
Abstract
Six α-monoglucosyl derivatives of ginsenoside Rg1 (G-Rg1) were synthesized by transglycosylation reaction of rice seed α-glucosidase in the reaction mixture containing maltose as a glucosyl donor and G-Rg1 as an acceptor. Their chemical structures were identified by spectroscopic analysis, and the effects of reaction time, pH, and glycosyl donors on transglycosylation reaction were investigated. The results showed that rice seed α-glucosidase transfers α-glucosyl group from maltose to G-Rg1 by forming either α-1,3 (α-nigerosyl)-, α-1,4 (α-maltosyl)-, or α-1,6 (α-isomaltosyl)-glucosidic linkages in β-glucose moieties linked at the C6- and C20-position of protopanaxatriol (PPT)-type aglycone. The optimum pH range for the transglycosylation reaction was between 5.0 and 6.0. Rice seed α-glucosidase acted on maltose, soluble starch, and PNP α-D-glucopyranoside as glycosyl donors, but not on glucose, sucrose, or trehalose. These α-monoglucosyl derivatives of G-Rg1 were easily hydrolyzed to G-Rg1 by rat small intestinal and liver α-glucosidase in vitro.
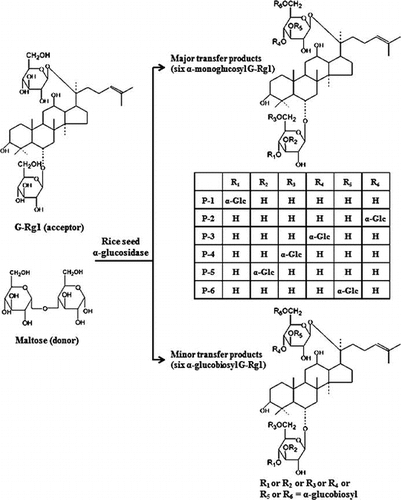
Graphical abstract
Chemical structures of transglycosylation products from maltose to ginsenoside Rg1 by rice seed α-glucosidase.
The root and their processed products of Panax ginseng C. A. Meyer, commonly known as ginseng, have been used as folk medicine to cure diseases and maintain health for thousands of years in the Oriental countries such as Korea, China, and Japan. Ginseng saponins, namely ginsenosides, have come to be regarded as major active components responsible for the pharmacological effects of ginseng. So far, more than 40 ginsenoside derivatives have been identified from roots and processed P. ginseng products.Citation1) The pharmacological effects of ginsenosides frequently depend on the type or site of sugar moieties linked in dammarane- or oleanane-type aglycones. Therefore, the structural modification may contribute to the multiple pharmacological effects of ginsenosides. Although various methods, such as mild acid hydrolysis,Citation2,3) alkaline cleavage,Citation4) and microbial and enzymatic degradation,Citation5–8) have been previously reported to modulate the structures of naturally occurring ginsenosides, microbial or enzymatic hydrolysis method is considered to be more favorable than conventional chemical methods. This is due to its performance under milder reaction conditions, simplicity of reaction step, and high regio- and diastereo-selectivities.Citation9)
Enzymatic transglycosylation by carbohydrases has also been used for the improvement of chemical and physiological properties of various compounds. The glycosylation of biological compounds such as sugars, vitamins, flavonoids, phenolics, saponins, and natural glycosides increased the solubility in water, stability against heat or oxidation stability, or physiological activities.Citation10–13) Glycosidase especially (also called glycoside hydrolases or glycosyl hydrolases) are distributed widely in almost all living organisms including microbial, plants, and animals. These enzymes catalyze the transglycosylation reaction; that is, they synthesize glycosides by the transfer of glycosyl units to other hydroxylated compounds, as well as catalyze the cleavage of glucosyl residue from the non-reducing ends of oligo- and polysaccharide chains.Citation13,14) There are a few reports dealing with regioselective glycosylation and acylation of ginsenosides by enzymatic methods.Citation14–16) In our previous report,Citation17,18) we reported the transglycosylation of G-Rb1, G-Rc, G-Re, and G-Rg1 by cyclomaltodextrin glucanotransferase (CGTase) from Bacillus (B) stearothermophilus, enzymatic synthesis of new β-xylosyl derivatives by Tricoderma viride β-glucosidase, and Aspergillus oryzae β-galactosidase. The glycosylation of ginsenosides have advantages over original compounds in their solubility in water and reduction of a bitter taste.Citation17)
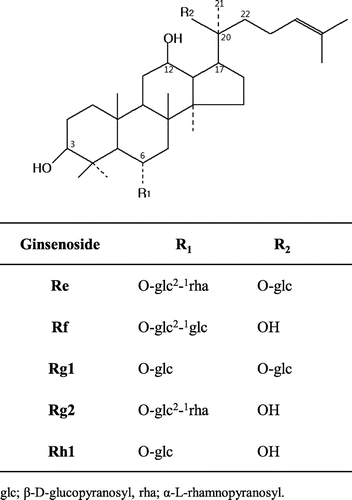
G-Rg1 [6-O-β-D-glucopyranosyl-20-O-β-D-glucopyranosyl-20(S)-protopanaxatriol] is one of the major protopanaxatriol (PPT)-type saponins (Fig. ) in root of P. ginseng and has been reported to possess various pharmacological activities such as estrogen-like activities,Citation19,20) cardioprotective effects,Citation21) hepatoprotective effects,Citation22) and neuroprotective effects.Citation23) During our investigation on the transglycosylation of ginseng saponins by various carbohydrases, commercial α-glucosidase prepared from rice seed was found to catalyze transglycosylation reaction of α-glucosyl moiety from maltose to G-Rg1. α-Glucosidase (EC 3.2.1.20) catalyzes the splitting of an α-glucosyl residue from the nonreducing terminal of substrate to liberate α-glucose and also catalyzes the transglycosylation to synthesize α-glucosylated compounds. It was also reported that rice seed α-glucosidase has transglycosylation properties, and there are a few studies dealing with the enzymatic synthesis of a more useful compounds using transglycosylation reaction of rice seed α-glucosidase obtained from commercial source.Citation24–26) Therefore, we report herein on the enzymatic formation of a series of new α-glucosyl derivatives of G-Rg1 by transglycosylation action of rice seed α-glucosidase, and the effects of reaction condition on the transglycosylation.
Fig. 1. Chemical structures of protopanaxatriol-type ginsenosides.
Materials and methods
Materials
G-Rg1 was isolated from fresh roots of P. ginseng by usual procedure.Citation27) The purified G-Rg1 was identified by comparison with spectral data,Citation17) and retention time by HPLC with an authentic sample. G-Rh1 and G-Rg2 were kindly donated by the Korea Ginseng Corporation Research Institute (Daejon, Republic of Korea). Commercial rice seed α-glucosidase type V (G-9259) was purchased from Sigma-Aldrich Co. (St. Louis, MO, USA) and was used without further purification. p-Nitrophenol (PNP), PNP α-D-glucopyranoside, maltose, soluble starch, sucrose, trehalose, and glucose were purchased from Sigma-Aldrich Co. (St. Louis, MO, USA). Silica gel 60 (70–230 mesh), pre-coated silica gel 60 F254 TLC plates (0.25 mm), and pyridine-d5 were purchased from Merck Co. (Darmstadt, Germany). Other reagents used were of analytical grade from commercial sources.
Preparation of crude enzyme containing α-glucosidase from rat’s small intestine and liver
The small intestine and liver of male Sprague–Dawley rats, weighing 230–250 g, were supplied from the Division of Biotechnology, Chonbuk National University, Iksan, Republic of Korea. The animals were euthanized under ether anesthesia and bled from the heart. Small intestine and liver regions were separately removed, rinsed with cold saline, and homogenized in four volumes of 0.1 M sodium phosphate buffer (pH 7.0) with a homogenizer. The homogenates were filtered through four layers of gauze to remove particles, and filtrate was centrifuged at 9000 × g for 20 min at 4 °C. The solid ammonium sulfate was added in supernatant at 30% saturation and then to eventually 90% saturation. After centrifugation at 9000 × g for 20 min at 4 °C, the precipitate was dissolved in the 10-mM sodium phosphate buffer (pH 7.0). After dialysis overnight, the solution was centrifuged at 9000 × g for 20 min at 4 °C, and the supernatant was lyophilized.
Assay of α-glucosidase activity
α-Glucosidase activity was assayed using PNP α-D-glucopyranoside as a substrate, according to the method of Iwata et al. Citation28) with slight modification. A reaction mixture (1.2 mL) containing 1.0 mL of PNP α-D-glucopyranoside (6 mM) in 0.1 M sodium acetate buffer (pH 5.3), 0.2 mL of diluted enzyme solution, was incubated for 30 min at 37 °C. The reaction was terminated by the addition of 1.0 mL of 0.1 M Na2CO3 solution. The released PNP was measured immediately using UV–visible spectrophotometer (UV-1601, Shimadzu, Kyoto, Japan) at 400 nm. One unit of α-glucosidase activity was defined as the amount of enzyme releasing 1 μmole of PNP per min under the assay condition.
Enzyme characterization
To investigate time course on transglycosylation by rice seed α-glucosidase, the reaction mixture (5.0 mL) containing 138 mg of G-Rg1 in 0.5 mL methanol, 138 mg of maltose, 7.1 or 14.2 U of rice seed α-glucosidase in 0.1 M sodium acetate buffer (pH 5.3) was incubated for 36 h at 50 °C with gentle shaking. The reaction mixture (0.5 mL) was withdrawn at regular time intervals and was extracted twice with 2.0 mL of water-saturated n-butanol. The n-butanol extract was concentrated to dryness in vacuo. The residue was dissolved in 1.0 mL of methanol and was applied on TLC and HPLC analyses.
The effect of pH on transglycosylation activity of rice seed α-glucosidase was examined in the following buffer (each at 0.1 M): sodium acetate (pH 4.0–5.5), sodium phosphate (pH 6.0 and pH 7.0), Tris-HCl (pH 8.0 and pH 9.0), and glycine-NaOH (pH 10.0). The 2.0 mL of reaction mixture containing 10 mg of G-Rg1 in 0.2 mL methanol, 10 mg of maltose, 2.5 U of rice seed α-glucosidase, and buffer solution was incubated for 2 h at 50 °C with gentle shaking. The effects of glycosyl donors were examined with the reaction mixtures (2.0 mL) containing 10 mg of G-Rg1 in 0.2 mL methanol, 10 mg of each glycosyl donor, 2.5 U of rice seed α-glucosidase, and 0.1 M sodium acetate buffer (pH 5.3). The mixture was incubated for 2 h at 50 °C with gentle shaking. The concentration of transglycosylation products in the reaction mixtures was calculated from the calibration curves of standard mixtures, and all experiments were performed in triplicate.
Hydrolysis of transglycosylation products by α-glucosidase from rat’s small intestine and liver
The reaction mixture (3.0 mL) containing 10.0 mg of each transglycosylation products of G-Rg1 and enzyme preparations from rat’s small intestine (1.0 U or 5.0 U as α-glucosidase) or liver (0.5 U), and 0.1 M sodium acetate buffer (pH 5.3) were incubated for 20 h at 50 °C, considering that the optimum temperature of transglycosylation reaction and maltose hydrolysis of rat intestinal α-glucosidase are between 50 and 60 °C.Citation26) The reaction mixture (0.3 mL) was withdrawn at regular time intervals, and was mixed with 0.7 mL of methanol to inactivate the enzyme, and then was applied on HPLC analysis.
TLC and HPLC analyses
TLC was performed on silica gel 60 F254 plates with chloroform–methanol–water (65:35:10, v/v/v, lower phase) as the developing solvent. After being sprayed with 10% sulfuric acid in ethanol, the spots on the TLC were detected by heating for 10 min at 110 °C. HPLC analysis was performed under the following conditions using Waters 600 controller and 600 pump, 717 plus auto-sampler, 486 UV detector (Milford, MA, USA): column, Halo C18 column (4.6 mm × 100 mm, 2.7 μm, Advanced Materials Technology, Inc., NC, USA); flow rate, 0.5 mL/min; solvent system, acetonitrile–water (19:81, v/v); detector, UV (203 nm).
Instrumental analysis
The melting point (mp) was measured with a Fisher-Johns mp apparatus (Cole-Parmer, Vernon Hills, IL, USA). Optical rotation value was measured in methanol with Jasco model DIP-1000 polarimeter (Jasco, Easton, MD, USA). Negative ESI-MS was performed on an Agilent 6410 triple quadrupole LC/MS (Agilent Technologies, Santa Clara, CA, USA). NMR spectra were taken on a Bruker model AMX 400 spectrometer (Bruker, Billerica, MA, USA) at 400 MHz (1H NMR) and 100 MHz (13C NMR) in pyridine-d5 with tetramethylsilane as an internal standard.
Isolation and identification of transglycosylation products
Rice seed α-glucosidase (28.4 U) was incubated with a mixture (50 mL) containing 1.4 g of maltose and 1.4 g of G-Rg1 in 0.1 M sodium acetate buffer (pH 5.3) for 2 h at 50 °C with gentle stirring. After heating for 10 min in a boiling water bath, the reaction mixture was extracted with water-saturated n-butanol. The extract was then concentrated to dryness in vacuo. The concentrate was chromatographed on a silica gel column using chloroform–methanol–water (80:20:2 → 60:35:10, v/v/v, lower phase). Further isolation and purification was performed using semi-prep HPLC using a μBondapak C18 column (7.8 mm × 300 mm, 10 μm, Waters, Milford, MA, USA) with isocratic 19% acetonitrile in distilled water, and UV detector (203 nm) to obtain the compounds in pure states. The yields of α-monoglucosyl-like compounds, P-1, P-2, P-3, P-4, P-5, and P-6 were 90, 36, 44, 32, 56, and 44 mg, respectively. α-Glucobiosyl-like compounds, 2G-1–2G-6, were isolated in the same manner, but were not obtained in sufficient amounts to be identified by the NMR spectroscopy.
Acid hydrolysis of transglycosylation products
G-Rg1 and transglycosylation products were hydrolyzed in 50% aqueous acetic acid for 2 h at 70 °C. The hydrolyzates were neutralized by the addition of Na2CO3 solution, extracted with water-saturated n-butanol, concentrated, and identified by comparison with Rf values on TLC with those of authentic samples.
Results and discussion
Transglycosylation to G-Rg1 by rice seed α-glucosidase
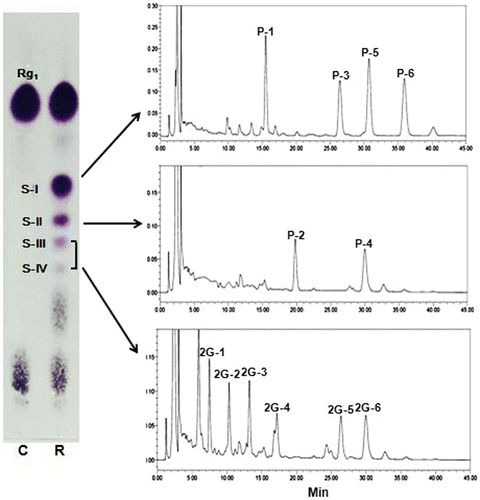
During our investigation on the glycosylation of ginseng saponins by various carbohydrases, rice seed α-glucosidase was found to catalyze transglycosylation reaction from maltose to G-Rg1. When rice seed α-glucosidase was incubated with maltose as a donor, and G-Rg1 as an acceptor, new spots having lower Rf values than that of G-Rg1 were detected on the TLC plate (Fig. ). These new spots were not observed when rice seed α-glucosidase was incubated in the reaction mixture containing G-Rg1 or maltose only. These results suggest that rice seed α-glucosidase catalyzed the transfer of α-glucosyl residues from maltose to G-Rg1 to form α-glucosyl-like compounds of G-Rg1.
Fig. 2. TLC and HPLC analyses of transglycosylation products from maltose to G-Rg1 by rice seed α-glucosidase.
Chemical structures of transglycosylation products
Each transglycosylated product formed by rice seed α-glucosidase was separated by silica gel column chromatography. Even though each of the separated compounds was detected as a single spot on a TLC plate, HPLC profiles showed that the single spot separated by the column chromatography was a mixture of two or four compounds (Fig. ). Considering that G-Rg1 used as an acceptor in this study have two β-glucosyl moieties in the PPT-type aglycone, these results suggest that it was due to the formation of various regioselective isomers. Subsequently, 4 compounds from spot I, and 2 compounds from spot II, and 6 compounds from the mixture of spot III and IV were isolated in a pure state by semi-prep HPLC chromatographic separation.
Compound P-1 was obtained as a white amorphous powder from n-butanol-methyethyl ketone (1:3, v/v); mp 210–212 °C (decomposed); [a]24D = +54.20° (c 0.1, MeOH). Negative ESI-MS spectrum of compound P-1 showed two quasi-molecular ion peaks at (m/z 961.7 [M−H]−, calcd. for C48H81O19: 961.5378) and (m/z 1007.6 [M−H+HCOOH]−-1-, calcd. for C49H83O21: 1007.5432). This result demonstrates the molecular formula of compound P-1 to be C48H82O19. 1H NMR spectrum of compound P-1 showed eight methyl signals assignable to an aglycone part at δH (pyridine-d5) 0.80, 1.02, 1.15, 1.60, 1.60, 1.61, 1.61, and 2.05 (all 3H, all s), three anomeric proton signals due to two β-glucosidic linkages at δH 4.90 (1H, d, J = 7.76 Hz, H-1 of 6-O-glucose), 5.18 (1H, d, J = 7.76 Hz, H-1 of 20-O-glucose), and one additional α-glucosidic linkage at δH 5.90 (1H, d, J = 3.78 Hz). Carbon-13 chemical shifts of compound P-1 were compared with those of G-Rg1. As shown in Table , 13C NMR spectrum showed an additional anomeric carbon signal at δC 103.0 due to α-glucosidic linkage with two anomeric carbon signals due to β-glucosidic linkages at δC 98.2 and 105.7. The carbon signal assignable to C4′ in glucose moiety linked at the C6-position of aglycone was shifted downfield by δC 9.6, while those of C3′ and C5′ was shifted upfield by δC 0.6, and downfield by δC 0.3, respectively. The carbon signals in β-glucose moiety linked at the C20-position of aglycone remained unshifted. It is well known that the treatment of ginsenosides in 50% aqueous acetic acid for 2 h at 70 °C leads to the elimination of sugar moieties, accompanied by subsequent dehydration and epimerization at C-20 position of aglycone in ginsenosides. This results in the production of 20 (R&S)-prosapogenin and ⊿20-prosapogenin.Citation29) The hydrolysis of G-Rg1 and compound P-1 in 50% aqueous acetic acid produced 20 (R&S)-prosapogenin and ⊿20-prosapogenin. As shown in Fig. , the Rf values on TLC of the prosapogenins obtained from compound P-1, after hydrolysis in 50% aqueous acetic acid, were lower than those from G-Rg1, but were similar to that of G-Rg2. These results showed that the α-glucosylation site was present at C4′ of glucose moiety linked at the C6-position of aglycone. Therefore, compound P-1 was identified as 6-O-[α-D-glucopyranosyl (glcp)-(1 → 4)-β-D-glcp]-20-O-β-D-glcp-20(S)-protopanaxatriol. This compound was previously reported as one of the transglycosylation products from dextrin and G-Rg1 by B. stearothermophilus CGTase.Citation17)
Table 1. 13C-NMR chemical shifts of α-monoglucoyl derivatives of G-Rg1 isolated from reaction mixture of maltose and G-Rg1 using rice seed α-glucosidase.
Fig. 3. TLC analysis of hydrolyzates of transglycosylation products in 50% aqueous acetic acid.
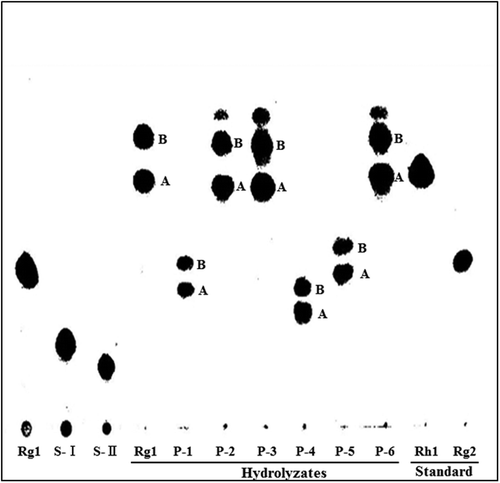
Compound P-2 was also obtained as a white amorphous powder from ethanol-ethyl acetate (1:1, v/v); mp 205–208 °C (decomposed); [a]24D = +52.11° (c 0.5, MeOH). ESI-MS spectrum of compound P-2 showed two quasi-molecular ion peaks at (m/z 961.7 [M−H]−, calcd. for C48H81O19: 961.5378) and (m/z 1007.7 [M−H + HCOOH]−, calcd. for C49H83O21: 1007.5432). 1H NMR spectrum of compound P-2 showed eight methyl signals assignable to an aglycone part at δH 0.80, 1.02, 1.15, 1.60, 1.60, 1.62, 1.62, and 2.05 (all 3H, all s), three anomeric proton signals due to two β-glucosidic linkages at δH 5.03 (1H, d, J = 7.75 Hz), 5.10 (1H, d, J = 7.62 Hz), and one α-glucosidic linkage at δH 5.47 (1H, d, J = 3.37 Hz). 13C-chemical shifts of compound P-2 showed an additional anomeric carbon signal at δC 100.4 due to α-glucosidic linkage with two anomeric carbon signals due to β-glucosidic linkages at δC 97.9 and 105.9, respectively. A carbon signal assignable to C5′ and C6′ in glucose moiety, linked at the C20-position of aglycone, was shifted downfield by δC 1.9 and 5.2, respectively. The carbon signals in β-glucose moiety linked at the C6-position of aglycone remained unshifted. The Rf values by TLC of the prosapogenins obtained from compound P-2 after hydrolysis in 50% aqueous acetic acid were the same as those from G-Rg1, and similar to that of G-Rh1 (Fig. ). These results show that the α-glucosylation site of compound P-2 was present at C6′ of glucose moiety, linked at the C20-position of aglycone. Compound P-2 was identified as 6-O-β-D-glcp-20-O-[α-D-glcp-(1 → 6)-(β-D-glcp)]-20(S)-protopanaxatriol.
Compound P-3 was obtained as a white amorphous powder from ethanol-ethyl acetate (1:2, v/v); mp 218–220 °C (decomposed); [a]24D = +57.29° (c 0.1, MeOH). ESI-MS spectrum of compound P-3 showed quasi-molecular ion peaks at (m/z 961.7 [M−H]−, calcd. for C48H81O19: 961.5378) and (m/z 1007.7 [M−H + HCOOH]−, calcd. for C49H83O21: 1007.5432), demonstrating the molecular formula of compound P-3 to be C48H82O19, which are the same as those of compounds P-1 and P-2. In 1H NMR spectrum, eight methyl signals were observed and assignable to an aglycone part at δH 0.81, 1.03, 1.17, 1.57, 1.58, 1.60, 1.63, and 2.09 (all 3H, all s), and three anomeric proton signals due to two β-glucosidic linkages at δH 4.95 (1H, d, J = 7.60 Hz) and δH 5.08 (1H, d, J = 7.89 Hz), and one α-glucosidic linkage at δH 5.88 (1H, d, J = 3.83 Hz). 13C-chemical shifts of compound P-3 showed an additional anomeric carbon signal at δC 102.9 due to α-glucosidic linkage with two anomeric carbon signals due to β-glucosidic linkages at δC 98.0 and 105.9, respectively. In 13C NMR chemical shifts, a carbon signal assignable to C4′ in glucose moiety, linked at the C20-position of aglycone, was shifted downfield by δC 9.7, while that of C3′ was shifted upfield by δC 2.6. The Rf values on TLC of the prosapogenins obtained from compound P-3 after hydrolysis in 50% aqueous acetic acid were the same as those from G-Rg1. These results imply that the α-glucosylation site of compound P-3 was also present at C4′ of glucose moiety, linked at the C20-position of aglycone. From these results, compound P-3 was determined as 6-O-β-D-glcp-20-O-[α-D-glcp-(1 → 4)-(β-D-glcp)]-20(S)-protopanaxatriol. This compound was already identified from the air-dried root of P. ginseng.Citation30)
Compound P-4 was obtained as a white amorphous powder from ethanol-ethyl acetate (1:1, v/v); mp 199–201 °C; [a]24D = +45.02° (c 1.0, MeOH) with a molecular formula C48H81O19 on the base of ESI-MS (m/z 961.3 [M−H]−, calcd. for C48H81O19: 961.5378) and 1007.7 [M−H+HCOOH]−, calcd. for C49H83O21: 1007.5432). In 1H NMR data, eight methyl signals assignable to an aglycone part were observed at δH 0.88, 0.98, 1.10, 1.54, 1.54, 1.57, 1.57, and 2.10, with three anomeric proton signals due to two β-glucosidic linkages at δH 5.08 (1H, d, J = 7.63 Hz), 5.17 (1H, d, J = 7.82 Hz), and one α-glucosidic linkage at δH 5.52 (1H, d, J = 3.74 Hz). 13C-chemical shifts of compound P-4 showed an additional anomeric carbon signal at δC 101.5 due to α-glucosidic linkage with two anomeric carbon signals, due to β-glucosidic linkages at δC 98.2 and 106.1, respectively. The 13C NMR chemical shifts of compound 4 revealed a carbon signal assignable to C6′ in glucose moiety linked at the C6-position of aglycone was shifted downfield by δC 6.3, while that of C5′ was shifted upfield by δC 0.5. The Rf value on TLC of the prosapogenin obtained from compound P-4 after acid hydrolysis was lower than those from acid hydrolyzates of G-Rg1 or compound P-1. From these results, it was apparent that the a-glucosylation site was present at C6′ of glucose moiety linked at the C6-position of aglycone. Compound P-4 was identified as 6-O-[α-D-glcp-(1 → 6)-β-D-glcp]-20-O-β-D-glcp-20(S)-protopanaxatriol.
Compound P-5: a white amorphous powder from n-butanol-methyethyl ketone (1:3, v/v); mp 208–210 °C; [a]24D = +46.60° (c 0.1, MeOH) with two quasi-molecular ion peaks at (m/z 961.7 [M−H]−, calcd. for C48H81O19: 961.5378) and (m/z 1007.7 [M−H+HCOOH]−, calcd. for C49H83O21: 1007.5432) in ESI-MS analyses, demonstrating the molecular formula to be C48H82O19. 1H NMR spectrum of compound P-5 showed eight methyl signals at δH 0.79, 1.04, 1.14, 1.50, 1.52, 1.60, 1,60, and 1.98 (all 3H, all s), three anomeric proton signals due to two β-glucosidic linkages at δH 4.94 (1H, d, J = 7.66 Hz) and 5.19 (1H, d, J = 7.76 Hz), and one additional α-glucosidic linkage at δH 5.92 (1H, d, J = 3.80 Hz). When 13C NMR chemical shifts of compound P-5 were compared with those of G-Rg1, a carbon signal assignable to C3′ in glucose moiety, linked at the C6-position of aglycone, was shifted downfield by δC 9.8, while those of C2′ and C4′ was shifted downfield slightly. The carbon signals in β-glucose moiety linked at the C20-position of aglycone remained unshifted. The Rf values on TLC of the prosapogenins obtained from this compound after acid hydrolysis were lower than those from G-Rg1, compound P-2, and P-3. From these results, the α-glucosylation site was present at C3′ of glucose moiety, linked at the C6-position of aglycone. Compound P-5 was identified as 6-O-[α-D-glcp-(1 → 3)-β-D-glcp]-20-O-β-D-glcp-20(S)-protopanaxatriol. This compound was previously reported as one of the transglycosylation products from dextrin and G-Rg1 by B. stearothermophilus CGTase,Citation17) and the presence of this compound in the air-dried root of P. ginseng was also reported.Citation30)
Compound P-6: a white amorphous powder from methanol-ethyl acetate (1:2, v/v); mp 220–223 °C (decomposed); [a]24D = +58.54° (c 1.0, MeOH). ESI-MS spectrum of compound P-6 showed quasi-molecular ion peaks at (m/z 961.7 [M−H]−, calcd. for C48H81O19: 961.5378) and (m/z 1007.7 [M−H + HCOOH]−, calcd. for C49H83O21: 1007.5432), demonstrating the molecular formula to be C48H82O19. In 1H NMR spectrum, eight methyl signals at δH 0.77, 1.00, 1.13, 1.55, 1.51, 1.61, 1.61, and 2.08 (all 3H, all s), three anomeric proton signals due to two β-glucosidic linkages at δH 5.02 (1H, d, J = 7.73 Hz) and 5.11 (1H, d, J = 7.73 Hz), and one α-glucosidic linkage at δH 5.92 (1H, d, J = 3.66 Hz). 13C-chemical shifts of compound P-2 showed an additional anomeric carbon signal at δC 101.9 due to α-glucosidic linkage with two anomeric carbon signals, due to β-glucosidic linkages at δC 98.1 and 106.0, respectively. In 13C NMR chemical shifts, a carbon signal assignable to C2′, C3′, and C4′ in glucose moiety, linked at the C20-position of aglycone was shifted downfield by δC 0.4, 9.3, and 0.3, respectively. The carbon signals in β-glucose moiety linked at the C6-position of aglycone remained unshifted, demonstrating α-glucosylation site was present at C3′ of glucose moiety linked at the C20-position of aglycone. The Rf values on TLC of the prosapogenins obtained from compound P-6 were the same as those from G-Rg1, compound P-2 or P-3, and very similar to that of G-Rh1. Compound P-6 was identified as 6-O-β-D-glcp-20-O-[α-D-glcp-(1 → 3)-(β-D-glcp)]-20(S)-protopanaxatriol. This compound is presented in a trace amount in the air-dried root of P. ginseng.Citation30) Proposed chemical structures of six α-monoglucosyl derivatives identified from reaction mixture of maltose and G-Rg1 in this study are shown in Fig. .
Fig. 4. Proposed chemical structures of transglycosylation products from maltose to G-Rg1 by rice seed α-glucosidase.
In addition, a mixture of spot III and spot IV on TLC plate in Fig. was isolated by column chromatography. Further separation was performed by semi-prep HPLC chromatography using a reverse phase column to obtain six compounds (2G-1 to 2G-6). These compounds commonly showed quasi-molecular ion peaks at m/z 1123.5 [M−H]−, demonstrating the molecular formula to be C54H92O24 in ESI-MS analysis (Table ). These values were consistent with the calculated molecular masses (calcd m/z 1123.54) of glucobiosyl derivatives of G-Rg1 produced by linking one additional glucose molecule in monoglucosyl derivatives of G-Rg1. These results suggested that rice seed α-glucosidase catalyzed not only regioselective formation of α-monoglucosyl derivatives of G-Rg1, but also formation of glucobiosyl derivatives of Rg1 in the presence of maltose and G-Rg1. However, regioselectivity of glucobiosyl derivatives were not determined because the products were not obtained in sufficient amounts to be identified by the NMR spectroscopy.
Table 2. ESI-LC/MS data of α-glucobiosyl G-Rg1-like compounds isolated from reaction mixture of maltose and G-Rg1 with rice seed α-glucosidase.
Effect of reaction condition on transglycosylation
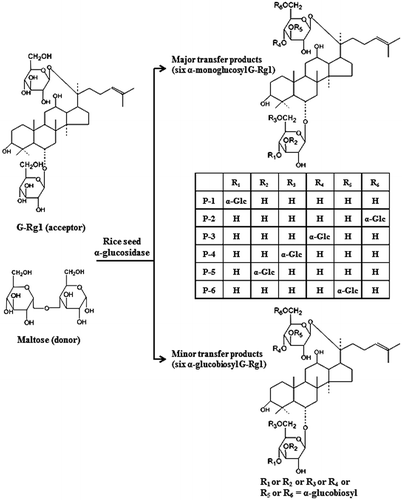
The effect of reaction time on the formation of transglycosylation products is shown in Fig. . A rapid formation of compound P-5 and P-6 occurred in the early stage (0.5–2.0 h) of the reaction, and thereafter decreased. Compound P-2 and P-4 successively increased during the reaction, and attained a maximum accumulation after 5–8 h reactions. These results showed that the transglycosylation ratio and the composition of reaction products were significantly influenced by reaction time. When reaction time was prolonged, the concentration of transglycosylation products were gradually decreased (Fig. ). The transglycosylation activity from maltose to G-Rg1 by rice seed α-glucosidase as a function of reaction pH is shown in Fig. , and the optimum pH ranges were between 5.0 and 5.5. These pH ranges were consistent with the optimum pH ranges for enzymatic syntheses of ascorbic acid 2-glucoside by transglycosylation with rice seed α-glucoside.Citation26)
Fig. 5. Time course of transglycosylation from maltose to G-Rg1 by rice seed α-glucosidase.
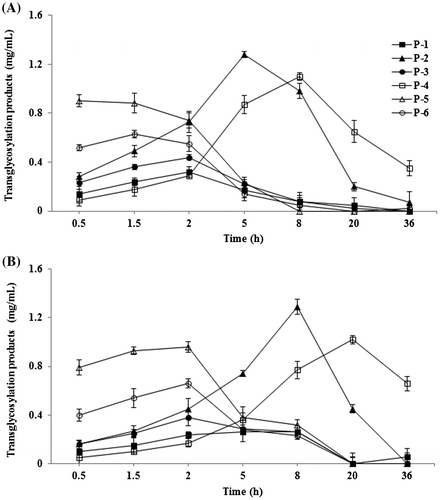
Fig. 6. Effects of pH value on the transglycosylation from maltose to G-Rg1 by rice seed α-glucosidase.
The effect of various sugars as glycosyl donor is given in Table . Rice seed α-glucosidase used maltose, soluble starch, and PNP α-D-glucopyranoside as a glucosyl donor, but not glucose, sucrose, and trehalose. This result also implies that rice seed α-glucosidase catalyzes transglycosylation reaction of α-glucosyl residue from maltose to C3′, C4′, or C6′ of β-glucose moieties linked at C6- and C20-positions of aglycone in G-Rg1 through α-1,3-glucosidic (α-nigerosyl), α-1,4-glucosidic (α-maltosyl), or α-1,6-glucosidic (α-isomaltosyl) linkages. Asano et al.Citation31) reported that rice α-glucosidase catalyzed the formation of PNP α-nigeroside, PNP α-maltoside, and PNP α-isomaltoside, when commercially available rice α-glucosidase was incubated in a reaction system containing PNP α-glucoside as an acceptor and maltose as a glucosyl donor. Plant α-glucosidase, buckwheat α-glucosidase catalyze formation of kojibiose, nigerose, and maltose from soluble starch as well as maltose,Citation32) and also spinach α-glucosidase I and IV catalyze formations of α-1,2, α-1,4, and α-1,6-glucosylated products in the reaction mixture containing 10% maltose.Citation33)
Table 3. Substrate specificity of rice seed α-glucosidase.
Hydrolysis of transglycosylation products by rat small intestine and liver α-glucosidases
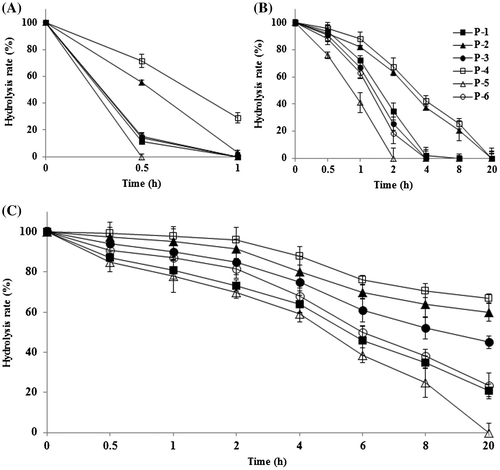
It is known that the cleavage of glycosidic linkages of ginsenosides after oral administration in human mainly takes place in the gastrointestinal tract caused either by gut microorganisms, intestinal enzymes, or gastric fluid, and the rate of glycosidic bond cleavage is closely related with the type and site of sugar moieties, and type of glycosidic bond.Citation34) Therefore, we investigated whether new α-monoglucosyl derivatives of G-Rg1 could be effectively hydrolyzed in vitro by rat small intestinal or rat liver α-glucosidase. As shown in Fig. , α-monoglucosyl derivatives of G-Rg1 were easily hydrolyzed by rat small intestinal or liver enzyme preparations containing α-glucosidase activity. These results imply that the compounds obtained in this study can be served as G-Rg1 sources through the hydrolysis by human small intestinal or liver α-glucosidase, and the subsequent absorption of released G-Rg1 after oral administration. The cleavage rate of α-monoglucosyl derivatives of α-1,6-glycosidic bond by rat small intestinal or liver α-glucosidase were slower than those for α-1,3- and α-1,4-glucosidic bonds, and α-glycosylation of G-Rg1 may influence absorption rate from human intestinal tract, metabolism, and pharmacological properties of G-Rg1. In fact, it is known that galactosylation can modulate the pharmacokinetics and biodistribution of natural glycosides by enhancing the hepatocyte uptake, and live targeting may have therapeutic implications due to the numerous postulated live-related modes of actions of ginsenosides.Citation14,35)
Fig. 7. Time course of G-Rg1 release from α-monoglucosyl derivatives of G-Rg1 by α-glucosidase preparation from rat’s small intestine and liver.
In conclusion, these results showed that rice seed α-glucosidase transfer α-glucosyl group from maltose to G-Rg1 by forming either α-1,3-, α-1,4- or α-1,6-glucosidic linkage in β-glucose moieties linked at the C6- and C20-position of aglycone. Although compounds P-3, P-5, and P-6 were previously reported to be presented with an extremely small amounts in air-dried root of P. ginseng,Citation30) compounds P-2 and P-4 are new compounds, which were not previously reported in natural ginseng, processed ginseng products, or any enzymatic transglycosylation products. Formation mechanism of these compounds in air-dried ginseng roots and their pharmacological activities are still unclear. Therefore, further studies on the pharmacological and physiological activities of these compounds are necessary, and new α-glucosyl derivatives of G-Rg1 obtained in this study will be used for these purposes.
Author contributions
MKK and YS conceived and designed the experiments. YHK carried out isolation and identification of products. MJK and GSS contributed HPLC analysis of products. YS, MJK, and YHK carried out characterization studies of enzyme. MKK prepared the manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
Funding
This work was supported by the Rural Development Administration, Republic of Korea [grant number PJ00985903].
References
- Ruan CC, Liu Z, Li X, et al. Isolation and characterization of a new ginsenoside from the fresh root of Panax ginseng. Molecules. 2010;15:2319–2325.10.3390/molecules15042319
- Han BH, Park MH, Han YN, et al. Degradation of ginseng saponins under mild acidic conditions. Planta Med. 1982;44:146–149.10.1055/s-2007-971425
- Kim MH, Lee YC, Choi SY, et al. The changes of ginsenoside patterns in red ginseng processed by organic acid impregnation pretreatment. J. Ginseng Res. 2011;35:497–503.10.5142/jgr.2011.35.4.497
- Chen YJ, Nose M, Ogihara Y. Alkaline cleavage of ginsenosides. Chem. Pharm. Bull. 1987;35:1653–1655.10.1248/cpb.35.1653
- Ko SR, Suzuki Y, Suzuki K, et al. Marked production of ginsenosides Rd, F2, Rg3, and compound K by enzymatic method. Chem. Pharm. Bull. 2007;55:1522–1527.10.1248/cpb.55.1522
- Park CS, Yoo MH, Noh KH, et al. Biotransformation of ginsenosides by hydrolyzing the sugar moieties of ginsenosides using microbial glycosidases. Appl. Microbiol. Biotechnol. 2010;87:9–19.10.1007/s00253-010-2567-6
- Yang XD, Yang YY, Ouyang DS, et al. A review of biotransformation and pharmacology of ginsenoside compound K. Fitoterapia. 2015;100:208–220.10.1016/j.fitote.2014.11.019
- Kim DH. Chemical diversity of Panax ginseng, Panax quinquifolium, and Panax notoginseng. J. Ginseng Res. 2012;36:1–15.10.5142/jgr.2012.36.1.1
- Wandrey C, Liese A, Kihumbu D. Industrial biocatalysis: past, present, and future. Org. Process Res. Dev. 2000;4:286–290.10.1021/op990101l
- Suzuki Y. Enzymatic synthesis of glycosides. J. Jpn Soc. Starch Sci. 1988;35:93–102.10.5458/jag1972.35.93
- Suzuki Y, Kim YH, Uchida K, et al. Enzymatic synthesis of glycosylated and phosphatidylated biologically active compounds. J. Appl. Glycosci. 1996;43:273–282. Japan.
- Riva S, Monti D, Luisetti M, et al. Enzymatic modification of natural compounds with pharmacological properties. Ann. N. Y. Acad. Sci. 1998;864:70–80.10.1111/nyas.1998.864.issue-1
- Thuan NH, Sohng JK. Recent biotechnological progress in enzymatic synthesis of glycosides. J. Ind. Microbiol. Biotechnol. 2013;40:1329–1356.10.1007/s10295-013-1332-0
- Danieli B, Falcone L, Monti D, et al. Regioselective enzymatic glycosylation of natural polyhydroxylated compounds: galactosylation and glucosylation of protopanaxatriol ginsenosides. J. Org. Chem. 2001;66:262–269.10.1021/jo001424e
- Gebhardt S, Bihler S, Schubert-Zsilavecz M, et al. Biocatalytic generation of molecular diversity: modification of ginsenoside Rb1 by β-1,4-galactosyltransferase and Candida antarctica lipase. Helv. Chim. Acta. 2002;85:1943–1959.10.1002/1522-2675(200207)85:7<1943::AID-HLCA1943>3.0.CO;2-T
- Teng R, Ang C, McManus D, et al. Regioselective acylation of ginsenosides by novozyme 435 to generate molecular diversity. Helv. Chim. Acta. 2004;87:1860–1872.10.1002/(ISSN)1522-2675
- Kim YH, Lee YG, Choi KJ, et al. Transglycosylation to ginseng saponins by cyclomaltodextrin glucanotransferases. Biosci. Biotechnol. Biochem. 2001;65:875–883.10.1271/bbb.65.875
- Ko SR, Suzuki Y, Kim YH, et al. Enzymatic synthesis of two ginsenoside Re-β-xylosides. Biosci. Biotechnol. Biochem. 2001;65:1223–1226.10.1271/bbb.65.1223
- Chan RYK, Chen WF, Dong A, et al. Estrogen-like activity of ginsenoside rg1 derived from Panax notoginseng. J. Clin. Endocrinol. Metab. 2002;87:3691–3695.10.1210/jcem.87.8.8717
- Lau WS, Chan RYK, Guo DA, et al. Ginsenoisde Rg1 exerts estrogen-like activities via ligand-independent activation of ERα pathway. J. Steroid Biochem. Mol. Biol. 2008;108:64–71.10.1016/j.jsbmb.2007.06.005
- Zhu D, Wu L, Li CR, et al. Ginsenoside Rg1 protects rat cardiomyocyte from hypoxia/reoxygenation oxidative injury via antioxidant and intracellular calcium homeostasis. J. Cell Biochem. 2009;108:117–124.
- Korivi M, Hou CW, Huang CY, et al. Ginsenoside-Rg1 protects the liver against exhaustive exercise-induced oxidative stress in rats. Evid. Based Complement. Altern. Med. 2012;2012:8 p. Article ID 932165.
- Fang F, Chen X, Huang T, et al. Multi-faced neuroprotective effects of ginsenoside Rg1 in an Alzheimer mouse model. Biochim. Biophys. Acta. 2012;1822:286–292.10.1016/j.bbadis.2011.10.004
- Muto N, Nakamura T, Yamamoto I. Enzymatic formation of a nonreducing L-ascorbic acid α-glucose: purification and properties of α-glucosidases catalyzing site-specific transglucosylation from rat small intestine. J. Biochem. 1990;107:222–227.
- Nakae T, Kometani T, Nishimura T, et al. Synthesis of monoacylglycerol monoglucoside by rice α-glucosidase and its foaming power. Food. Sci. Technol. Res. 1999;5:214–219.10.3136/fstr.5.214
- Muto N, Suga S, Fujii K, et al. Formation of a stable ascorbic acid 2-glucoside by specific transglucosylation with rice seed α-glucosidase. Agric. Biol. Chem. 1990;54:1697–1703.10.1271/bbb1961.54.1697
- Sanada S, Kondo N, Shoji J, et al. Studies on the saponins of ginseng. I. Structures of ginsenoside-Ro, -Rb1, -Rb2, -Rc and -Rd. Chem. Pharm. Bull. 1974;22:421–428.10.1248/cpb.22.421
- Iwata H, Suzuki T, Takahashi K, et al. Critical importance of α-glucosidase contained in rice kernel for alcohol fermentation of rice polish. J. Biosci. Bioeng. 2002;93:296–302.10.1016/S1389-1723(02)80031-7
- Kaku T, Kawashima Y. Isolation and characterization of ginsenosdie-Rg2, 20R-prosapogenin, 20S-prosapogenin and delta 20–prosapogenin. Chemical studies on saponins of Panax ginseng C. A. Meyer, third report. Arzneim. Forsch. 1980;30:936–943.
- Zhu GY, Li YW, Hau DKP, et al. Protopanaxatriol-type ginsenosides from the root of Panax ginseng. J. Agric. Food Chem. 2011;59:200–205.10.1021/jf1037932
- Asano N, Tanaka K, Matsui K. Enzymic synthesis of p-nitrophenyl α-glucobiosides by use of native and immobilized rice α-glucosidase. Carbohydr. Res. 1991;217:255–261.10.1016/0008-6215(91)84137-4
- Chiba S, Shimomura T. Transglucosylation of α-glucosidase. J. Jpn. Soc. Starch Sci. 1979;26:59–67.10.5458/jag1972.26.59
- Sugimoto M, Furui S, Sasaki K, et al. Transglucosylation activities of multiple forms of α-glucosidase from spinach. Biosci. Biotechnol. Biochem. 2003;67:1160–1163.10.1271/bbb.67.1160
- Tawab MA, Bahr U, Karas M, et al. Degradation of ginsenosides in humans after oral administration. Drug Metab. Dispos. 2003;31:1065–1071.10.1124/dmd.31.8.1065
- Marshall D, Pedley RB, Melton RG, et al. Galactosylated streptavidin for improved clearance of biotinylated intact and F(ab’)2 fragments of an anti-tumour antibody. Br. J. Cancer. 1995;71:18–24.10.1038/bjc.1995.5