
Abstract
White-rot basidiomycete Coriolopsis gallica HTC is one of the main biodegraders of poplar. In our previous study, we have shown the strong capacity of C. gallica HTC to degrade lignocellulose. In this study, equal amounts of total RNA fromC. Gallica HTC cultures grown in different conditions were pooled together. Illumina paired-end RNA sequencing was performed, and 13.2 million 90-bp paired-end reads were generated. We chose the Merged Assembly of Oases data-set for the following blast searches and gene ontology analyses. The reads were assembled de novo into 28,034 transcripts (≥ 100 bp) using combined assembly strategy MAO. The transcripts were annotated using Blast2GO. In all, 18,810 transcripts (≥100 bp) achieved BLASTX hits, of which, 7048 transcripts had GO term and 2074 had ECs. The expression level of 11 lignocellulolytic enzyme genes from the assembled C. gallica HTC transcriptome were detected by real-time quantitative polymerase chain reaction. The results showed that expression levels of these genes were affected by carbon source and nitrogen source at the level of transcription. The current abundant transcriptome data allowed the identification of many new transcripts in C. gallica HTC. Data provided here represent the most comprehensive and integrated genomic resources for cloning and identifying genes of interest from C. gallica HTC. Characterization of C. gallica HTC transcriptome provides an effective tool to understand mechanisms underlying cellular and molecular functions of C. gallica HTC.
Lignocellulose is mainly composed of cellulose, hemicellulose, and lignin. It is a renewable biomass resource and is abundantly distributed worldwide. Numerous micro-organisms degrade lignin at different degrees. Of these, white-rot fungi are the most effective.Citation1–3) Because lignocellulolytic enzymes produced by white-rot fungi degrade lignin and aromatic pollutants effectively; white-rot fungi have important value and broad applications in biopulping, biobleaching of pulp, and environmental protection. White-rot fungi produce C. gallica HTC produces lignin peroxidase, manganese-dependent peroxidase, hemicellulase, cellulase, and laccase.Citation4) The researcher compared this fungus with more than 40 white-rot fungi and proved that C. gallica HTC could degrade lignocellulose more effectively than other fungi, such as Lentinula, Phanerochaete, and Pleurotus.Citation5) Because of its capacity to degrade lignin and several refractory organics efficiently, C. gallica can be applied for the utilization of plant biomass and bioremediation of organic pollutants.Citation5) Some progress has been made in the research on C. gallica; however, the research level had certain disparity in contrast to other fungi and the main study was in the gene engineering research, such as mutational screening of strain, gene cloning, and gene expression.Citation6,7) It is difficult to obtain systematic genetic information using these methods because they are complicated, time-consuming, and expensive.
Despite the continuous research on lignocellulolytic enzymes, no information about genome and transcriptome of C. gallica is reported to date. This makes in-depth research on C. gallica lignocellulolytic enzymes more difficult. Since 2005, use of next-generation high-throughput sequencing has greatly reduced the cost of genome and transcriptome sequencing.Citation8,9) Improvement in sequencing technologies and corresponding assembly tools has facilitated various studies, including identification of genes under natural selection, reconstruction of demographic history, and large-scale inference of phylogeny.Citation10,11) This in turn has promoted the sequencing of C. gallicatranscriptome. However, because many assemblers are available, it is unclear as to which assembler should be used for high-quality de novo transcriptome assembly. Transcriptome assembly is difficult despite the rapid advancements in assemblers that can efficiently handle more reads.Citation8,12) Quality of de novo transcriptome assembly is highly dependent on the overlap length of user-defined sequence.Citation8) Different assemblers have different applicability and performance.Citation13,14) Researchers usually chose only one assembler to assemble transcriptome.Citation15–17) However, new assembly strategies such as merging contigs obtained using multiple assemblies can provide better results.Citation18,19) In this study, we evaluated and compared the performance of two strategies: single assembly strategy and combined assembly strategy. Performance of the assembly strategies was evaluated based on the total number of contigs, number of contigs of ≥ 1000 bp, N50 length, maximum contig length, accuracy, sensitivity, rate of mapping short reads onto the contigs, and open-reading frames (ORFs). After assembly and comparison, genes encoding lignocellulolytic enzymes were identified and their transcription levels were investigated under different culture conditions using real-time quantitative polymerase chain reaction (PCR). Results of the present study will help us better understand molecular mechanisms underlying cellular metabolism and identify new genes. Moreover, these results will serve as a valuable resource for future genetic and genomic studies on C. gallica.
Materials and methods
Fungal species and media
The fungal strain of Coriolopsis gallica HTC(Heze Teacher College, stain isolated location) was (the preservation number is CCTCC NO: 2689) used in this study. We selected 24 kinds of liquid media (Table S1). Different kinds of nitrogen source media were improved by Kirk basic culture medium,Citation2) and each medium contained 20 g of glucose, 2 g of KH2PO4, 0.5 g of MgSO4·7H2O, 0.1 g of CaCl2, and 1000 mL of trace element solution per litter(Table S2). After activation on potato dextrose agar (PDA) plate, the strain of C. gallica HTC was inoculated in the 24 kinds of liquid media and cultured statically at 28 °C 15 d. Each collected sampleof C. gallica HTC which was cultured under different conditions were snap-frozen immediately in nitrogen and stored at −80 °C until further processing.
RNA extraction and transcriptome sequencing
Each sample’s total RNAs were extracted using the Trizol®Reagent (Invitrogen, Carlsbad, California, USA), and treated with DNase I (Fermentas, Pittsburgh PA, USA) according to the manufacturer’s instructions. RNA concentration was measured using Qubit® fluorometer (Invitrogen, Carlsbad, California, USA).
To obtain a comprehensive list of transcripts, equal amount of total RNA from each sample was pooled together and the mixed RNA was sent to Beijing Genomics Institute (Shenzhen, China) (http://www.genomics.cn) for transcriptome sequencing.
De novo transcriptome assembly
In order to obtain the best assembled results, we employed two assembly strategies to assemble transcriptome of C. gallicaHTC: one single assembler assembly strategy and one combined assembly strategy. The single assembler assembly strategy included four commonly used assemblers, Trinity,Citation20) Oases,21) Edena,Citation22) and SOAPdenovo,Citation23,24) The combined de novo transcriptome assembly strategy was an integrated strategy which merged the three multiple Parameters denovo assemblers, Oases, Edena, SOAPdenovo. It included Integrated DeNovo Transcriptome Assembly (IDTA), Merged Assembly of Oases (MAO), Merged Assembly of Edena (MAE), and Merged Assembly of SOAPdenovo (MAS).Citation25) All the works of assemblies were run on a 64-bit Linuxsystem (Ubuntu 10.10) with 32G physical memory.
Evaluation and comparison of each assembly result
To further assess the quality of these assemblies, some evaluation criteria were used. First, sensitivity and accuracy was considered to evaluate the quality of the assemblies. The 159 known protein sequences of C. gallica HTC and its related species available in NCBI were considered as “gold standard” reference in this study, and used to blast against each assembly by tBLASTN. Based on the blast results, we calculated sensitivity, accuracy, and the average of them of each assembly, respectively.Citation25)
To further assess the assembly, we mapped the reads onto the final assembled transcripts using the Bowtie programCitation26) available at the Galaxy website (http://main.g2.bx.psu.edu/).Citation27,28) First of all, build indexed file for each assembled transcript using Bowtie 2 (version Bowtie-2.0.0-beta5), and then map the read onto each assembly. Moreover, we evaluated the assemblies by scanning ORF with ORF prediction perl script from Trinity package.
After evaluating the assembly results, we compared the results of the two assembly strategies, Trinity,Citation20) Oases,Citation21) Edena,Citation22) and SOAPdenovoCitation23,24) and IDTA, MAO, MAE, MAS,Citation25) with respecting to the several well-known basic criteria, N50 length, maximum contig length, average contig length, and contig number (total contig and ≥ 1000 bp).
Then, to further compare this two assembly strategies, we also compared the result of the sensitivity and accuracy, the mapping onto assembly and the ORF prediction of each assembly strategies.
Functional annotation and classification
The final best assembled transcripts were submitted for homology and annotation searches using Blast2GO softwarev2.4.4.Citation29) For BLASTX against the non-redundant database which was loaded from NCBI, the threshold was set to E-value ≤ 10−3 and we run annotation with default parameters. GO classification was achieved using WEGO software (http://wego.genomics.org.cn/cgi-bin/wego/index.pl).Citation30)
Identification and expression analysis of genes encoding lignocellulolytic enzymesin C. gallica HTC
To verify the accuracy of assembly, we observed sequences of particular contigs with a complete ORF as representative examples. According to the annotated result, we here isolated the sequences of laccase and manganese peroxidase with complete ORF that have not yet been identified in C. gallica HTC. These particular transcripts were selected for reverse transcription polymerase chain reaction (RT-PCR) validation. Primers were designed according to assembled transcripts using Primer Premier 5.0 (PREMIER Biosoft. International, CA, USA) and gene amplification was implemented using KOD-Plus-Neo DNA polymerase (Toyobo Co., Ltd., Osaka Street, Japan). After TA-cloning, positive clones were identified by Sanger sequencing.
After verifying the accuracy of assembly, we screened sequences of laccase, manganese peroxidase, lignin peroxidase, and cellulase to analysis their expression at transcriptional level under different culture conditions. We selected four liquid medium, TG, AG, 2%T, 0.1%G, as research conditions. Total RNA from the four liquid medium was reverse-transcribed into cDNA by reverse transcription kit(Takara Biotechnology-Dalian Co., Ltd., Dalian, China), respectively. Fluorescence quantitative PCR kit used in this experiment is SYBR® Premix Ex Taq TMⅡ (Takara Biotechnology-Dalian Co., Ltd., Dalian, China) and the analysis method is 2−ΔΔCT.Citation31) The gpd gene, encoding the glyceraldehyde-3-phosphate dehydrogenase gene, acted as reference genes to normalize data differences. According to the gene sequences chosen from C. gallica HTC transcriptome library, primer pairs were designed and the oligonucleotide sequences for qPCR analysis were listed in Table , which were used to amplify 100–300 bp of the target genes. These genes chosen fromC. gallica HTC transcriptome library were analyzed by qPCR (Bio-Rad Laboratories, Inc., Hercules, CA, USA) with SYBR Green as fluorescent dye. The amplification mixture (total volume 20 μL) contained 10 μL of 2 × SYBR®Premix Ex Taq TMⅡ (Takara Biotechnology-Dalian Co., Ltd., Dalian, China), 1.6 μL of forward and reverse primers (16 μmol/L), and 2 μL of cDNA (serial dilution). The qPCR reaction conditions were as follows: step 1, 95 °C for 30 s (hot-start activation); step 2, 95 °C for 5 s (denaturation); step 3, 56, 57, and 58 °C for 30 s (annealing) and 72 °C for 30 s for extension,40 cycles of step 2 and 3; step 4, melt curve, heat from 65 to 95 °C with a ramp speed of 0.5 °C per 5 s, giving rise to melting curves to confirm the specificity of the primer pairs in each experiment. Data were analyzed with Bio-Rad CFX Manager 3.0 software. The value of Ct decreased correspondingly with the increase of the concentration of template. So, the Ct value was opposite to the expression level of mRNA.
Table 1. Primers of RT-PCR and real-time quantitative PCR.
Results
RNA sequencing and de novo transcriptome assembly
Somegenes are presumably not expressed under one condition, and analysis of various conditions can increase the coverage.Citation32) Therefore, in order to obtain a comprehensive transcriptome of C. gallica HTC, total RNA was isolated fromC. gallica HTC cultured in 24 different liquid media. The total RNA was isolated from C. gallica HTC cultured in 24 different liquid media. The total RNA was sequenced using Illumina NGS platform GAII to obtain 1.33 × 107 90-bp pair-end reads corresponding to 1.19 × 109 nucleotides, with 58% GC content across all bases. Before the assembly, read quality was assessed using FastQC software (http://www.bioinformatics.bbsrc.ac.uk/projects/download.html).
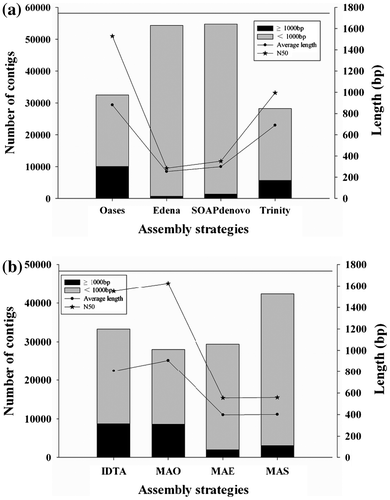
As mentioned above, we used two de novo assembly strategies, i.e. single and combined assembly strategies, to obtain a better assembly of C. gallica HTC transcriptome. First, 28,228 sequences (19,445,586 bp) were generated from reads that were de novo assembled into contigs using Trinity under default parameters, i.e. average contig length of 689 bp, N50 length of 995 bp, and maximum contig length of 7067 bp (Table ). It has been generally accepted that larger values of performance criteria are associated with better assembly performance. The assembly results of the three short sequence de novo assemblers were extensively varied at different kmer values. The data of contigs longer than 1000 bp were chosen to assess the assembly quality. This was because more number of long length contigs indicated better assembly performance.
Table 2. Statistics of de novo assembly output of C. gallica HTC transcriptome.
In the result of Oases assembly, when the kmer length was 21, the criteria values of the number of long length contigs (≥ 1000 bp), N50 length, and average contig length seemed the best. Edena assembly produced the best values of the number of long length contigs (≥ 1000 bp), N50length, average contig length, and maximum contig length, with a kmer length of 45. For SOAPdenovo assembly, the optimal parameters were kmer length of 47 and p value of 8. The obtained results indicated that 21 was the best kmer length for Oases assembly; 45 was the best kmer length for Edena assembly; and 47 and 8 were the best kmer length and p value, respectively, for SOAP de novo assembly.
We obtained 32,520 contigs (28,652,066 bp) using the Oases assembly; of these, 10,044 were long length contigs, with an average contig length of 881 bp, N50 length of 1529 bp, and maximum contig length of 8607 bp. Further, we obtained 53,606 contigs (13,557,719 bp) using Edena assembly; of these, 711 were long length contigs, with average contig length of 253 bp, N50 length of 285 bp, and maximum contig length of 3524 bp. In all, 54,736 sequences (116,389,293 bp) were generated with SOAPdenovo assembly and 1386 long length contigs (≥1000 bp), 299 bp for average contig length, 351 bp for N50, 4253 bp for maximum contig length.
For IDTA strategy, we selected five better assemblies from each of the three short sequences obtained using the de novo assemblers (k = 21, 23, 25, 27, and 29 for Oases; k = 45, 43, 47, 49, and 51 for Edena; and K47p8, K47p4, K45p8, K47p16, and K51p16 for SOAPdenovo) and merged these 15 assemblies. This generated 683,348 contigs (270.31 Mbp). Next, we reassembled these contigs using CAP3Citation30) to reduce redundancy and generate longer sequences and obtained 32,849 sequences (26,553,720 bp in total) with length of ≥ 100 bp after the final assembly. Of these, 8729 were long length contigs; further, average length, N50 length, and maximum length of each contig were 808, 1551, and 10,985 bp, respectively.
In the assembly of MAO, the five better assemblies, which we selected, were when the kmer length was 21, 23, 25, 27, and 29 for Oases. MAO assembly produced 28,034 contigs (25,448,177 bp). Of these, 8627 were long length contigs, with an average contig length of 908 bp, N50 length of 1624 bp, and maximum contig length of 10,963 bp. For MAE and MAS assemblies, 5 better assemblies were kmer lengths of 45, 43, 47, 49, and 51 for Edena and K47p8, K47p4, K45p8, K47p16, K51p16 for SOAPdenovo, respectively. Sequence statistics of all these assembly strategies are listed in Table .
After obtaining assemblies using each of these assemblers, the results were compared using basic criteria (Fig. ). In the single assembly strategy, Oases assembly was better than other assemblies were. N50 lengths, average lengths, and number of long length contigs obtained using Oases (1529 bp, 881 bp, and 10,044, respectively) were higher than those obtained using Trinity Edena and SOAPdenovo. In the combined assembly strategy, MAO assembly appeared to be the best. Although the number of long length contigs (8627) was slightly lower than that obtained using IDTA, N50 length (1624 bp), and average length (907 bp) were higher than those obtained using other assemblies, including single assembly strategies. Therefore, these results indicated that MAO assembly performed better than all other assemblies did.
Fig. 1. Comparison of de novo assembly quality among the different programs with Basic statistics.
Evaluation of the quality of transcriptome assembly
Previous reports have shown that criteria such as number of contigs, N50 length, average contig length, and maximum contig length are not sufficient to assess the quality of the assemblies.Citation33) In this study, we used sensitivity, accuracy, mapping rate, and ORF prediction to assess the quality of the obtained assemblies.
First, local BLAST similarity search was performed using 159 “gold standard” sequences as queries to BLAST against the assembled sequences. The average sensitivity and accuracy of Trinity was higher than that of other single assemblies. The sensitivity of MAO was the highest, while its accuracy was the lowest.
Next, reads were mapped back to the assembled transcripts that were ≥ 100 bp in length using Bowtie to evaluate their quality.Citation26) Mapping value was 96.37% for Trinity, which was higher than that for other single assemblies. Further, 97.64% reads were mapped to IDTA transcripts. This value was higher than that for any other assembly, including those for single assembly strategies. In the MAO assembly, 89.23% reads were mapped to the transcripts. Further, transcripts that were ≥ 100 bp in length were used for subsequent analysis.
Sequence and ORF lengths are important indexes for evaluating the contiguity of a sequence. ORFs of the eight assembled transcripts were predicted using PERL script from the Trinity package. ORFs with lengths more than 1499, 1199, 899, 599, and 299 bp were used as a reference. Results showed that the number of ORFs obtained using the MAO assembly was much higher than that obtained using other seven assemblies (Fig. ). Remarkably, in the MAO assembly, approximately 85.09% (8107), 67.52% (4817), and 54.94% (2917) ORFs had lengths of more than 899, 1199, and 1499 bp, respectively.
Fig. 2. ORF prediction of the two assembly strategies.
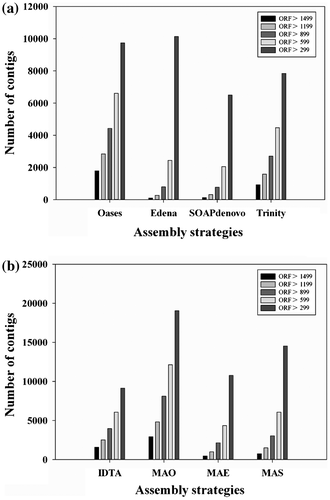
Comparison of assembly statistics indicated that the MAO assembly was better than other assemblies. It should be noted that IDTA has an expected advantage of assembling long length sequences over the other seven assembly strategies. In addition, Oases and Trinity include plural variants in a singular transcript, whereas MAO reduces redundancy using CAP3. Therefore, MAO seemed better for subsequent analysis, and the MAO program was selected for assembling the complete data-set.
Functional annotation and classification
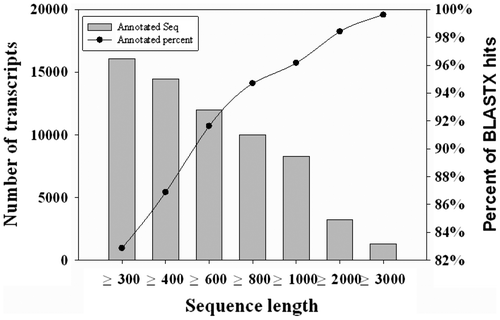
First, non-redundant database was loaded from NCBI. Assembly result of MAO (≥ 100 bp) was used to perform local BLAST. Sequence similarity search was conducted using Blast2GO.Citation29) Of 28,034 transcripts having lengths of ≥ 100 bp, 18,810 (67.10%) showed significant BLASTX hits and matched 10,793 unique protein accessions. Longer transcripts had higher percentage of BLASTX hits. For example, > 80% contigs having lengths of ≥ 300 bp and > 90% contigs having lengths of ≥ 600 bp showed significant BLASTX hits (Fig. ). Further, BLASTX hit percentage of transcripts having lengths of ≥ 3000 bp was 99.62% (Fig. ). More than 80% (15,239) sequences showed sequence identity of ≥ 70% and 280 sequences showed sequence identity of 100%.Of 10,793 unique protein accessions, 60.56% (6536) corresponded to one transcript, 23.19% (2503) corresponded to two transcripts, and 16.25% (1754) corresponded to more than two transcripts.
Fig. 3. Distribution of transcripts in length and percentage of transcripts with BLASTX hits.
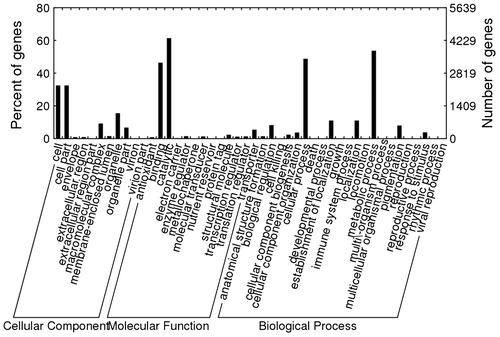
Functions of the assembled transcripts were classified using gene ontology assignments.Citation34) In all, 19,675 annotations were associated with 7048 transcripts. Fig. provides a summary of the classification of annotated C. gallica HTC sequences at level 2. For the classification of cellular components, clusters related to “cell” and “cell part” were relatively larger (32.5 and 32.5%, respectively) than other clusters of cellular components, such as “macromolecular complex,” “organelle part,” and “membrane-enclosed lumen.” For the classification of molecular functions, “catalytic activity,” and “binding” were higher (61.4 and 46.4%, respectively). For the classification of biological process, cluster sizes of “metabolic process” (53.7%) and “cellular process” (48.8%) were relatively larger than those of other clusters were. In all, 2074 transcripts were annotated using 22, 11 enzyme codes (ECs), which included 473 unique ECs.
Fig. 4. Gene ontology distribution for the C. gallica HTC.
Identification and expression of genes encoding lignocellulolytic enzymes in C. gallica
To verify the accuracy of the assembly, we observed three Mnp sequences with a single complete ORF as a reference. After primer designing, electrophoresis, and sequence alignment, three new genes were obtained. Sequence identities between Sanger sequencing results and the assembled transcripts were higher than 90%.
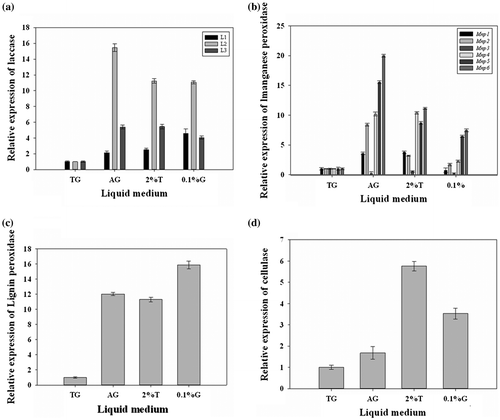
After verification, three laccase genes (L1, L2, and L3), six manganese peroxidase genes (Mnp1, Mnp2, Mnp3, Mnp4, Mnp5, and Mnp6), one lignin peroxidase gene (Lip1), and one cellulase gene (Cel1) were screened and their transcriptional levels under four different culture conditions were analyzed using real-time quantitative PCR. For L1, L2, L3, Mnp2, Mnp4, Mnp5, Mnp6, Lip1, and Cel1, the results of real-time quantitative PCR showed that increase in tryptone concentration from 1% to 2% increased the expression of the lignocellulolytic genes.When other conditions of the culture medium remained unchanged and the glucose was in the ratio of 0.1%, the expression level of gene was higher than when that was 2%. When other conditions of the culture medium remained unchanged and the nitrogen source was ammonium tartrate, the expression level of gene was higher than that when tryptone was used as the nitrogen source. However, these changes in expression were not observed with Mnp1 and Mnp3 (Fig. ). When the glucose content changed the expression changing trends of Mnp1 and Mnp3 were opposite to that of the other lignocellulolytic enzyme. Furthermore, expression levels of Mnp3 were low in AG and 2% T culture media. All the results indicated that the expression and regulation of lignocellulolytic enzyme genes influenced by carbon source and nitrogen source at the level of transcription.
Fig. 5. Quantitative real-time RT-PCR analysis of lignocellulolytic enzyme genes under different culture conditions.
Discussion
To compare these assembly strategies further, we assessed the assembly quality using several metrics. The results of these assessments showed that MAO assembly was better than other assemblies were. Values of sensitivity and accuracy were low in general (0.4–0.6). For each assembly, MAO achieved higher sensitivity than other assemblers did; however, its accuracy was the lowest. Because there is always a trade-off between contiguity and accuracy, average value of sensitivity and accuracy was used as the metrics.Citation35) The average value of sensitivity and accuracy of Trinity assembly was the highest (0.59), while that of MAO assembly was 0.57. Approximately 98% reads obtained using the IDTA assembly was mapped; this percentage was low for the MAO assembly. Results of mapping showed that the read utilization rate of the combined assembly strategy was higher than that of the single assembly strategy, indicating that the combined assembly strategy largely used original reads. Further, we evaluated the ORFs of the assembled sequences. MAO assembly showed the best results irrespective of the total number of ORFs or the proportion of ORFs. Based on the above comparisons, we concluded that the single assembly strategy was inferior to the combined assembly strategy. In the single assembly strategy, Edena and SOAPdenovo could assemble more number of total contigs; however, the average length of these contigs was very low. Oases could assemble more number of long length contigs. Further, Trinity was more user-friendly and time-saving than other assemblers, and its accuracy was better. In the combined assembly strategy, MAO performed better in general (such as N50, average length, and ORF). It should be noted that Oases and Trinity assemblies produced large number of variants, while MAO assembly reduced redundancy and clustered the transcripts. Moreover, it is widely accepted that increase in contig and ORF lengths is beneficial for obtaining more significant genes, thus making the subsequent experiments easier. Slight increase in N50 length could be of great significance for some genes.
C. gallica HTC has a strong capacity to degrade lignocellulose. This degradation is mainly performed by different lignocellulolytic enzymes such as laccase, manganese-dependent peroxidase, and lignin peroxidase. In our study, we screened three laccase genes, six manganese-dependent peroxidase genes, one lignin peroxidase gene, and one cellulase gene from the assembled C. gallica HTC transcriptome database. Expression levels of these lignocellulolytic enzymes were affected by different culture conditions. For further analysis the separated genes from C. gallica transcriptome database, this study analyzed the transcriptional responses of the genes in different culture conditions. We studied the expression level of lignocellulolytic enzymes in four different culture conditions to analyze the influences of different factors on the expression. The results showed that concentrations of tryptone and glucose significantly affected the expression levels of these genes. When the concentration of tryptone increased, expression levels of L1, L2, L3, Mnp1, Mnp2, Mnp4, Mnp5, Mnp6, Lip1, and Cel1 increased. This result was similar to that reported for laccase gene in our previous study.Citation36) When the concentration of glucose decreased, expression levels of L1, L2, L3, Mnp2, Mnp4, Mnp5, Mnp6, Lip1, and Cel1 increased, while that of Mnp1 decreased. Furthermore, use of ammonium tartrate as the nitrogen source increased the expression levels ofL1, L2, L3, Mnp1, Mnp2, Mnp4, Mnp5, Mnp6, Lip1, and Cel1 compared with those obtained using tryptone as the nitrogen source. Similar results were reported by Sun et al. used enzyme assays to analyze the effects of different nitrogen sources on the expression of lignocellulolytic enzymes of C. gallica. Remarkably, expression characteristics of Mnp3 were opposite to those of other genes.Citation5) These results suggested that the expression and regulation of genes encoding lignocellulolytic enzymes were affected by carbon and nitrogen sources at the transcriptional level and the different genes showed different transcriptional responses. Thus, different culture conditions may have important effects on the expressions and activities of genes encoding lignocellulolytic enzymes. These results provide important basis to further study the expression and regulation of C. gallica genes encoding lignocellulolytic enzymes.
Conclusions
In this study, we successfully performed de novo transcriptome sequencing using an Illumina short-read sequencer and well-developed assembly programs. The optimal assembly strategy, i.e. MAO, produced 28,034 transcript sequences as a transcriptome assembly in C. gallica HTC. Blast2GO obtained 18,810 BLASTX hits and 10,793 uniqueprotein accessions. In all, 11 genes encoding lignocellulolytic enzymes were screened from the assembled C. gallica HTC transcriptome. The present data can help increase thepublic database of fungal species. Moreover, RNA sequencing together with combined de novo transcriptome assembly strategies used in the present study could facilitate future genetic analysis of non-model plants.
Authors contribution
Xuemei Tan conceived the ideas and coordinated the overall project. Yuehong Chen conducted the main experiments, performed data analysis, drafted, and revised the manuscript. Qinghua Cao participated in the design of the study, prepared the experimental materials, and conducted the experiments of RNA extraction. Xiang Tao offered help in bioinformatics. Huanhuan Shao, Kun Zhang, and Yizheng Zhang help to conceive the ideas and offered help in qPCR and provided useful suggestions. All authors read and approved the final manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
Funding
Research support was provided by grants from the Nature Science Foundation of China with project [grant number 30470984].
Notes
Abbreviations: Coriolopsis gallica, C. gallica; HTC, Heze Teacher College,stain isolated location); GO, Gene Ontology; CCTCC, China Center for Type Culture Collection; IDTA, Integrated DeNovo Transcriptome Assembly; MAO, Merged Assembly of Oases; MAE, Merged Assembly of Edena; MAS, Merged Assembly of SOAPdenovo
Related Research Data
References
- Yao Y, Sakamoto T, Honda Y, et al. The white-rot fungus pleurotus ostreatus transformant overproduced intracellular cAMP and laccase. Biosci. Biotechnol. Biochem. 2013;77:2309–2311.10.1271/bbb.130470
- Kirk TK, Farrell RL. Enzymatic “combustion”: the microbial degradation of lignin. Annu. Rev. Microbiol. 1987;41:465–501.
- Misumi K, Sugiura T, Yamaguchi S, et al. Cloning and transcriptional analysis of the gene encoding 5-aminolevulinic acid synthase of the white-rot fungus Phanerochaete sordida YK-624. Biosci. Biotechnol. Biochem. 2011;75:178–180.10.1271/bbb.100674
- Calvo AM, Copa-Patino JL, Alonso O, et al. Studies of the production and characterization of laccase activity in the basidiomycete Coriolopsis gallica , an efficient decolorizer of alkaline effluents. Arch. Microbiol. 1998;171:31–36.10.1007/s002030050674
- Sun X, Zhang R, Zhang Y. Production of lignocellulolytic enzymes by Trametes gallica and detection of polysaccharide hydrolase and laccase activities in polyacrylamide gels. J. Basic Microbiol. 2004;44:220–231.10.1002/(ISSN)1521-4028
- Carbajo JM, Junca H, Terrón MC, et al. Tannic acid induces transcription of laccase gene cglcc1 in the white-rot fungus Coriolopsis gallica. Can. J. Microbiol. 2002;48:1041–1047.10.1139/w02-107
- Díaz R, Saparrat MC, Jurado M, et al. Biochemical and molecular characterization of Coriolopsis rigida laccases involved in transformation of the solid waste from olive oil production. Appl. Microbiol. Biotechnol. 2010;88:133–142.10.1007/s00253-010-2723-z
- Surget-Groba Y, Montoya-Burgos JI. Optimization of de novo transcriptome assembly from next-generation sequencing data. Genome Res. 2010;20:1432–1440.10.1101/gr.103846.109
- Wang L, Li P, Brutnell TP. Exploring plant transcriptomes using ultra high-throughput sequencing. Brief Funct Genomics. 2010;9:118–128.10.1093/bfgp/elp057
- Luca F, Hudson RR, Witonsky DB, et al. A reduced representation approach to population genetic analyses and applications to human evolution. Genome Res. 2011;21:1087–1098.10.1101/gr.119792.110
- Smith SA, Wilson NG, Goetz FE, et al. Resolving the evolutionary relationships of molluscs with phylogenomic tools. Nature. 2011;480:364–367.10.1038/nature10526
- Haas BJ, Zody MC. Advancing RNA-Seq analysis. Nat. Biotechnol. 2010;28:421–423.10.1038/nbt0510-421
- Zhang W, Chen J, Yang Y, et al. A practical comparison of de novo genome assembly software tools for next-generation sequencing technologies. PLoS ONE. 2011;6:e17915.10.1371/journal.pone.0017915
- Lin Y, Li J, Shen H, et al. Comparative studies of de novo assembly tools for next-generation sequencing technologies. Bioinformatics. 2011;27:2031–2037.10.1093/bioinformatics/btr319
- Schafleitner R, Tincopa L, Palomino O, et al. A sweet potato gene index established by de novo assembly of pyrosequencing and Sanger sequences and mining for gene-based microsatellite markers. BMC Genomics. 2010;11:2–10.10.1186/1471-2164-11-604
- Birol I, Jackman SD, Nielsen CB, et al. De novo transcriptome assembly with ABySS. Bioinformatics. 2009;25:2872–2877.10.1093/bioinformatics/btp367
- Shi CY, Yang H, Wei CL, et al. Deep sequencing of the Camellia sinensis transcriptome revealed candidate genes for major metabolic pathways of tea-specific compounds. BMC Genomics. 2011;12:2–14.10.1186/1471-2164-12-131
- Martin J, Bruno VM, Fang Z, et al. Rnnotator: an automated de novo transcriptome assembly pipeline from stranded RNA-Seq reads. BMC Genomics. 2010;11:2–8.10.1186/1471-2164-11-663
- Kumar S, Blaxter ML. Comparing de novo assemblers for 454 transcriptome data. BMC Genomics. 2010;11:2–12.10.1186/1471-2164-11-571
- Grabherr MG, Haas BJ, Yassour M, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011;29:644–652.10.1038/nbt.1883
- Schulz MH, Zerbino DR, Vingron M, et al. Oases: robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics. 2012;28:1086–1092.
- Hernandez D, François P, Farinelli L, et al. De novo bacterial genome sequencing: millions of very short reads assembled on a desktop computer. Genome Res. 2008;18:802–809.10.1101/gr.072033.107
- Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–829.10.1101/gr.074492.107
- Li R, Zhu H, Ruan J, et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2010;20:265–272.10.1101/gr.097261.109
- Tao X, Gu YH, Wang HY, et al. Digital gene expression analysis based on integrated de novo transcriptome assembly of sweet potato [Ipomoea batatas (L.) Lam]. PLoS ONE. 2012;7:e36234.10.1371/journal.pone.0036234
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25-1–R25-10.10.1186/gb-2009-10-3-r25
- Goecks J, Nekrutenko A, Taylor J, et al. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010;11:2–13.10.1186/gb-2010-11-8-r86
- Blankenberg D, Kuster GV, Coraor N, et al. A Web‐Based Genome Analysis Tool for Experimentalists. Curr. Protoc. Mol. Biol. 2010;19:1–33.
- Conesa A, Götz S, García-Gómez JM, et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–3676.10.1093/bioinformatics/bti610
- Huang X, Madan A. CAP3: a DNA sequence assembly program. Genome Res. 1999;9:868–877.10.1101/gr.9.9.868
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2− ΔΔCT method. Methods. 2001;25:402–408.10.1006/meth.2001.1262
- Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009;10:57–63.10.1038/nrg2484
- Garg R, Patel RK, Tyagi AK, et al. De novo assembly of chickpea transcriptome using short reads for gene discovery and marker identification. DNA Res. 2011;18:53–63.10.1093/dnares/dsq028
- Fiedler T, Hudder A, McKay S, et al. The transcriptome of the early life history stages of the California Sea Hare Aplysia californica. Comp. Biochem. Physiol. Part D Genomics Proteomics. 2010;5:165–170.10.1016/j.cbd.2010.03.003
- Paszkiewicz K, Studholme DJ. De novo assembly of short sequence reads. Brief Bioinform. 2010;11:457–472.10.1111/fml.1985.29.issue-1-2
- Zhang XG, He C, Zhang YZ. Cloning of laccase gene from a constructed cDNA library of Trametes gallica. Chin. J. Biochem. Mol. Biol. 2009;6:528–533.