
Abstract
Aplog-1 is a simplified analog of debromoaplysiatoxin (DAT) with potent tumor-promoting and proinflammatory activities. Aplog-1 and DAT exhibited anti-proliferative activities against several human cancer cell lines, whereas aplog-1 did not have tumor-promoting nor proinflammatory activities. We have recently found 10-methyl-aplog-1 (1) to have strong anti-proliferative activity compared with aplog-1. To further investigate the structural factors involved in the tumor-promoting, proinflammatory, and anti-proliferative activities, two dimethyl derivatives of aplog-1 (2, 3) were synthesized, where two methyl groups were installed at positions 4 and 10 or 10 and 12. 10,12-Dimethyl-aplog-1 (2) had stronger inhibitory effects on the growth of several human cancer cell lines than 1 and DAT, but exhibited no tumor-promoting and proinflammatory activities. In contrast, 4,10-dimethyl-aplog-1 (3) displayed weak tumor-promoting and proinflammatory activities along with anti-proliferative activity similar to that of 1 and DAT. Compound 2 would be the optimized seed for anticancer drugs among the simplified analogs of DAT.
Natural products have received considerable attention for their chemotherapeutic potential in the treatment of human diseases.Citation1) Of course, however, natural products themselves are not optimized for medicinal use, and, thus, often work both beneficially and adversely. Furthermore, their natural scarcity and structural complexity make it difficult to ensure sufficient supply for therapeutic use and study on structural optimization. Recent studies clarified the pleiotropic nature of natural bioactive ligands, that is, many natural products can be considered as “a bunch of keys” with multiple cellular targets.Citation2) Efforts to identify the structural factor (the particular key) required for each desirable effect may contribute to the development of medicinal leads from natural products. The function-oriented synthesis of simplified analogs of natural products represents one of the promising strategies to create new medicinal leads as exemplified by eribulin mesylate (Halaven®)Citation3) and picolog,Citation4) which are simplified analogs of anticancer compounds, halichondrin B,Citation5) and bryostatin 1 (bryo-1),Citation6) respectively.
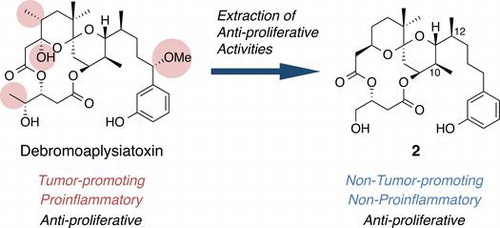
We are particularly interested in naturally occurring tumor promoters such as phorbol esters, teleocidins, and aplysiatoxins (ATX) (Fig. ), because they exhibit pleiotropic effects including tumor-promoting, proinflammatory, and anti-proliferative activities.Citation7) The main targets of these compounds are protein kinase C (PKC) isozymes,Citation8) a key enzyme family consisting of at least ten isozymes that are involved in the signal transduction pathways regulating cellular events, such as proliferation, differentiation, inflammation, and apoptosis.Citation9,10) PKC isozymes are also attractive targets in the treatment of Alzheimer’s diseaseCitation11) and acquired immunodeficiency syndrome (AIDS),Citation12) in addition to cancer.Citation13) Since the tumor promoters shown in Fig. are potent activators of PKC isozymes, they may have a potential as therapeutic leads for these intractable diseases. In fact, 12-O-tetradecanoylphorbol 13-acetate (TPA) and ingenol 3-angelate have been evaluated in clinical trials for some cancers regardless of the expected adverse effects of proinflammatory and tumor-promoting activities.Citation14,15) Like most of the natural products, they can be considered as “a bunch of keys,” and appropriate structural modification to and/or the simplification of these tumor promoters may decouple desirable functions from adverse effects. The finding that bryo-1, a potent PKC activator isolated from marine bryozoan Bugula neritina,Citation6) showed little tumor-promoting activityCitation16) encouraged us to develop tumor promoter-based anticancer lead compounds. In spite of its promising activity, availability of bryo-1 is limited due to its extremely low yields (ca. 1.3 ppm) from natural sourcesCitation17) and total synthesis of bryo-1 required 46 steps.Citation18) Therefore, synthetically more accessible PKC ligands without tumor-promoting activity would be necessary as bryo-1 surrogates.
Fig. 1. Structures of protein kinase C (PKC) activators occurring in nature.
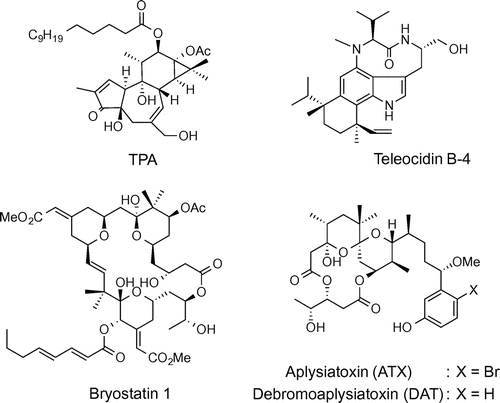
We recently developed aplog-1 (Fig. ), a simple analog of ATX and debromoaplysiatoxin (DAT),Citation19) potent tumor promoters and highly proinflammatory principles from sea hares,Citation7) as a possible surrogate of bryo-1.Citation20,21) Aplog-1 inhibited the proliferation of several human cancer cell lines with a potency similar to that of bryo-1, but did not exhibit tumor-promoting or proinflammatory activities in mouse skin. However, its anti-proliferative activity was too weak to be applicable as an anticancer lead. Therefore, the next step was to improve the anti-proliferative activity of aplog-1. The first approach was to modify the phenolic side chain. Our comprehensive structure–activity study on the side chain of aplog-1 indicated that optimal hydrophobicity (log p = 4.0–4.5) existed, and that the contribution of this moiety to the anti-proliferative activity was not so high.Citation22) This finding prompted us to focus on the spiroketal moiety of aplog-1.Citation23,24) The introduction of a chiral methyl group on the spiroketal moiety at position 10 of aplog-1 markedly enhanced its anti-proliferative potency as well as its affinity for the C1 domains of PKC isozymes, whereas this derivative (1) hardly enhanced tumorigenesis in the mouse skin or inflammation in the mouse ear. The introduction of a bromine atom into the phenol ring of 1 (21-bromo-10-methyl-aplog-1) slightly improved anti-proliferative activity, and did not enhance tumorigenesis in vivo even at 8.5 nmol application in the mouse, and hardly induced inflammation in the mouse ear at 170 nmol (Supplemental material).
Fig. 2. Structures of aplog-1, 10-methyl-aplog-1 (1), 10,12-dimethyl-aplog-1 (2), and 4,10-dimethyl-aplog-1 (3).
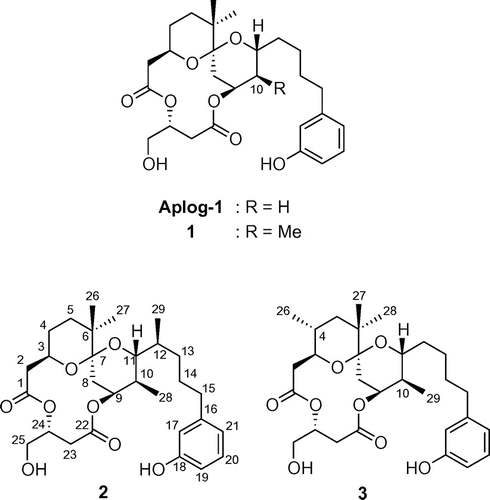
Fig. 3. “Fingerprints” against 26 human cancer cell lines of 10,12-dimethyl-aplog-1 (2), bryostatin-1, cisplatin, and methotrexate.
Fig. 4. EBV-EA production induced by TPA, DAT, 2, and 3.
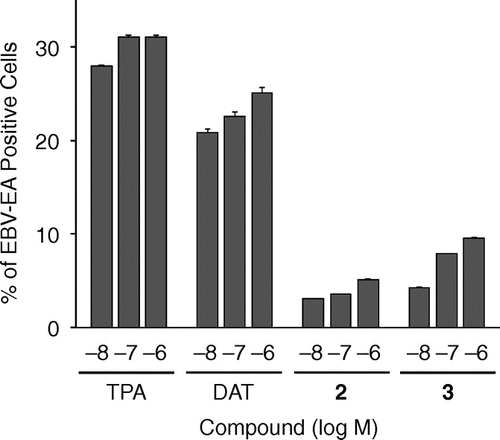
Fig. 5. Change in the relative weight of the mouse ear 24 h after the application of TPA, DAT, 1, 2, and 3.
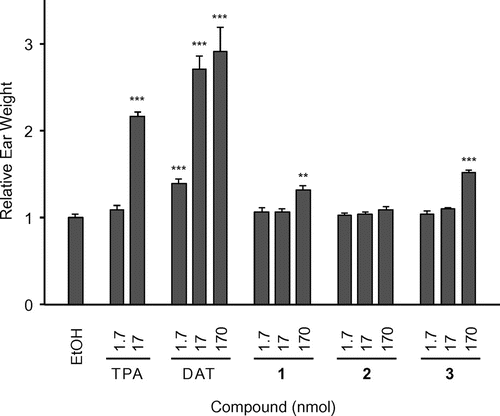
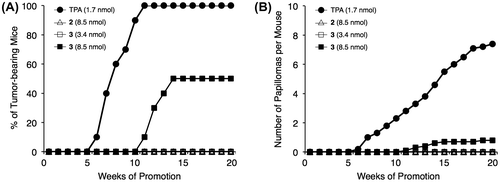
Fig. 6. Tumor-promoting activities of TPA (1.7 nmol), 2 (8.5 nmol), and 3 (3.4 and 8.5 nmol).
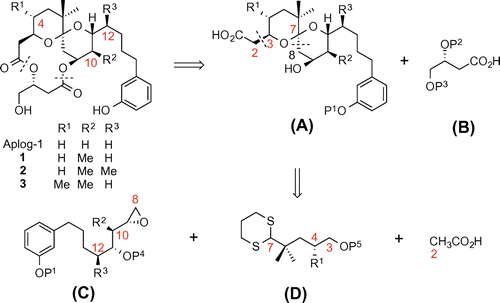
To further clarify the structural factors related to the tumor-promoting, proinflammatory, and anti-proliferative activities of DAT, and to optimize 1 as a medicinal lead, we synthesized two novel dimethyl derivatives of aplog-1 (2 and 3, Fig. ), in which the methyl groups were installed in the spiroketal moiety at positions 10 and 12 or 4 and 10 because these substituents may be responsible for the strong binding of DAT to PKC isozymes. Their biological data indicated that the difference in the position of a methyl group drastically changed the quality of the biological activities. We succeeded in the extraction of the anti-proliferative activities from pleiotropic DAT with tumor-promoting, proinflammatory, and anti-proliferative activities by the synthesis of 10,12-dimethyl-aplog1 (2), which would be a promising lead for anticancer drugs.
Results and discussion
Synthesis of 2 and 3
The macrocyclic structure of 2 and 3 can be constructed through convergent synthetic routes such as total syntheses of ATXCitation25) and 3-deoxy-DAT.Citation26) Scheme shows the retrosynthetic analysis of 1–3 along with that of aplog-1. The dissection of these compounds at the two-ester linkages gave subunits A and B. Fragment A provided fragments C and D, which were named as an epoxide unit and a dithiane unit, respectively. In many cases, a methyl group unexpectedly and markedly changes the reactivity of synthetic intermediates via steric hindrance and/or conformational changes, making it more difficult to obtain methyl-substituted derivatives. For example, in the synthesis of 2 (Scheme ), our initial attempts to construct the corresponding epoxide unit 11 using Smith’s iodocarbonate cyclization,Citation27) as previously reported for the synthesis of aplog-1,Citation20) resulted in an unsatisfactory yield and poor 1,3-syn-selectivity presumably because of some steric influence of the C-10 methyl group of fragment C. Moreover, in the synthesis of 4-methyl-aplog-1,Citation24) the C-4 methyl group hampered the proper formulation of the C-3 asymmetric center, and increased the stability of an undesired intermediate with a double anomeric spiroketal, which forced us to find another strategy or optimization of the reaction conditions.
Scheme 1. Retrosynthetic analysis of aplog-1, 10-methyl-aplog-1 (1), 10,12-dimethyl-aplog-1 (2), and 4,10-dimethyl-aplog-1 (3).
Scheme 2. Synthesis of 10,12-dimethyl-aplog-1 (2).a
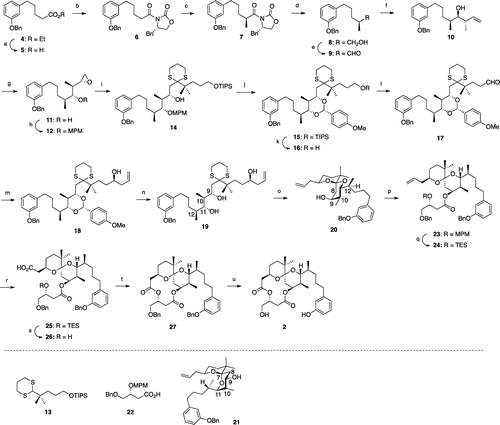
The assembly of a C8–C21 unit (fragment C) containing an anti, anti, syn-stereotetrad, which is required for the synthesis of 2, was particularly challenging because a reliable method has not yet been identified. We employed vanadium-catalyzed hydroxyl-directed epoxidation of the corresponding homoallylic alcohol (10). The synthesis of 2 was initiated from a previously reported ester (4).Citation20,28) After the alkaline hydrolysis of the ester group, the resultant acid was subjected to the Evans’ protocolCitation29); the α-chiral methyl group (S-configuration) was successfully constructed in 7. The removal of the Evans auxiliary using sodium borohydride provided an alcohol, which was oxidized to give aldehyde 9. Substrate-matched Brown’s asymmetric crotylationCitation30) produced the desired homoallylic alcohol (10) with an anti, syn-stereotriad in sufficient diastereoselectivity (>80% de). The treatment of tert-butyl hydroperoxide and catalytic amount of vanadyl acetylacetonate successfully gave the desired epoxide (11) as the main product. At this step, 11 was easily separated from any undesirables by medium pressure silica gel chromatography. The chiral HPLC analysis confirmed that the enantiomeric purity of 11 was greater than 95% (Supplemental material). The hydroxyl group was protected as an MPM ether to give the novel epoxide 12 with an anti, anti, syn-stereotetrad.
The coupling of 12 with the known dithiane 13Citation20) provided 14 according to the protocol of Ide and Nakata.Citation31) Oxidation of the MPM group by DDQ efficiently protected the secondary hydroxyl group as an anisylidene acetal, followed by deprotection of the TIPS group to give the primary alcohol 16. Following the oxidation of 16, Maruoka’s asymmetric allylation yielded the homoallylic alcohol 18. However, the acid hydrolysis of 18 gave the triol 19 at a moderate yield (27, 62% based on recovered 18), presumably because the two methyl groups at positions 10 and 12 restricted the orientation of a hydroxyl group at position 11 to stabilize the six-membered acetal moiety. The recovered acetal was, thus, recycled several times to increase the yield of 19. Cleavage of the dithiane moiety in 19 using Selectfluor® provided a desired single anomeric spiroketal (20) at excellent yields because the undesired double anomeric spiroketal (21) was unstable due to 1,3-diaxial interactions between the hydrogen atom at position 8 and the methyl group at position 10, and between the side chain at position 11 and the side chain at position 7, and the hydrogen atom at position 9. The subsequent construction of the diolide ring from 20 was achieved by procedures similar to those used for the synthesis of 10-methyl-aplog-1 (1).Citation24) Compound 2 was obtained in 22 steps from 4 with an overall yield of 0.63%.
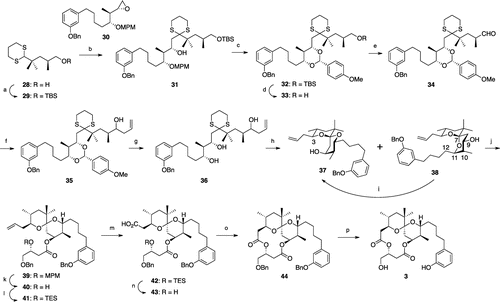
The synthesis of 4,10-dimethyl-aplog-1 (3) (Scheme ) began with the coupling of a Kishi’s dithiane (29)Citation25) and a known epoxide (30)Citation24) to give 31, which was converted to an anisylidene acetal (32) in a manner similar to the synthesis of 15. After the oxidation of 33, attempts to install an allyl group stereoselectively into the aldehyde employing Keck’s or Maruoka’s method proved to be unsuccessful, as reported in the synthesis of 4-methyl-aplog-1,Citation24) and this may have been because of steric hindrance around the aldehyde group. Therefore, a Grignard reaction was employed to produce a homoallylic alcohol (35) as a 1:1 diastereomeric mixture. Detachment of the anisylidene acetal and subsequent hydrolytic cleavage of the dithiane moiety gave three spiroketals (37, 38, and a diastereomer) in a ratio of approximately 1:1.2:2. Unlike the synthesis of 2, the undesired double anomeric spiroketal (38) was also obtained because the contribution of 1,3-diaxial interaction between the side chain at position 11 and the substituent at position 7, and the hydrogen atom at position 9 in 38 was low due to the lack of the methyl group at position 12. Subsequent construction of the diolide ring from 37 was achieved by procedures similar to those used for the synthesis of 1.Citation24) Compound 3 was obtained in 16 steps from 30 with an overall yield of 3.5%.
Scheme 3. Synthesis of 4,12-dimethyl-aplog-1 (3).a
Anti-proliferative activities of 2 and 3 against 39 human cancer cell lines
The analogs (2 and 3) were initially evaluated for their anti-proliferative activities in growth inhibition tests against 39 human cancer cell lines established by Yamori et al.Citation32) Growth inhibitory activity was expressed as GI50 (M), the concentration required to inhibit cell growth by 50% from an untreated control. The average log GI50 value for all 39 human cancer cells was expressed as MG-MID (mean-graph mid-point value). The MG-MIDs of 1, 2, 3, and DAT were −5.24, −5.51, −5.27, and −5.22, respectively. Ten cell lines with log GI50 values less than −6.00 in 2 are listed in Table . The average log GI50 values of 1, 2, 3, and DAT for these cell lines were −6.12, −6.73, −6.37, and −5.91, respectively. The log GI50 values for the other 29 cell lines are summarized in the Supplemental material.
Table 1. Growth inhibition of 1, 2, 3, and debromoaplysiatoxin (DAT) against aplog-sensitive human cancer cell lines.
As shown in Table , the inhibitory effects of 3 on the growth of half of the cell lines listed in Table were weaker than those of 1. On the other hand, the growth inhibitory activities of 2 for these cell lines, except for HCC2998 and NCI-H460 were stronger than those of 1. These results indicated that methyl groups not only at position 10 but also at position 12 played an important role in enhancing anti-proliferative activities.
Fig. shows the efficacy profiles (fingerprints) of 2, bryo-1, and two anticancer drugs against 26 cancer cell lines, information on which was available from the NCI/NIH public database.Citation33) Additional information is incorporated in the Supplemental material. The profiles of aplogs (1–3) were completely different from those of any type of anticancer agents previously developed, and these aplogs were highly specific to several cancer cell lines including HBC-4, MDA-MB-231, HCC2998, and NCI-H460, whereas the specificity of other anticancer agents were lower. These results suggested their unique mode of action and potency as new anticancer agents.
Tumor-promoting activities of 2 and 3
Compounds 2 and 3 may exhibit tumor-promoting activities because two additional methyl groups at positions 4 and 10 or 10 and 12 in aplog-1 have been suggested to increase the hydrophobicity of these molecules.Citation34,35) We evaluated the tumor-promoting activities of 2 and 3 by testing the induction of the Epstein–Barr virus early antigen (EBV-EA).Citation36,37) EBVs are under the strict control of host human B lymphoblastoid Raji cells. However, treatments with tumor promoters such as TPA activate these cells to produce the early antigen, which can be detected using an indirect immunofluorescence technique. EBV-EA-inducing activities were expressed as the percentage of EBV-EA-positive cells. DAT potently induced EBV-EA similar to TPA at 10−8 M, whereas 2 and 3 as well as 1 without tumor-promoting activityCitation24) more weakly induced EBV-EA (Fig. ). The induction by 2 with methyl groups at positions 10 and 12 was weaker than that by 3 with methyl groups at positions 4 and 10. These results suggested that neither 2 nor 3 might act as tumor promoters.
Previous studies indicated that chronic inflammation was closely related to tumor promotion.Citation38) Therefore, we evaluated the proinflammatory activities of 1–3 in the mouse ear (Fig. ). The ears of ICR mice were treated with various doses of each drug. Proinflammatory activity was expressed as an increase in relative weight of the ears by treatment with each drug. Although 1 and 3 weakly induced inflammation at 170 nmol, 2 was not proinflammatory even at this concentration. These results suggested that the methyl group at 12 suppressed this activity, and may be related to our previous finding in which 12,12-dimethyl-aplog-1 was found to be an antitumor promoter, similar to that of bryo-1.Citation39) The proinflammatory activity of DAT was significantly stronger than that of TPA.
The in vivo tumor-promoting activities of 2 and 3 were investigated in a two-stage carcinogenesis experiment on the mouse skin (Fig. ). The skin on the back of ICR mice was treated with a single dose of 390 nmol of 7,12-dimethylbenz[a]anthracene (DMBA) and, one week later, with 8.5 nmol of 2 or 3 (fivefold excess of TPA and DAT) or 3.4 nmol of 3 twice a week. In the positive control experiment using TPA (1.7 nmol), the first tumor appeared in week 6, and the percentage of tumor-bearing mice reached 100% by week 11. The number of papillomas/mouse was 7.4 in week 20. DAT also exhibited potent tumor-promoting activity, albeit weaker than that of TPA, and this activity was dose dependent at 1.7 and 3.4 nmol (the percentage of tumor-bearing mice: 80 and 90%; the number of papillomas/mouse: 2.4 and 4.7, respectively), as previously reported.Citation40) The activity of DAT was similar to that reported previously.Citation41) Although the application of 2 (8.5 nmol) with methyl groups at positions 10 and 12 did not induce any papillomas, 3 (8.5 nmol) with methyl groups at positions 4 and 10 significantly induced the formation of papillomas. The application of 3 (3.4 nmol) was negative, suggesting that the tumor-promoting activity of 3 was very weak. Since 1 with a methyl group at position 10 was inactive at 8.5 nmol application, as reported previously,Citation24) these results suggested that the introduction of a methyl group at position 4 increased tumor-promoting activity. Moreover, the tumor-promoting activities correlated well with proinflammatory activities on mouse ears.
Since the hydrophobicity of a molecule was one of the factors that enhanced the tumor-promoting activities of phorbol esters and teleocidins,Citation34,35) a log P value for each derivative was estimated by the HPLC method, as recommended by OECD.Citation42,43) The retention time of each derivative on a reversed-phase column correlated with log P using appropriate reference compounds with known log P values. The log P value of 2 (4.5) was higher than that of 3 (3.9), and was similar to that of DAT (4.4), even though they had an equal number of methyl substituents. The log P values of aplog-1 and 1 were 3.3 and 3.6, respectively. These results were important because the hydrophobicity of a molecule was not the sole factor required for PKC activators to exhibit tumor-promoting and proinflammatory activities.
In order to explain the high log P value of 2, an Nuclear Overhauser Effect (NOE) experiment was carried out in both CDCl3 and CD3OD. A significant NOE was detected between the H-2β and H-17 signals of 2, which implied a CH/π interaction between the β-hydrogen at C-2 and the aromatic ring. In addition, the chemical shift of the phenolic hydroxyl group of 2 (δ 6.77) was higher than that of 1 (δ 6.35) in CDCl3, suggesting that the methyl group at position 12 stabilized the hydrogen bond between the carbonyl oxygen at C-1 and the phenolic hydroxyl group. Therefore, the log P value of 2 may at least in part be explained by the intramolecular hydrogen bond.
Binding affinity for PKC isozymes by 2 and 3
Previous studies indicated that the activation of PKCs was responsible for the anti-proliferative, proinflammatory, and tumor-promoting activities of naturally occurring PKC ligands such as TPA (Fig. ).Citation10) To investigate the reason for the difference in these biological activities between DAT and aplogs, the ability to bind to conventional and novel PKC isozymes was evaluated. Since commercially available recombinant PKC isozymes occasionally do not work properly, the synthetic C1 domains (C1A and C1B) of PKCs were employed for the binding assay.Citation44,45) The binding constants of some PKC ligands against these C1 domains were almost equal to those of whole PKC isozymes, as reported previously.Citation24,45) The C1A peptides were used as conventional PKC surrogates, while the C1B peptides were employed as novel PKC surrogates because tumor promoters mainly bind to these domains.Citation45–48)
We recently reported that tumor promoters such as phorbol 12,13-dibutyrate (PDBu) and DAT bound potently to both cPKCs and nPKCs, whereas non-tumor-promoting compounds such as 1 and bryo-1 exhibited some selectivity for nPKCs other than PKCε, that is, PKCδ, η, and θ.Citation24) The affinity profiles of 2 and 3 for cPKCs and nPKCs were almost equal to that of 1 (Table ), supporting the absence or weakness of their proinflammatory and tumor-promoting activities from those of DAT. Binding for cPKCs, especially PKCα may be responsible for tumor-promoting and proinflammatory activities rather than anti-proliferative activity against several cancer cell lines. The activation of PKCα drove the signaling pathway to produce a proinflammatory protein (COX-2Citation49)) and proliferative factor (cyclin D1Citation50)), and PKCα was also involved in the anti-apoptotic protein Bcl-2-mediated chemoresistance.Citation51) Moreover, abnormally activated PKCα activated AP-1, a transcriptional factor facilitating mitogenic signals.Citation52) However, the significant difference observed in the proinflammatory and tumor-promoting activities of 2 and 3 could not be explained by binding selectivity for PKC isozymes. More detailed experiments including the expression levels of each PKC isozyme in cells when treated with these aplogs are necessary. Since 2 did not show any tumor-promoting or proinflammatory activity, its combination with DAT may clarify the mechanism underlying adverse effects other than anti-proliferative activity.
Table 2. Values of Ki for the inhibition of [3H]PDBu binding by 1, 2, 3, and debromoaplysiatoxin (DAT).
Conclusions
10,12-Dimethyl-aplog-1 (2) exhibited more potent anti-proliferative activity against several cancer cell lines than 10-methyl-aplog-1 (1), but did not exhibit any tumor-promoting nor proinflammatory activity. Since 1 weakly induced inflammation, 2 is the first analog that retains only the anti-proliferative activity of multifunctional DAT. In contrast, the installation of methyl groups at positions 4 and 10 enhanced tumor-promoting and proinflammatory activities. It is quite noteworthy that the difference in the position of a methyl group drastically changed the quality of the biological activities. We have recently established the large-scale synthesis of 1,Citation53) and this strategy would also be applicable to that of 2. Since the methoxy group in the side chain and the hydroxyl group at position 3 of DAT were involved in tumor promotion,Citation40) and since the methyl group at position 25 of aplog-1 decreased the anti-proliferative activity,Citation54) 2 would be the optimized seed for anticancer drugs among the simplified analogs of DAT.
Experimental
General remarks
The following spectroscopic and analytical instruments were used: Digital Polarimeter, DIP-1000 (Jasco, Tokyo, Japan); 1H and 13C NMR, AVANCE III 400 and AVANCE III 500 (Ref. TMS, Bruker, Germany); HPLC, Model 600E with a Model 2487 UV detector (Waters, Tokyo, Japan); HR-FAB-MS, JMS-600H and JMS-700 (JEOL, Tokyo, Japan); HR-ESI-TOF-MS, Waters Xevo G2-SQtof (Waters, Tokyo, Japan). HPLC was carried out on YMC-packed ODS-A AA12S05–1520WT and SIL SL12S05–2510WT (Yamamura Chemical Laboratory, Kyoto, Japan), and CHIRALCEL OJ-RH (Daicel Corporation, Osaka, Japan). Wakogel C-200 (silica gel, Wako Pure Chemical Industries, Osaka, Japan) and YMC A60–350/250 gel (ODS, Yamamura Chemical Laboratory, Kyoto, Japan) were used for column chromatography. [3H]PDBu (18.7 or 17.2 Ci/mmol) was purchased from PerkinElmer Life Sciences Research Products (Boston, MA, USA). The PKC C1 peptides were synthesized as reported previously.Citation44) All other chemicals and reagents were purchased from chemical companies and used without further purification. The purity of the final compounds was greater than 95% as judged by HPLC.
Measurements of cell growth inhibition
A panel of 39 human cancer cell lines established by Yamori and colleaguesCitation32) according to the NCI method with modifications was employed, and cell growth inhibitory activity was measured as reported previously. In brief, the cells were plated in 96-well plates in RPMI 1640 medium supplemented with 5% fetal bovine serum and allowed to attach overnight. The cells were incubated with each test compound for 48 h. Cell growth was estimated by the sulforhodamine B assay. The 50% growth inhibition (GI50) parameter was calculated as reported previously.Citation55) Absorbance for the control well (C) and the test well (T) was measured at 525 nm along with that for the test well at time 0 (T0). Cell growth inhibition (% growth) by each concentration of drug (10−8, 10−7, 10−6, 10−5, and 10−4 M) was calculated as 100[(T − T0)/(C − T0)] using the average of duplicate points. By processing these values, each GI50 value, defined as 100[(T − T0)/(C − T0)] = 50, was determined.
EBV-EA induction test
Human B-lymphoblastoid Raji cells (5 × 105/mL) were incubated at 37 °C under a 5% CO2 atmosphere in 1 mL of RPMI 1640 medium (supplemented with 10% fetal bovine serum) with 4 mM sodium n-butyrate (a synergist) and 10, 100, or 1000 nM of each test compound for the induction test. Each test compound was added as 2 μL of a DMSO solution (5, 50, and 500 μM stock solution) along with 2 μL of DMSO; the final DMSO concentration was 0.4%. After 48 h of incubation, smears were made from the cell suspension, and the EBV-EA-expressing cells were stained by a conventional indirect immunofluorescence technique with an NPC patient’s serum (a gift from Kobe University) and FITC-labeled anti-human IgG (DAKO, Glostrup, Denmark) as reported previously.Citation36) In each assay, at least 500 cells were counted and the proportion of the EA-positive cells was recorded. Cell viability exceeded 60% in all experiments.
Mouse ear edema test
A solution of each test compound in EtOH (20 μL) or EtOH was applied to the right ear only of ICR mice (Sankyo Lab Service, Tokyo, Japan) by means of a micropipette. A volume of 10 μL was delivered to both the inner and outer surfaces of the ear. After 24 h, a disk (1-cm diameter) was obtained from the ear and weighed. Each group consisted of at least three mice. Statistical analysis was performed using R software (version 3.1.2; R Foundation for Statistical Computing, Vienna, Austria).
Two-stage carcinogenesis experiment
The back of each six-week-old female ICR mice (Sankyo Lab Service, Tokyo, Japan) was shaved with surgical clippers. From a week after initiation by a single application of 390 nmol of DMBA in 0.1 mL acetone, 1.7 nmol of TPA in 0.1 mL acetone, 3.4 nmol of 2 in 0.1 mL acetone, or 8.5 nmol of 2 or 3 in 0.1 mL acetone was applied twice a week from week 1 to 20. Each group consisted of ten mice.
Inhibition of specific binding of [3H]PDBu to PKC C1 peptides
The binding of [3H]PDBu to the PKC C1 peptides was evaluated by the procedure of Sharkey and Blumberg56) with modifications as reported previouslyCitation45) using 50 mM Tris-maleate buffer (pH 7.4 at 4 °C), 10–20 nM PKC C1 peptides, 20 nM [3H]PDBu (18.7 or 17.2 Ci/mmol), 50 μg/mL 1,2-dioleoyl-sn-glycero-3-phospho-l-serine (Sigma or Funakoshi), 3 mg/mL bovine γ-globulin (Sigma), and various concentrations of inhibitors. Binding affinity was evaluated based on the concentration required to cause 50% inhibition of the specific binding of [3H]PDBu, the IC50, which was calculated by log–probit regression analysis. The inhibition constant, Ki, was calculated by the method of Sharkey and Blumberg.Citation56) Although we used each PKC C1 peptide in the range of 10–20 nM, the concentration of the properly folded peptide was estimated to be about 3 nM on the basis of Bmax values of Scatchard analyses reported previously.Citation45) Therefore, the concentration of free PDBu will not markedly vary over the dose–response curve.
Authors contributions
M. K. and K. I. designed the study. M. K. and H. T. carried out the experiments. K. S. and H. N. supplied the sample of debromoaplysiatoxin. M. K. shared the responsibility for the writing of the manuscript with R. C. Y. and K. I.
Disclosure statement
No potential conflict of interest was reported by the authors.
Funding
This work was supported by JSPS KAKENHI [grant number 23102011] to K.I. and R.C.Y., and 25.2518 to M.K.
Supplemental material
Synthetic procedures of 2 and 3, spectral data of 21-bromo-10-methyl-aplog-1, 2, and 3 as well as their synthetic intermediates, and their efficacy profiles (fingerprints) against 26 human cancer cell lines along with representative anticancer drugs, and HPLC profiles of 2, 3, and 11. The supplemental material for this paper is available at http://dx.doi.org/10.1080/09168451.2015.1091718.
Supplemental_material2015.7.25part3.compressed.pdf
Download PDF (1.7 MB)Supplemental_material2015.7.25part1.compressed.pdf
Download PDF (2 MB)Supplemental_material2015.7.25part2.compressed.pdf
Download PDF (2.1 MB)Acknowledgments
We thank the Screening Committee for Anticancer Drugs supported by a Grant-in-Aid for Scientific Research on Innovative Areas (“Scientific Support Programs for Cancer Research”) from the Ministry of Education, Culture, Sports, Science and Technology, Japan. Mass measurements were partly carried out with the JEOL JMS-700 MS spectrometer in the joint Usage/Research Center (JURC) at the Institute for Chemical Research, Kyoto University, Japan.
References
- Newman DJ, Cragg GM. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod. 2012;75:311–335.
- Ueda M. Chemical biology of natural products on the basis of identification of target proteins. Chem. Lett. 2012;41:658–666.
- Towle MJ, Salvato KA, Budrow J, et al. In vitro and in vivo anticancer activities of synthetic macrocyclic ketone analogues of halichondrin B. Cancer Res. 2001;61:1013–1021.
- De Christopher BA, Fan AC, Felsher DW, et al. “Picolog”, a synthetically-available bryostatin analog, inhibits growth of MYC-induced lymphoma in vivo. Oncotarget. 2012;3:58–66.
- Hirata Y, Uemura D. Halichondrins—antitumor polyether macrolides from a marine sponge. Pure Appl. Chem. 1986;58:701–710.
- Pettit GR, Herald CJ, Doubek DL, et al. Isolation and structure of bryostatin 1. J. Am. Chem. Soc. 1982;104:6846–6848.
- Fujiki H, Sugimura T. New classes of tumor promoters: teleocidin, aplysiatoxin, and palytoxin. Adv. Cancer Res. 1987;49:223–264.
- Castagna M, Takai Y, Kaibuchi K, et al. Direct activation of calcium-activated, phospholipid-dependent protein kinase by tumor-promoting phorbol esters. J. Biol. Chem. 1982;257:7847–7851.
- Nishizuka Y. Protein kinase C and lipid signaling for sustained cellular responses. FASEB J. 1995;9:484–496.
- Newton AC. Protein kinase C: structural and spatial regulation by phosphorylation, cofactors, and macromolecular interactions. Chem. Rev. 2001;101:2353–2364.
- Alkon DL, Sun MK, Nelson TJ. PKC signaling deficits: a mechanistic hypothesis for the origins of Alzheimer’s disease. Trends Pharmacol. Sci. 2007;28:51–60.
- McKernan LN, Momjian D, Kulkosky J. Protein kinase C: one pathway towards the eradication of latent HIV-1 reservoirs. Adv. Virol. 2012;2012:805347.
- Fährmann M. Targeting protein kinase C (PKC) in physiology and cancer of the gastric cell system. Curr. Med. Chem. 2008;15:1175–1191.
- Schaar D, Goodell L, Aisner J, et al. A phase I clinical trial of 12-O-tetradecanoylphorbol-13-acetate for patients with relapsed/refractory malignancies. Cancer Chemother. Pharmacol. 2006;57:789–795.
- Ogbourne SM, Hampson P, Lord JM, et al. Proceedings of the first international conference on PEP005. Anticancer Drugs 2007;18:357−362.
- Hennings H, Blumberg PM, Pettit GR, et al. Bryostatin 1, an activator of protein kinase C, inhibits tumor promotion by phorbol esters in SENCAR mouse skin. Carcinogenesis. 1987;8:1343–1346.
- Schaufelberger DE, Koleck MP, Beutler JA, et al. The large-scale isolation of bryostatin 1 from Bugula neritina following current good manufacturing practices. J. Nat. Prod. 1991;54:1265–1270.
- Keck GE, Poudel YB, Cummins TJ, et al. Total synthesis of bryostatin 1. J. Am. Chem. Soc. 2011;133:744–747.
- Kato Y, Scheuer PJ. Aplysiatoxin and debromoaplysiatoxin, constituents of the marine mollusk Stylocheilus longicauda. J. Am. Chem. Soc. 1974;96:2245–2246.
- Nakagawa Y, Yanagita RC, Hamada N, et al. A simple analogue of tumor-promoting aplysiatoxin is an antineoplastic agent rather than a tumor promoter: development of a synthetically accessible protein kinase C activator with bryostatin-like activity. J. Am. Chem. Soc. 2009;131:7573–7579.
- Irie K, Yanagita RC. Synthesis and biological activities of simplified analogs of the natural PKC ligands, bryostatin-1 and aplysiatoxin. Chem. Rec. 2014;14:251–267.
- Kamachi H, Tanaka K, Yanagita RC, et al. Structure–activity studies on the side chain of a simplified analog of aplysiatoxin (aplog-1) with anti-proliferative activity. Bioorg. Med. Chem. 2013;21:2695–2702.
- Nakagawa Y, Kikumori M, Yanagita RC, et al. Synthesis and biological evaluation of the 12,12-dimethyl derivative of aplog-1, an anti-proliferative analog of tumor-promoting aplysiatoxin. Biosci. Biotechnol. Biochem. 2011;75:1167–1173.
- Kikumori M, Yanagita RC, Tokuda H, et al. Structure–activity studies on the spiroketal moiety of a simplified analogue of debromoaplysiatoxin with antiproliferative activity. J. Med. Chem. 2012;55:5614–5626.
- Park PU, Broka CA, Johnson BF, et al. Total synthesis of debromoaplysiatoxin and aplysiatoxin. J. Am. Chem. Soc. 1987;109:6205–6207.
- Toshima H, Suzuki T, Nishiyama S, et al. Synthetic study on aplysiatoxins, highly inflammatory agents and tumor promoters: synthesis of 3-deoxydebromoaplysiatoxin. Tetrahedron Lett. 1989;30:6725–6728.
- Duan JJW, Smith AB III. Iodine monobromide (IBr) at low temperature: enhanced diastereoselectivity in electrophilic cyclizations of homoallylic carbonates. J. Org. Chem. 1993;58:3703–3711.
- Murphy WS, Wattanasin S. Intramolecular alkylation of phenols. Part 5. A regiospecific anionic ring closure of phenols via quinone methides. J. Chem. Soc. Perkin Trans. 1980;1:1567–1577.
- Evans DA, Kim AS. (S)-4-Benzyl-2-oxazolidinone. In: LO Paquette, editor. Encyclopedia of reagents for organic synthesis. New York, NY: John Wiley and Sons; 1995. Vol. 1, p.345−356.
- Brown HC, Jadhav PK. Asymmetric carbon-carbon bond formation via B-allyldiisopinocampheylborane. Simple synthesis of secondary homoallylic alcohols with excellent enantiomeric purities. J. Am. Chem. Soc. 1983;105:2092–2093.
- Ide M, Nakata M. Room-temperature metallation of 2-substituted 1,3-dithiane derivatives and subsequent coupling with 2,3-disubstituted oxiranes. Bull. Chem. Soc. Jpn. 1999;72:2491–2499.
- Yamori T, Matsunaga A, Sato S, et al. Potent antitumor activity of MS-247, a novel DNA minor groove binder, evaluated by an in vitro and in vivo human cancer cell line panel. Cancer Res. 1999;59:4042–4049.
- NCI/NIH. Developmental therapeutics program. http://dtp.nci.nih.gov/ (accessed on 2015 Feb 2).
- Wang QJ, Fang TW, Fenick D, et al. The lipophilicity of phorbol esters as a critical factor in determining the pattern of translocation of protein kinase C δ fused to green fluorescent protein. J. Biol. Chem. 2000;275:12136–12146.
- Irie K, Hagiwara N, Tokuda H, et al. Structure-activity studies of the indole alkaloid tumor promoter teleocidins. Carcinogenesis. 1987;8:547–552.
- Ito Y, Yanase S, Fujita J, et al. A short-term in vitro assay for promoter substances using human lymphoblastoid cells latently infected with Epstein-Barr virus. Cancer Lett. 1981;13:29–37.
- zur Hausen H, Bornkamm GW, Schmidt R, et al. Tumor initiators and promoters in the induction of Epstein—Barr virus. Proc. Natl. Acad. Sci. USA. 1979;76:782−785.
- Philip M, Rowley DA, Schreiber H. Inflammation as a tumor promoter in cancer induction. Semin. Cancer. Biol. 2004;14:433–439.
- Irie K, Kikumori M, Kamachi H, et al. Synthesis and structure–activity studies of simplified analogues of aplysiatoxin with antiproliferative activity like bryostatin-1. Pure Appl. Chem. 2012;84:1341–1351.
- Yanagita RC, Kamachi H, Kikumori M, et al. Effects of the methoxy group in the side chain of debromoaplysiatoxin on its tumor-promoting and anti-proliferative activities. Bioorg. Med. Chem. Lett. 2013;23:4319–4323.
- Suganuma M, Fujiki H, Tahira T, et al. Estimation of tumor promoting activity and structure-function relationships of aplysiatoxins. Carcinogenesis. 1984;5:315–318.
- Klein W, Kördel W, Weiß M, et al. Updating of the OECD test guideline 107 “partition coefficient n-octanol/water”: OECD laboratory intercomparison test on the HPLC method. Chemosphere. 1988;17:361–386.
- Organization for Economic Cooperation and Development. Test No. 117: Partition Coefficient (n-octanol/water), HPLC Method. In OECD guidelines for the testing of chemicals. Paris: OECD; c2004 [revised 2004 Nov 23; cited 2015 Sep 29]. Available from: http://www.oecd-ilibrary.org/environment/test-no-117-partition-coefficient-n-octanol-water-hplc-method_9789264069824-en. 2004.
- Irie K, Oie K, Nakahara A, et al. Molecular basis for protein kinase C isozyme-selective binding: the synthesis, folding, and phorbol ester binding of the cysteine-rich domains of all protein kinase C isozymes. J. Am. Chem. Soc. 1998;120:9159–9167.
- Shindo M, Irie K, Nakahara A, et al. Toward the identification of selective modulators of protein kinase C (PKC) isozymes: establishment of a binding assay for PKC isozymes using synthetic C1 peptide receptors and identification of the critical residues involved in the phorbol ester binding. Bioorg. Med. Chem. 2001;9:2073–2081.
- Quest AFG, Bell RM. The regulatory region of protein kinase Cγ. Studies of phorbol ester binding to individual and combined functional segments expressed as glutathione S-transferase fusion proteins indicate a complex mechanism of regulation by phospholipids, phorbol esters, and divalent cations. J. Biol. Chem. 1994;269:20000–20012.
- Raghunath A, Ling M, Larsson C. The catalytic domain limits the translocation of protein kinase Cα in response to increases in Ca2+ and diacylglycerol. Biochem. J. 2003;370:901–912.
- Szállási Z, Bögi K, Gohari S, et al. Non-equivalent roles for the first and second zinc fingers of protein kinase Cδ. Effect of their mutation on phorbol ester-induced translocation in NIH 3T3 cells. J. Biol. Chem. 1996;271:18299–18301.
- Wang HQ, Kim MP, Tiano HF, et al. Protein kinase C-α coordinately regulates cytosolic phospholipase A2 activity and the expression of cyclooxygenase-2 through different mechanisms in mouse keratinocytes. Mol. Pharamacol. 2001;59:860–866.
- Soh JW, Weinstein IB. Roles of specific isoforms of protein kinase C in the transcriptional control of cyclin D1 and related genes. J. Biol. Chem. 2003;278: 34709–34716.
- Arnott CH, Scott KA, Moore RJ, et al. Tumour necrosis factor-α mediates tumour promotion via a PKCα- and AP-1-dependent pathway. Oncogene. 2002;21:4728–4738.
- Jiffar T, Kurinna S, Suck G, et al. PKCα mediates chemoresistance in acute lymphoblastic leukemia through effects on Bcl2 phosphorylation. Leukemia 2004;18:505−512.
- Kikumori M, Yanagita RC, Irie K. Improved and large-scale synthesis of 10-methyl-aplog-1, a potential lead for an anticancer drug. Tetrahedron. 2014;70:9776–9782.
- Hanaki Y, Kikumori M, Ueno S, et al. Structure–activity studies at position 27 of aplog-1, a simplified analog of debromoaplysiatoxin with anti-proliferative activity. Tetrahedron. 2013;69:7636–7645.
- Monks A, Scudiero D, Skehan P, et al. Feasibility of a high-flux anticancer drug screen using a diverse panel of cultured human tumor cell lines. JNCI J. Natl. Cancer Inst. 1991;83:757–766.
- Sharkey NA, Blumberg PM. Highly lipophilic phorbol esters as inhibitors of specific [3H]phorbol 12,13-dibutyrate binding. Cancer Res. 1985;45:19–24.