.10.1016/S0965-1748(97)00003-9
- Suzuki K, Ojima T, Nishita K. Purification and cDNA cloning of a cellulase from abalone Haliotis discus hannai. Eur. J. Biochem. 2003;270:771–778.10.1046/j.1432-1033.2003.03443.x
- Kim N, Choo YM, Lee KS, et al. Molecular cloning and characterization of a glycosyl hydrolase family 9 cellulase distributed throughout the digestive tract of the cricket Teleogryllus emma. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2008;150:368–376.10.1016/j.cbpb.2008.04.005
- Lee SJ, Kim SR, Yoon HJ, et al. cDNA cloning, expression, and enzymatic activity of a cellulase from the mulberry longicorn beetle, Apriona germari. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2004;139:107–116.
- Lee SJ, Lee KS, Kim SR, et al. A novel cellulase gene from the mulberry longicorn beetle, Apriona germari: gene structure, expression, and enzymatic activity. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2005;140:551–560.10.1016/j.cbpc.2004.12.003
- McNabb DS, Xing Y, Guarente L. Cloning of yeast HAP5: a novel subunit of a heterotrimeric complex required for CCAAT binding. Genes Dev. 1995;9:47–58.10.1101/gad.9.1.47
- Dowzer CEA, Kelly JM. Analysis of the creA gene, a regulator of carbon catabolite repression in Aspergillus nidulans. Mol. Cell. Biol. 1991;11:5701–5709.