
Abstract
Patients with severe Wernicke–Korsakoff syndrome (WKS) associated with vitamin B1 (thiamine) deficiency (TD) show enduring impairment of memory formation. The mechanisms of memory impairment induced by TD remain unknown. Here, we show that hippocampal degeneration is a potential microendophenotype (an endophenotype of brain disease at the cellular and synaptic levels) of WKS in pyrithiamine-induced thiamine deficiency (PTD) mice, a rodent model of WKS. PTD mice show deficits in the hippocampus-dependent memory formation, although they show normal hippocampus-independent memory. Similarly with WKS, impairments in memory formation did not recover even at 6 months after treatment with PTD. Importantly, PTD mice exhibit a decrease in neurons in the CA1, CA3, and dentate gyrus (DG) regions of the hippocampus and reduced density of wide dendritic spines in the DG. Our findings suggest that TD induces hippocampal degeneration, including the loss of neurons and spines, thereby leading to enduring impairment of hippocampus-dependent memory formation.
Graphical abstract
PTD mice showed deficits in hippocampus-dependent memory and loss of neurons and spines in the hippocampus.
Thiamine (vitamin B1), a water-soluble vitamin, was identified initially as oryzanin from rice bran.Citation1) Thiamine is highly concentrated in the brain, skeletal muscle, heart, liver, and kidney.Citation2) Thiamine pyrophosphate, which is the active form of vitamin B1, functions as a coenzyme for enzymes involved in glucose and amino acid metabolism.Citation3,4) Thiamine is known to play important roles in the central nervous system. In humans, thiamine deficiency (TD) is associated with Wernicke–Korsakoff syndrome (WKS), a neurodegenerative disease that is characterized by neuropathological impairments, neurological deficits including ataxia and oculomotor disturbance, and cognitive dysfunction such as memory impairments.Citation5–7)
Importantly, severe WKS cases have enduring memory impairment, although treatment with thiamine at the early stage of symptomatic TD largely reverses the classical neurological signs such as ophthalmoplegia, nystagmus, and gait ataxia.Citation8–14) Neuropathological changes have been identified in the thalamus and mammillary nuclei of the hypothalamus, periaqueductal gray matter, brainstem nuclei, particularly the superior and inferior colliculi, and anterior cerebellar vermis of WKS cases.Citation9,10,15,16) However, the mechanisms underlying the severe memory deficits observed in WKS cases remain unclear.
The rodent model of WKS is induced by feeding a thiamine-deficient diet combined with daily injections of pyrithiamine, a thiamine antagonist (pyrithiamine-induced thiamine deficiency; PTD).Citation17,18) Similar to WKS cases, PTD rodents exhibit neurologic signs such as ataxia, loss of the righting reflex, and seizures, neuropathologic changes in several brain regions including the thalamus, mammillary bodies, and brainstem nuclei, and memory impairment.Citation15,19–22)
Short-term memory (STM) is labile and converted into stable long-term memory (LTM) through a process known as memory consolidation.Citation23–25) The consolidation of episodic memory depends on the function of the hippocampus.Citation26) There is abundant evidence that damage to the hippocampus causes severe anterograde amnesia in which new memories cannot be formed.Citation27,28) Importantly, a few studies have reported that PTD rats show impairment in spatial and avoidance memories, both of which are formed in a hippocampus-dependent manner, in Morris water maze and inhibitory avoidance tasks, respectively.Citation29–31) Nevertheless, the impact of PTD on the hippocampus has not been examined in detail.
Microendophenotypes are phenotypes of brain disorders at the level of molecular dynamics, neurons and synapses, and neural circuits.Citation32) In this study, we tried to identify the microendophenotypes of the memory deficits observed in PTD mice, a mouse model of WKS. We first performed hippocampus-dependent and -independent memory tasks and found that PTD mice show deficits in the formation of hippocampus-dependent LTM. We next examined damage to the hippocampus caused by PTD at the cellular and spine levels and found that PTD mice show decreased numbers of neurons and wide dendritic spines in the hippocampus, suggesting that neurodegeneration of the hippocampus at the cellular and synaptic levels is a possible microendophenotype of the memory impairment caused by PTD.
Materials and methods
Mice
All experiments were conducted according to the Guide for the Care and Use of Laboratory Animals, Japan Neuroscience Society and Tokyo University of Agriculture. All animal experiments performed in this study were approved by the Animal Care and Use Committee of Tokyo University of Agriculture. Male C57BL/6N mice were obtained from Charles River (Yokohama, Japan). Heterozygous Thy1-EGFP line M mice were maintained by crossing with C57BL/6N mice.Citation33) The mice were housed in cages of 5 or 6, maintained on a 12 h light/dark cycle, and allowed ad libitum access to pellet food and water. The mice were at least 8 weeks of age at the start of the experiments, and all behavioral procedures were conducted during the light phase of the cycle. All experiments were conducted blind to the treatment condition of the mice.
PTD treatment
PTD was induced by dietary thiamine deprivation and treatment with a thiamine antagonist, as described previously.Citation17,34) The mice were divided randomly into PTD and control groups. To habituate the animals to a powdered diet, both groups were given a powdered thiamine-containing diet (control diet; Oriental Yeast Co., Ltd., Tokyo, Japan; Table ) ad libitum for 1 week. The PTD group received a powdered TD diet (Oriental Yeast Co., Ltd.; Table ) ad libitum during the PTD treatment period (days 0−11) and daily intraperitoneal (i.p.) injections of the thiamine antagonist pyrithiamine hydrobromide (5 μg in 0.1 mL of saline/10 g body weight; Sigma, MO, USA) at days 1−10. At the end of PTD treatment (day 11), when the last sign (seizures) of TD was observed in the PTD group, the PTD mice received a single i.p. thiamine injection (1 mg in 0.1 mL of saline/10 g body weight; Sigma) and the control diet was resumed. The control group received the control diet ad libitum and daily i.p. saline injections (0.1 mL/10 g body weight) at days 1−10.
Table 1. Composition of control and TD diets.
Rotarod test
The rotarod test was performed as described previously.Citation35) The mice were placed on a rotating drum (3 cm in diameter; O’Hara & Co., Ltd., Tokyo, Japan). The drum was initially rotated at a speed of 4 rpm after which it was accelerated gradually to 40 rpm over the course of 5 min. The amount of time that a mouse remained on the accelerating rod (latency to fall) was recorded as an indicator of their motor coordination.
Contextual fear conditioning task
The mice were trained and tested in conditioning chambers (17.5 × 17.5 × 15 cm) that had a stainless steel grid floor through which a footshock could be delivered.Citation36–38) Training consisted of placing the mice in the chamber and delivering an unsignaled footshock (2 s duration, 0.4 mA) 148 s later, and the mice were returned to their home cage at 30 s after the footshock (training). Memory was assessed at 30 min and 24 h later by calculating the percentage of time spent freezing during 5 min when re-placed in the training context (test). Freezing behavior (defined as a complete lack of movement, except for respiration) was measured automatically as described previously (O’Hara & Co., Ltd.).Citation39)
Morris water maze task
The Morris water maze task was performed as described previously.Citation40–42) The mice were trained with 2 trials per day at an interval of 1 min for 4 days. The mice were trained at approximately the same time every day. In the probe test at 24 h after the last training session on day 4, the platform was removed and the mice were allowed to swim for 1 min. We measured the time that the mice spent in each quadrant (opposite [OP], adjacent right [AR], target quadrant [TQ], and adjacent left [AL]).
Social recognition task
The social recognition task was performed as described previously.Citation40,42–44) Adult mice were placed into individual plastic cages, identical to those in which they were normally housed (30 × 17 × 12 cm), in the experimental room. After a period of 60 min, a juvenile mouse was placed into a cage with an adult mouse for 3 min (training). The duration of the adult’s social investigation behavior was quantified using a stopwatch. Social investigation was defined as described previously.Citation45) Memory was assessed 24 h later by recording the length of social investigation time exhibited by the subject to the same juvenile (test) for the same duration as training (3 min). A recognition index was calculated as the ratio of the social investigation times during the test and training sessions.
Cued fear conditioning task
A similar procedure was used for cued fear conditioning as described previously.Citation46) The mice were placed in the training context, and 2 min later, a 30 s tone (70 dB) co-terminated with a 2 s footshock (0.4 mA), and then the mice were returned to their home cage at 30 s after the footshock (training). After 24 h, the mice were placed in a different chamber, and 2 min later, the tone was replayed for 3 min (test). Cued fear memory was assessed as the percentage of time the mice spent freezing during the 3 min tone.
Conditioned taste aversion task
The conditioned taste aversion task was performed as described previously.Citation43) After moving to individual cages, the mice were given access to water from 2 bottles for 60 min, and then returned to their home cages. On day 2, the mice were given access to water for 45 min. On days 3 and 4, water access was limited to 30 min. On day 5 (the day of conditioning), the mice were given access to 2 bottles containing a 0.1% saccharin solution for 15 min. After 40 min, the mice were injected (2% of body weight) with saline or 0.1 M LiCl, and then returned to their cages. To prevent dehydration, the mice were given access to water for 15 min at 2 h after injection. On day 6 (the test day), the mice were presented with the saccharin solution and water in separate bottles (bottle positions counterbalanced across cages). To determine consumption, the bottles were weighed before and after testing. An aversion index was calculated as follows: saccharin solution consumed/(saccharin solution consumed + water consumed).
Open field test
The open field test was performed as described previously.Citation35,36) The mice were placed into the center of a square open field chamber (50 × 50 × 40 cm) that was surrounded by white walls. The total length of the path traveled (total distance) and the time spent in the center square (30 × 30 cm; %center) were measured over the course of 5 min using an automatic monitoring system (O’Hara & Co., Ltd.).
Immunohistochemistry
Immunohistochemistry was performed as described previously.Citation40,47) After anesthetization, mice were perfused with 4% paraformaldehyde. The brains were then removed, fixed overnight, transferred to 30% sucrose, and stored at −80 °C. Coronal sections (30 μm) were generated using a cryostat. Free floating sections were treated with 1% H2O2 and then incubated overnight with a mouse monoclonal anti-NeuN primary antibody (1:500; Millipore, MA, USA) in a blocking solution (TBS buffer plus 10% goat serum albumin). Subsequently, the sections were washed with PBS and then incubated with horseradish peroxidase-conjugated donkey anti-rabbit IgG (1:500; Jackson ImmunoResearch, PA, USA). NeuN signals were amplified with TSA-FCM (Invitrogen, CA, USA). The sections were mounted on slides and coverslipped using mounting medium (Merck, Darmstadt, Germany). Fluorescence images were acquired using a confocal microscope (TCS SP8; Leica, Wetzlar, Germany) and LAS AF software (Leica) using a 40× (for the hippocampus) or 20× (for the medial prefrontal cortex [mPFC] and amygdala) objective. Confocal 2 μm z-stack images were obtained. Equal cut-off thresholds were applied to all slices. Structures were defined anatomically according to the atlas of Paxinos and Franklin.Citation48) Quantification of NeuN-immunoreactive (IR) cells in sections of the dorsal hippocampus (50 × 50 μm/sub region; bregma between −1.46 and −1.82 mm), amygdala (100 × 100 μm/sub region; bregma between −1.22 and −1.34 mm), and mPFC (100 × 100 μm/sub region; bregma between +2.10 and +1.98 mm) was performed using an analog counter (for the hippocampus) and a computerized image analysis system (for the amygdala and mPFC; WinROOF version 5.6 software; Mitani Corporation, Fukui, Japan). NeuN-IR cells were counted bilaterally with a fixed sample window across at least 3 sections by an experimenter blind to the treatment condition.
Dendritic spine analysis
After anesthetization, mice were perfused with 4% paraformaldehyde. The brains were then removed, fixed overnight, transferred to 30% sucrose, and stored at −80 °C. Coronal sections (30 μm) were generated using a cryostat. Fluorescence images were acquired using a confocal microscope (TCS SP8; Leica) and LAS AF software (Leica). Equal cut-off thresholds were applied to all slices. Segments were imaged under a 63×/1.4 N.A. oil-immersion objective and 15× zoom. All confocal stacks were acquired at 512 × 512 pixel resolution with a z-step of 0.5 μm. The settings for pinhole size and gain were optimized initially and remained constant throughout imaging to ensure the images were digitized under consistent illumination. These parameters resulted in dendritic segments of 12.3 μm in length. Spine density was calculated as the number of spines divided by dendritic segment length. Spine head width was defined as the maximum diameter of the spine head. Data were additionally segregated into narrow and wide spines using a median split based on head width.Citation49)
Data analysis
One-way or two-way factorial or repeated analysis of variance (ANOVA) followed by Newman–Keuls post hoc comparison were used to analyze the effects of PTD, time, and drugs. A paired t-test was used to analyze differences in the time spent in the TQ compared with the other quadrants in the Morris water maze task and to analyze differences in social investigation times within each group between training and test in the social recognition task. All values in the text and figure legends represent the mean ± standard error of the mean (SEM).
Results
Body weight loss and ataxia induced by PTD treatment and recovery from PTD
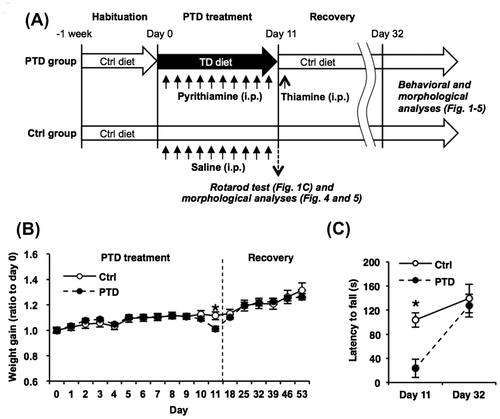
PTD treatment induces acute physical deficits in mice including body weight loss, ataxia, and seizures.Citation17) We first examined the effects of PTD and recovery from PTD on body weight and motor coordination. The mice were fed the TD diet (Table ) with daily systemic injections of pyrithiamine for 10 days (days 1–10; PTD; Fig. (A)) and then received a single systemic injection of thiamine at day 11, followed by refeeding the control diet for more than 3 weeks (recovery treatment; Fig. (A)). Two-way repeated measures ANOVA comparing body weight gain revealed a significant PTD (control vs. PTD mice)×time (days 0–32) interaction (PTD, F[1, 17] = 0.01, p > 0.05; time, F[14, 238] = 85.00, p < 0.05; PTD × time, F[14, 238] = 6.92, p < 0.05; Fig. (B)). Newman–Keuls post hoc test revealed that PTD mice showed significantly less weight gain than control mice at day 11 (p < 0.05; Fig. (B)), although PTD mice had shown normal body weight gain compared to the control group from day 18 (p > 0.05; Fig. (B)). Two-way repeated measures ANOVA comparing latency to fall revealed a significant PTD × time (days 11 and 32) interaction (PTD, F[1, 16] = 5.58, p < 0.05; time, F[1, 16] = 15.64, p < 0.05; PTD × time, F[1, 16] = 3.79, p < 0.05; Fig. (C)). PTD mice showed a significantly shorter latency to fall than control mice at day 11 (p < 0.05; Fig. (C)), although they showed normal latency to fall at day 32 (p > 0.05; Fig. (C)). It is important to note that seizures were observed in the PTD mice at day 11, but they disappeared immediately after thiamine injection (data not shown). These results indicated that PTD treatment induced significant body weight loss and abnormal motor coordination, but these deficits were ameliorated by the recovery treatment for less than 3 weeks, suggesting that severe and acute physical deficits are induced by PTD, but are improved by the recovery treatment. According to these observations, the following behavioral analyses were performed using PTD mice that had recovered for more than 3 weeks after PTD.
Fig. 1. Body weight loss and ataxia by PTD treatment and its rescue by recovery treatment.
PTD mice display impairment of hippocampus-dependent LTM formation
PTD rats have shown impaired memory performance in the Morris water maze and passive avoidance tasks,Citation29–31) both of which are hippocampus-dependent.Citation50,51) Therefore, we examined whether PTD mice that had recovered from PTD for more than 3 weeks had impaired hippocampus-dependent learning and memory.
We first performed the contextual fear conditioning task. In this task, the mice learn an association between context (conditioned stimulus; CS) and electrical footshock-induced fear (unconditioned stimulus; US) and form a hippocampus-dependent aversive memory.Citation52) The mice were trained with a single footshock (0.4 mA), and behavioral freezing was assessed at 30 min (test 1) and 24 h (test 2) after training. Two-way repeated measures ANOVA revealed a significant PTD × time (test 1 and test 2) interaction (PTD, F[1, 22 = 6.48, p < 0.05; time, F[1, 22] = 0.02, p > 0.05; PTD × time, F[1, 22] = 7.27, p < 0.05; Fig. (A)). Newman–Keuls post hoc test revealed that PTD mice showed significantly less freezing than the control group during test 2, but showed normal freezing during test 1. These results indicated that PTD mice showed an impairment of 24 h LTM (p < 0.05; Fig. (A)), but showed normal 2 h STM (p > 0.05; Fig. (A)), suggesting that PTD mice show impairment in the consolidation of contextual fear memory without affecting STM.
Fig. 2. Impaired hippocampus-dependent LTM formation in PTD mice.
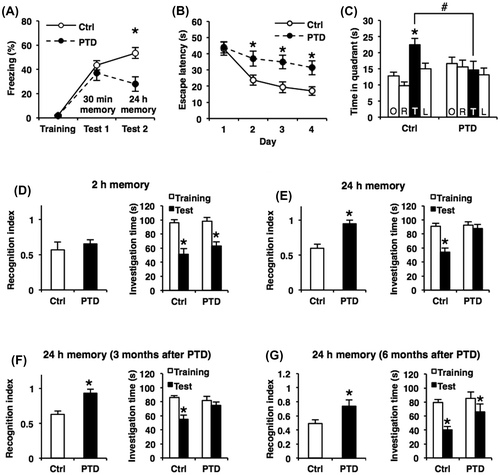
We next performed the Morris water maze task. In this task, the animals learn to escape to a hidden platform in a swimming pool using spatial cues located around the pool and form a hippocampus-dependent spatial memory. The mice were trained with 2 trials per day for 4 days (at 1 min intervals). Two-way repeated measures ANOVA revealed significant effects of PTD and time (days 1–4) (PTD, F[1, 36] = 8.76, p < 0.05; time, F[3, 108] = 14.59, p < 0.05; PTD × time, F[3, 108] = 2.38, p > 0.05; Fig. (B)). Newman–Keuls post hoc test revealed that PTD mice showed significantly longer escape latency than control mice at days 2–4 (p < 0.05; Fig. (B)). These results indicated that PTD mice show impairment of memory performance. To measure the formation of spatial memory, the mice were given a probe trial in which the platform was removed from the pool at 24 h after the last training session on day 4. A paired t test revealed that control mice searched selectively in the TQ compared with the other quadrants (TQ vs. OP, AR, and AL, p < 0.05; Fig. (C)), while PTD mice failed to do so (p > 0.05; Fig. (C)). Importantly, PTD mice showed significantly lower time spent in the TQ than control mice (Newman–Keuls post hoc test following one-way ANOVA, F[1, 36] = 5.56, p < 0.05; Fig. (C)). Consistent with the results shown in Fig. (B), these results indicated that PTD mice failed to form spatial memory. Collectively, these observations indicated that PTD mice show impaired spatial memory formation.
We finally performed the social recognition task. In this task, the mice form a hippocampus-dependent non-aversive social memory.Citation42,53) This task measures the difference in time taken to investigate a juvenile male mouse by comparing between the first and second exposures for 3 min. The second exposure was performed at 2 (STM) or 24 h (LTM) after the first exposure. We assessed the recognition index (the ratio of the second investigation time relative to the first investigation time). One-way ANOVA revealed a significant effect of PTD when memory was tested at 24 h (F[1, 24] = 20.12, p < 0.05; Fig. (E)), but not at 2 h (F[1, 23] = 0.49, p > 0.05; Fig. (D)), after training. Newman–Keuls post hoc test revealed that PTD mice showed a significantly worse LTM recognition index (p < 0.05; Fig. (E)) but normal STM recognition index (p > 0.05; Fig. (D)) compared to control mice. Consistently, PTD mice failed to reduce investigation time during test compared to training when tested at 24 h (p > 0.05; Fig. (E)), but not at 2 h (p < 0.05; Fig. (D)), after training, although the control group did reduce this significantly at both time points (p < 0.05; Fig. (D) and (E)). These results indicated that PTD mice display impaired LTM, but showed normal STM, of social recognition, suggesting that PTD mice show impairment in the consolidation of social recognition memory.
WKS patients show no recovery from amnesia even when they have recovered from TD. Therefore, we examined if the impaired memory performance observed in PTD mice recovered at 3–6 months after PTD treatment. The mice were trained and tested at 3 and 6 months after PTD. PTD mice still showed significantly worse recognition indices of 24 h LTM than control mice at 3 and 6 months (Newman–Keuls post hoc test following one-way ANOVA, 3 months, F[1, 20] = 14.69, p < 0.05; 6 months, F[1, 20] = 6.27, p < 0.05; Fig. (F) and (G)). Consistently, PTD mice failed to reduce investigation time when trained and tested at 3 and 6 months, although PTD mice formed weak LTM at 6 months after PTD (3 months, p > 0.05; 6 months, p < 0.05; Fig. (F) and (G)), whereas control mice did reduce investigation time (p < 0.05; Fig. (F), (G)). These results indicated that PTD mice showed chronic impairment of memory formation even at 3–6 months after PTD.
Collectively, our observations suggest that PTD mice show deficits in the consolidation of hippocampus-dependent memory (contextual, spatial, and social recognition memories). Importantly, our observation that PTD mice showed impaired memory formation even at 6 months after PTD suggests that PTD mice display memory deficits similar to those of WKS patients.
PTD mice display normal amygdala-dependent LTM formation
We examined the effects of PTD on memory formation that is independent of hippocampal function. To do this, we first performed the amygdala-dependent cued (tone) fear conditioning task.Citation52) In this task, a tone was paired with a footshock (0.4 mA), and the mice learn the association of tone (CS) and fear (US). The tone was replayed in a different chamber at 24 h after training and freezing was assessed. One-way ANOVA revealed no significant effect of PTD (F[1, 44] = 0.17, p > 0.05; Fig. (A)). This result indicated that PTD mice showed normal LTM of cued fear.
Fig. 3. Normal amygdala-dependent LTM formation in PTD mice.
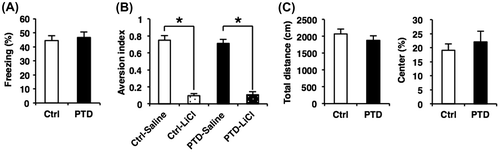
We next performed the conditioned taste aversion task that is amygdala- and insular cortex-dependent.Citation54,55) This task examines the formation of associative memory between saccharin (CS) and a malaise-inducing agent (i.p. injection of LiCl; US). The mice were given access to the saccharin solution and 40 min later, they were injected with saline or 0.1 M LiCl. At 24 h after conditioning, the mice were presented with the saccharin solution and water in separate bottles and the aversion index (the ratio of saccharin solution consumption relative to total solution consumption) was assessed. Two-way ANOVA revealed a significant effect of drug (saline and LiCl) (PTD, F[1, 36] = 0.10, p > 0.05; drug, F[1, 36] = 216.44, p < 0.05; PTD × drug, F[1, 36] = 0.33, p > 0.05; Fig. (B)). Newman–Keuls post hoc test revealed that both LiCl-injected control and PTD mice showed a significantly lower aversion index than saline-injected groups at 24 h after conditioning (p < 0.05; Fig. (B)) and comparable aversion indices (one-way ANOVA, F[1, 18] = 0.07, p > 0.05; Fig. (B)). These results indicated that PTD mice showed normal LTM of conditioned taste aversion.
Taken together, these observations suggested that PTD treatment does not affect the formation of amygdala-dependent LTM and that PTD treatment impairs hippocampus-dependent LTM.
It is important to note that PTD mice showed normal locomotor activity and anxiety-related behavior (time spent in the center of field) in the open field test compared to the control group (one-way ANOVA, total distance, F[1, 18] = 0.95, p > 0.05; % center, F[1, 18 = 0.47, p > 0.05; Fig. (C)). Furthermore, our results (Fig. (D)–(G)) also indicated that PTD mice displayed normal social investigation time during training in the social recognition task compared with control mice, suggesting that PTD mice show normal social behavior. Collectively, these observations strongly suggest that the memory impairment observed in PTD mice is not attributable to abnormal locomotor and/or emotional behavior (social and anxiety-related behavior).
PTD mice display neuronal loss in the hippocampus
Our findings that PTD mice show impairment of hippocampus-dependent memory formation suggest that PTD induces hippocampal dysfunction. To examine this possibility, we first examined the effects of PTD on the number of hippocampal neurons using immunohistochemistry. We measured the number of NeuN, a neuronal marker, -IR cells in the hippocampus, amygdala, and mPFC just after (day 11) and at 3 weeks after (day 32) PTD treatment. One-way ANOVA at day 11 revealed significant effects of PTD in the CA1, CA3, and dentate gyrus (DG) regions of the hippocampus (CA1, F[1, 10] = 6.34, p < 0.05; CA3, F[1, 10] = 5.47, p < 0.05; DG, F[1, 10] = 6.41, p < 0.05; Fig. (A)), but not in the lateral (LA), basolateral (BLA), and central (CeA) regions of the amygdala or prelimbic (PL) and infralimbic (IL) regions of the mPFC (LA, F[1, 9] = 1.83, p > 0.05; BLA, F[1, 9] = 1.74, p > 0.05; CeA, F[1, 9] = 1.80, p > 0.05; PL, F[1, 8] = 0.01, p > 0.05; IL, F[1, 8] = 0.14, p > 0.05; Fig. (B) and (C)). Newman–Keuls post hoc test revealed that PTD mice showed significantly less NeuN-IR cells in the CA1, CA3, and DG regions of the hippocampus than control mice (p < 0.05; Fig. (A)). These results indicated that PTD treatment decreased the number of neurons in the hippocampus without affecting the amygdala and mPFC.
Fig. 4. Hippocampal neuronal loss in PTD mice.
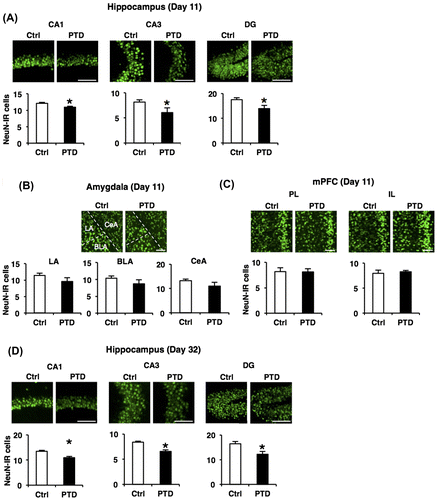
Importantly, PTD mice still displayed significantly less NeuN-IR cells in the CA1, CA3, and DG regions of the hippocampus than control mice at day 32 (at 3 weeks after PTD treatment; Newman–Keuls post hoc test following one-way ANOVA, CA1, F[1, 6] = 11.73, p < 0.05; CA3, F[1, 6] = 18.60, p < 0.05; DG, F[1, 6] = 6.50, p < 0.05; Fig. (D)). These results indicated that the hippocampal neuron loss observed in PTD mice did not recover even at 3 weeks after PTD. Collectively, these observations indicate that PTD causes chronic hippocampus-specific neuron loss.
PTD mice display decreased dendritic spine density on hippocampal DG neurons
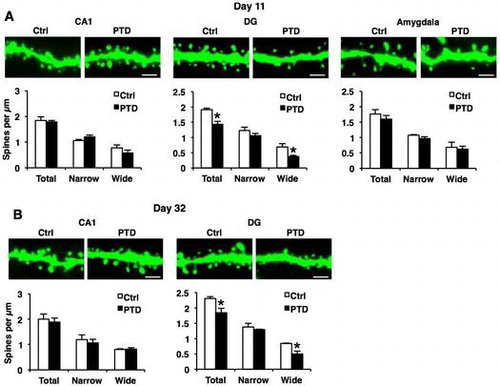
Dendritic spine morphology is important for memory formation and storage.Citation56–59) It is possible that PTD causes abnormal dendritic spine morphology in hippocampal neurons. To examine this, we analyzed the density and morphology of dendritic spines in the hippocampal and amygdala neurons of PTD mice using Thy1-EGFP line M mice (Thy1-GFP mice) that express EGFP in sparse neurons.Citation33) Thy1-GFP mice were analyzed just after (day 11) and 3 weeks (day 32) after PTD treatment. One-way ANOVA comparing spine density at day 11 revealed a significant effect of PTD only in hippocampal DG dendrites (total, F[1, 4] = 20.02, p < 0.05; Fig. (A)), but not in hippocampal CA1 and amygdala dendrites (CA1, total, F[1, 4] = 0.13, p > 0.05; amygdala, total, F[1, 4] = 0.81, p > 0.05; Fig. (A)). Newman–Keuls post hoc test revealed that PTD mice showed a significant decrease in spine density on DG dendrites compared with control mice (p < 0.05; Fig. (A)). Furthermore, analyses of spine width revealed a significant decrease in wide spines, but not narrow spines, in DG dendrites in PTD mice (Newman–Keuls post hoc test following one-way ANOVA, DG, narrow, F[1, 4] = 1.52, p > 0.05; wide, F[1, 4] = 8.74, p < 0.05; CA1, narrow, F[1, 4] = 2.90, p > 0.05; wide, F[1, 4] = 1.64, p > 0.05; amygdala, narrow, F[1, 4] = 2.66, p > 0.05; wide, F[1, 4] = 0.10, p > 0.05; Fig. (A)). These results indicate that PTD mice show a decrease in spine density, especially wide spines, in hippocampal DG neurons.
Fig. 5. Decreased dendritic spine density on hippocampal DG neurons in PTD mice.
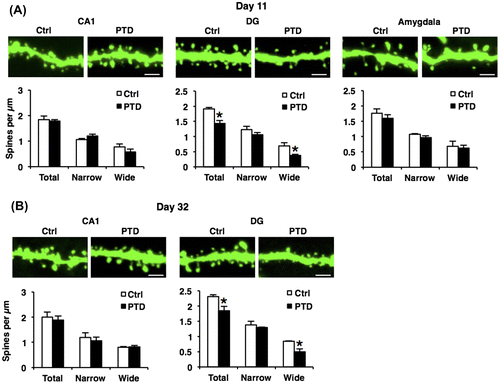
Similarly with the results shown in Fig. , PTD mice still displayed significant decreases in total and wide spines in DG dendrites compared with control mice at day 32 (Newman–Keuls post hoc test following one-way ANOVA, DG: total, F[1, 4] = 8.45, p < 0.05; narrow, F[1, 4] = 0.69, p > 0.05; wide, F[1, 4] = 11.73, p < 0.05; CA1: total, F[1, 4] = 0.17, p > 0.05; narrow, F[1, 4] = 0.29, p > 0.05; wide, F[1, 4] = 0.10, p > 0.05; Fig. (B)). Collectively, these observations indicate that PTD causes chronic loss of wide spines in hippocampal DG neurons.
Discussion
In this study, we examined the mechanisms underlying the memory impairment observed in PTD mice, a mouse model of WKS, and identified the microendophenotype of memory impairment in PTD mice. PTD mice showed impairment in the formation of LTM in three different hippocampus-dependent memory tasks at 3 weeks after PTD, although severe and acute physical deficits such as body weight loss and ataxia were reversed at this time point. Importantly, PTD mice still showed deficits in LTM formation at 6 months after PTD. In contrast, PTD mice show normal formation of LTM in two hippocampus-independent tasks. Interestingly, PTD mice showed significant decreases in the number of NeuN-positive neurons in the CA1, CA3, and DG areas of the hippocampus and wide dendritic spines of excitatory neurons in the DG. These observations suggest that TD induces degeneration of the hippocampus, thereby impairing the formation of hippocampus-dependent memory. From these observations, we propose that this hippocampal degeneration at the cellular and synaptic levels is a possible microendophenotype for the induction of memory impairment in PTD mice.Citation32)
Importantly, damage to the hippocampus causes anterograde and retrograde amnesia; new memories cannot be formed and recently formed memories cannot be retrieved, but remote memories that were formed before the hippocampus was damaged can be retrieved. WKS cases exhibit both amnesia, shown by poor performance to form a memory, and are unable to recall recently formed memories,Citation60,61) although they show intact immediate, primary, and working memory. In this study, we found that PTD mice show severe deficits in the formation of hippocampus-dependent memories, but show normal hippocampus-independent memories. A few studies using rodents have also shown that PTD rats exhibit deficits in spatial and inhibitory avoidance memories, both of which are hippocampus-dependent, supporting our observations.Citation29–31) Collectively, the evidence observed in WKS cases and rodent models raises the possibility that TD leads to hippocampus-dependent deficits in memory formation.
WKS patients show neuropathological changes in the medial thalamus and mammillary bodies, periaqueductal gray matter, brainstem nuclei, superior and inferior colliculi, and anterior cerebellar vermis.Citation9,10,15) However, in this study, we found neuron loss and deceases in wide dendritic spines in the hippocampus of PTD mice even at 3 weeks after PTD treatment, although other brain regions, such as the amygdala and mPFC, both of which also play critical roles in memory performance, did not show these abnormalities. Several previous studies using PTD or TD rats detected degenerating fibers and terminals in the hippocampal CA1, CA2, CA3, and DG regions at 1 week after more than 14 days of PTD using silver-stained sections,Citation62) neuronal degeneration and loss of the hippocampal CA3 region using hematoxylin-eosin stained sections and light microscopic examination,Citation63) and hyperintense regions in the dorsal and ventral hippocampus using magnetic resonance imaging.Citation34) Collectively, these findings using PTD or TD rodents suggest that TD causes degeneration of the hippocampus, thereby impairing memory performance. Hippocampus contains a high density of N-methyl-D-aspartate receptorsCitation64,65) and thereby, is highly sensitive to excitotoxic neurodegeneration.Citation66,67) Therefore, it is possible that this high sensitivity to excitotoxicity leads to loss of neurons and spines in the hippocampus of PTD mice. Moreover, seizures and neurodegenerative diseases such as Alzheimer’s disease causes dendritic spine abnormality including spine loss in the DG neurons,Citation68–70) raising the possibility that dendritic spines in DG neurons are susceptible to pathological damages by PTD. It is important to examine whether neural degeneration at the cellular, synaptic, and spine levels occurs in the hippocampus of WKS cases.
WKS cases have been shown to recover quickly from ocular and ataxia abnormalities following treatment with parenteral thiamine (after days, weeks, and 1–2 months). In contrast, the amnestic symptoms in WKS patients recover more slowly and incompletely; a year or so is required for the maximum degree of recovery.Citation61,71–74) Similarly to WKS, we observed that PTD mice showed deficits in memory formation even at 6 months after PTD treatment, although abnormalities of motor performance and body weight recovered quickly (~3 weeks). Thus, the impaired memory performance induced by TD is more permanent than the other symptoms, suggesting that TD more severely damages the brain regions required for the performance of learning and memory. Collectively, our observations at the behavioral level strongly suggest that TD leads to long-term dysfunction of the brain regions required for memory performance, supporting our conclusion that TD causes hippocampal degeneration.
Dendritic spines form the postsynaptic component of most excitatory synapses.Citation75) Spine density and structure are thought to reflect and/or determine connectivity between neurons at the synapse and contribute to learning and memory as well as synaptic function.Citation56–58) Importantly, previous studies suggest that neuronal degeneration and loss in the hippocampus correlate with impairments of hippocampal long-term potentiation, a synaptic model for LTM,Citation76,77) and hippocampus-dependent memory.Citation78,79) Additionally, a reduction of spine density was observed in hippocampal neurons in mouse models of Alzheimer’s disease, suggesting that a reduction in spine density in the hippocampal DG correlates with memory impairment.Citation80–82) On the other hand, spine-head volume reflects the strength of the synapse. Wide spines, such as mushroom spines, make these synapses functionally strongerCitation83–85) and have been suggested to be stable “memory spines” that are used store a memory.Citation86,87) Our findings that PTD mice show a loss of wide spines in the DG of the hippocampus suggest that this abnormality of dendritic spines in the hippocampus is linked with impairments of memory performance observed in PTD mice.
Previously, we have shown the impairment of memory performance such as deficits in the formation of LTM in mice using pharmacology and genetics.Citation36,38,41,44,46,88) In the present study, we showed that PTD mice showed impairment in the formation of hippocampus-dependent memories. Importantly, we found these deficits in memory performance using the C57BL/6N mouse strain and identical experimental procedures for the memory tasks. Therefore, we emphasize that the memory impairment observed in PTD mice is comparable with pharmacological and genetic models showing significant and severe deficits in learning and memory. It is also important to note that mice showing comparable loss of body weight by fasting for 48 h did not show impaired memory performance and hippocampal degeneration (data not shown), suggesting that the memory deficits in PTD mice are not attributed to a side effect of TD via lowering food intake.
In this study, we have shown that TD leads to severe impairment of hippocampus-dependent memory formation and neuron loss in the CA1, CA3, and DG regions and spine loss in the DG of the hippocampus. From these observations, we suggest that these impairments of hippocampal function are possible microendophenotypes of TD that cause deficits in memory performance.
Author contributions
SK is responsible for the hypothesis development and overall design of the research and experiment, and supervised the experimental analyses. HI and SK co-wrote the paper. SH assisted in writing the paper. TK performed morphological experiments. SO, TK, KN, and TW performed behavioral experiments. HI, TK, SO, KN, and SH analyzed data. TW assisted in preparing the control and TD diets. All authors read and approved this paper.
Disclosure statement
No potential conflict of interest was reported by the authors.
Funding
This work was supported by The Science Research Promotion Fund, The Promotion and Mutual Aid Corporation for Private Schools of Japan, Grant-in-Aids for Scientific Research (A) [15H02488], Scientific Research (B) [23300120, 20380078]; Challenging Exploratory Research [24650172, 26640014]; Grant-in-Aids for Scientific Research on Priority Areas-Molecular Brain Science [18022038, 22022039]; Grant-in-Aid for Scientific Research on Innovative Areas (Research in a proposed research area) [24116008, 24116001, 23115716]; Core Research for Evolutional Science and Technology (CREST), Japan; The Sumitomo Foundation, Japan, The Naito Foundation, Japan; and the Takeda Science Foundation, Japan.
References
- Suzuki U, Shimamura T, Odake S. Über Oryzanin, ein Bestandteil der Reiskleie und seine physiologische Bedeutung [Over Oryzanin, a component of the rice bran and its physiological meaning]. Imp. Univ. Tokyo; 1913.
- Singleton CK, Martin PR. Molecular mechanisms of thiamine utilization. Curr. Mol. Med. 2001;1:197–207.10.2174/1566524013363870
- Devlin TM. Textbook of biochemistry. Hoboken: Wiley; 2011.
- Butterworth RF, Kril JJ, Harper CG. Thiamine-dependent enzyme changes in the brains of alcoholics: relationship to the Wernicke–Korsakoff syndrome. Alcohol. Clin. Exp. Res. 1993;17:1084–1088.10.1111/acer.1993.17.issue-5
- Halliday G, Cullen K, Harding A. Neuropathological correlates of memory dysfunction in the Wernicke–Korsakoff syndrome. Alcohol Alcohol Suppl. 1994;2:245–251.
- Thomson AD. Mechanisms of vitamin deficiency in chronic alcohol misusers and the development of the Wernicke–Korsakoff syndrome. Alcohol Alcohol Suppl. 2000;35:2–7.
- Fattal-Valevski A, Azouri-Fattal I, Greenstein YJ, et al. Delayed language development due to infantile thiamine deficiency. Dev. Med. Child Neurol. 2009;51:629–634.10.1111/dmcn.2009.51.issue-8
- Caine D, Halliday GM, Kril JJ, et al. Operational criteria for the classification of chronic alcoholics: identification of Wernicke’s encephalopathy. J. Neurol. Neurosurg. Psychiatry. 1997;62:51–60.10.1136/jnnp.62.1.51
- Victor M, Adams RD, Collins GH. The Wernicke–Korsakoff syndrome. A clinical and pathological study of 245 patients, 82 with post-mortem examinations. Contemporary Neurology Series. Oxford: Oxford University Press; 1971.
- Victor M, Adams RD, Collins GH. The Wernicke–Korsakoff Syndrome: and related neurologic disorders due to alcoholism and malnutrition. Philadelphia (PA): FA Davis Company; 1989.
- Victor M, Herman K, White EE. A psychological study of the Wernicke–Korsakoff syndrome. Results of Wechsler-Bellevue intelligence scale and Wechsler memory scale testing at different stages in the disease. Q. J. Stud. Alcohol. 1959;20:467–479.
- Butters N, Cermak LS. Alcoholic Korsakoff’s syndrome: an information-processing approach to amnesia. San Diego (CA): Academic Press; 2014.
- Kopelman MD. The Korsakoff syndrome. Br. J. Psychiatry. 1995;166:154–173.10.1192/bjp.166.2.154
- Talland G. Deranged memory: a psychonomic study of the amnesic syndrome. San Diego (CA): Academic Press; 1965.
- Langlais PJ, Zhang SX, Savage LM. Neuropathology of thiamine deficiency: an update on the comparative analysis of human disorders and experimental models. Metab. Brain Dis. 1996;11:19–37.10.1007/BF02080929
- Wang X, Wang B, Fan Z, et al. Thiamine deficiency induces endoplasmic reticulum stress in neurons. Neuroscience. 2007;144:1045–1056.10.1016/j.neuroscience.2006.10.008
- Calingasan NY, Gandy SE, Baker H, et al. Novel neuritic clusters with accumulations of amyloid precursor protein and amyloid precursor-like protein 2 immunoreactivity in brain regions damaged by thiamine deficiency. Am. J. Pathol. 1996;149:1063–1071.
- Eliash S, Dror V, Cohen S, et al. Neuroprotection by rasagiline in thiamine deficient rats. Brain Res. 2009;1256:138–148.10.1016/j.brainres.2008.11.097
- Dror V, Eliash S, Rehavi M, et al. Neurodegeneration in thiamine deficient rats-A longitudinal MRI study. Brain Res. 2010;1308:176–184.10.1016/j.brainres.2009.10.032
- Watanabe I. Pyrithiamine-induced acute thiamine-deficient encephalopathy in the mouse. Exp. Mol. Pathol. 1978;28:381–394.10.1016/0014-4800(78)90012-6
- Troncoso JC, Johnston MV, Hess KM, et al. Model of Wernicke’s encephalopathy. Arch. Neurol. 1981;38:350–354.10.1001/archneur.1981.00510060052007
- Langlais PJ, Savage LM. Thiamine deficiency in rats produces cognitive and memory deficits on spatial tasks that correlate with tissue loss in diencephalon, cortex and white matter. Behav. Brain Res. 1995;68:75–89.10.1016/0166-4328(94)00162-9
- Abel T, Lattal KM. Molecular mechanisms of memory acquisition, consolidation and retrieval. Curr. Opin. Neurobiol. 2001;11:180–187.10.1016/S0959-4388(00)00194-X
- McGaugh JL. Memory–a century of consolidation. Science. 2000;287:248–251.10.1126/science.287.5451.248
- Silva AJ, Kogan JH, Frankland PW, et al CREB and memory. Annu. Rev. Neurosci. 1998;21:127–148.10.1146/annurev.neuro.21.1.127
- Burgess N, Maguire EA, O’Keefe J. The human hippocampus and spatial and episodic memory. Neuron. 2002;35:625–641.10.1016/S0896-6273(02)00830-9
- Squire LR, Wixted JT. The cognitive neuroscience of human memory since H.M. Annu. Rev. Neurosci. 2011;34:259–288.10.1146/annurev-neuro-061010-113720
- Squire LR. Memory and the hippocampus: a synthesis from findings with rats, monkeys, and humans. Psychol. Rev. 1992;99:195–231.10.1037/0033-295X.99.2.195
- Carvalho FM, Pereira SRC, Pires RGW, et al. Thiamine deficiency decreases glutamate uptake in the prefrontal cortex and impairs spatial memory performance in a water maze test. Pharmacol. Biochem. Behav. 2006;83:481–489.10.1016/j.pbb.2006.03.004
- Nakagawasai O, Tadano T, Niijima F, et al. Immunohistochemical estimation of rat brain somatostatin on avoidance learning impairment induced by thiamine deficiency. Brain Res. Bull. 2000;51:47–55.10.1016/S0361-9230(99)00201-4
- Langlais PJ, Mandel RJ, Mair RG. Diencephalic lesions, learning impairments, and intact retrograde memory following acute thiamine deficiency in the rat. Behav. Brain Res. 1992;48:177–185.10.1016/S0166-4328(05)80155-X
- Kida S, Kato T. Microendophenotypes of psychiatric disorders: phenotypes of psychiatric disorders at the level of molecular dynamics, synapses, neurons, and neural circuits. Curr. Mol. Med. 2015;15:111–118.10.2174/1566524015666150303002128
- Feng G, Mellor RH, Bernstein M, et al. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51.10.1016/S0896-6273(00)00084-2
- Pfefferbaum A, Adalsteinsson E, Bell RL, et al. Development and resolution of brain lesions caused by pyrithiamine- and dietary-induced thiamine deficiency and alcohol exposure in the alcohol-preferring rat: a longitudinal magnetic resonance imaging and spectroscopy study. Neuropsychopharmacology. 2007;32:1159–1177.10.1038/sj.npp.1301107
- Hasegawa S, Furuichi T, Yoshida T, et al. Transgenic up-regulation of alpha-CaMKII in forebrain leads to increased anxiety-like behaviors and aggression. Mol. Brain. 2009;2:6.10.1186/1756-6606-2-6
- Inaba H, Tsukagoshi A, Kida S. PARP-1 activity is required for the reconsolidation and extinction of contextual fear memory. Mol. Brain. 2015;8:63.10.1186/s13041-015-0153-7
- Mamiya N, Fukushima H, Suzuki A, et al. Brain region-specific gene expression activation required for reconsolidation and extinction of contextual fear memory. J. Neurosci. 2009;29:402–413.10.1523/JNEUROSCI.4639-08.2009
- Suzuki A, Josselyn SA, Frankland PW, et al. Memory reconsolidation and extinction have distinct temporal and biochemical signatures. J. Neurosci. 2004;24:4787–4795.10.1523/JNEUROSCI.5491-03.2004
- Anagnostaras SG, Josselyn SA, Frankland PW, et al. Computer-assisted behavioral assessment of pavlovian fear conditioning in mice. Learn. Mem. 2000;7:58–72.10.1101/lm.7.1.58
- Ishikawa R, Kim R, Namba T, et al. Time-dependent enhancement of hippocampus-dependent memory after treatment with memantine: implications for enhanced hippocampal adult neurogenesis. Hippocampus. 2014;24:784–793.10.1002/hipo.22270
- Kim R, Moki R, Kida S. Molecular mechanisms for the destabilization and restabilization of reactivated spatial memory in the Morris water maze. Mol. Brain. 2011;4:9.10.1186/1756-6606-4-9
- Suzuki A, Fukushima H, Mukawa T, et al. Upregulation of CREB-mediated transcription enhances both short- and long-term memory. J. Neurosci. 2011;31:8786–8802.10.1523/JNEUROSCI.3257-10.2011
- Fukushima H, Maeda R, Suzuki R, et al. Upregulation of calcium/calmodulin-dependent protein kinase iv improves memory formation and rescues memory loss with aging. J. Neurosci. 2008;28:9910–9919.10.1523/JNEUROSCI.2625-08.2008
- Nomoto M, Takeda Y, Uchida S, et al. Dysfunction of the RAR/RXR signaling pathway in the forebrain impairs hippocampal memory and synaptic plasticity. Mol. Brain. 2012;5:8.10.1186/1756-6606-5-8
- Thor DH, Holloway WR. Social memory of the male laboratory rat. J. Comp. Physiol. Psychol. 1982;96:1000–1006.10.1037/0735-7036.96.6.1000
- Kida S, Josselyn S A, de Ortiz SP, et al. CREB required for the stability of new and reactivated fear memories. Nat. Neurosci. 2002;5:348–355.10.1038/nn819
- Fukushima H, Zhang Y, Archbold G, et al. Enhancement of fear memory by retrieval through reconsolidation. Elife. 2014;3:e02736.
- Paxinos G, Franklin KB. The mouse brain in stereotaxic coordinates. San Diego (CA): Academic Press; 1997.
- Fitzgerald PJ, Pinard CR, Camp MC, et al. Durable fear memories require PSD-95. Mol. Psychiatry. 2015;20:901–912.10.1038/mp.2014.161
- Morris R. Developments of a water-maze procedure for studying spatial learning in the rat. J. Neurosci. Methods. 1984;11:47–60.10.1016/0165-0270(84)90007-4
- Stubley-Weatherly L, Harding JW, Wright JW. Effects of discrete kainic acid-induced hippocampal lesions on spatial and contextual learning and memory in rats. Brain Res. 1996;716:29–38.10.1016/0006-8993(95)01589-2
- Phillips RG, LeDoux JE. Differential contribution of amygdala and hippocampus to cued and contextual fear conditioning. Behav. Neurosci. 1992;106:274–285.10.1037/0735-7044.106.2.274
- Kogan JH, Franklandand PW, Silva AJ. Long-term memory underlying hippocampus-dependent social recognition in mice. Hippocampus. 2000;10:47–56.10.1002/(ISSN)1098-1063
- Berman DE, Dudai Y. Memory extinction, learning anew, and learning the new: dissociations in the molecular machinery of learning in cortex. Science. 2001;291:2417–2419.10.1126/science.1058165
- Josselyn SA, Kida S, Silva AJ. Inducible repression of CREB function disrupts amygdala-dependent memory. Neurobiol. Learn. Mem. 2004;82:159–163.10.1016/j.nlm.2004.05.008
- Hausser M. Diversity and dynamics of dendritic signaling. Science. 2000;290:739–744.10.1126/science.290.5492.739
- Spruston N. Pyramidal neurons: dendritic structure and synaptic integration. Nat. Rev. Neurosci. 2008;9:206–221.10.1038/nrn2286
- Lefebvre JL, Sanes JR, Kay JN. Development of dendritic form and function. Annu. Rev. Cell Dev. Biol. 2015;31:741–777.10.1146/annurev-cellbio-100913-013020
- Segal M. Dendritic spines, synaptic plasticity and neuronal survival: activity shapes dendritic spines to enhance neuronal viability. Eur. J. Neurosci. 2010;31:2178–2184.10.1111/(ISSN)1460-9568
- Parkin AJ, Leng NR. Neuropsychology of the amnesic syndrome. Hove: Psychology Press; 1993.
- Kopelman MD, Thomson AD, Guerrini I, et al. The Korsakoff syndrome: clinical aspects, psychology and treatment. Alcohol Alcohol. 2009;44:148–154.10.1093/alcalc/agn118
- Langlais PJ, Zhang SX. Cortical and subcortical white matter damage without Wernicke’s encephalopathy after recovery from thiamine deficiency in the rat. Alcohol Clin. Exp. Res. 1997;21:434–443.
- Ikarashi Y, Iizuka S, Imamura S, et al. Effects of Yokukansan, a traditional Japanese medicine, on memory disturbance and behavioral and psychological symptoms of dementia in thiamine-deficient rats. Biol. Pharm. Bull. 2009;32:1701–1709.10.1248/bpb.32.1701
- Greenamyre JT, Young AB, Penney JB. Quantitative autoradiographic distribution of L-[3H]glutamate-binding sites in rat central nervous system. J. Neurosci. 1984;4:2133–2144.
- Monaghan DT, Cotman CW. Distribution of N-methyl-D-aspartate-sensitive L-[3H]glutamate-binding sites in rat brain. J. Neurosci. 1985;5:2909–2919.
- Nadler JV, Perry BW, Cotman CW. Intraventricular kainic acid preferentially destroys hippocampal pyramidal cells. Nature. 1978;271:676–677.10.1038/271676a0
- Choi DW. Glutamate neurotoxicity and diseases of the nervous system. Neuron. 1988;1:623–634.10.1016/0896-6273(88)90162-6
- Tejada J, Garcia-Cairasco N, Roque AC. Combined role of seizure-induced dendritic morphology alterations and spine loss in newborn granule cells with mossy fiber sprouting on the hyperexcitability of a computer model of the dentate gyrus. PLoS Comput. Biol. 2014;10:e1003601.10.1371/journal.pcbi.1003601
- Singh SP, He X, McNamara JO, et al. Morphological changes among hippocampal dentate granule cells exposed to early kindling-epileptogenesis. Hippocampus. 2013;23:1309–1320.10.1002/hipo.22169
- Roy DS, Arons A, Mitchell TI, et al. Memory retrieval by activating engram cells in mouse models of early Alzheimer’s disease. Nature. 2016;531:508–512.10.1038/nature17172
- Baddeley AD, Warrington EK. Amnesia and the distinction between long- and short-term memory. J. Verbal Learn. Verbal Behav. 1970;9:176–189.10.1016/S0022-5371(70)80048-2
- Lishman WA. Organic psychiatry: the psychological consequences of cerebral disorder. Hoboken (NJ): Wiley-Blackwell; 1987.
- Zubaran C, Fernandes JG, Rodnight R. Wernicke–Korsakoff syndrome. Postgrad. Med. J. 1997;73:27–31.10.1136/pgmj.73.855.27
- Day E, Bentham P, Callaghan R, et al. Thiamine for Wernicke–Korsakoff syndrome in people at risk from alcohol abuse. Cochrane Database Syst. Rev. 2004;1; CD004033.
- Harris KM. Structure, development, and plasticity of dendritic spines. Curr. Opin. Neurobiol. 1999;9:343–348.10.1016/S0959-4388(99)80050-6
- Teyler TJ, Discenna P. Long-term potentiation as a candidate mnemonic device. Brain Res. 1984;7:15–28.10.1016/0165-0173(84)90027-4
- Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39.10.1038/361031a0
- Serenó L, Coma M, Rodríguez M, et al. A novel GSK-3beta inhibitor reduces Alzheimer’s pathology and rescues neuronal loss in vivo. Neurobiol. Dis. 2009;35:359–367.10.1016/j.nbd.2009.05.025
- Tomiyama T, Matsuyama S, Iso H, et al. A mouse model of amyloid beta oligomers: their contribution to synaptic alteration, abnormal tau phosphorylation, glial activation, and neuronal loss in vivo. J. Neurosci. 2010;30:4845–4856.10.1523/JNEUROSCI.5825-09.2010
- Lanz TA, Carter DB, Merchant KM. Dendritic spine loss in the hippocampus of young PDAPP and Tg2576 mice and its prevention by the ApoE2 genotype. Neurobiol. Dis. 2003;13:246–253.10.1016/S0969-9961(03)00079-2
- Jacobsen JS, Wu CC, Redwine JM, et al. Early-onset behavioral and synaptic deficits in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. 2006;103:5161–5166.10.1073/pnas.0600948103
- Perez-Cruz C, Nolte MW, van Gaalen MM, et al. Reduced spine density in specific regions of CA1 pyramidal neurons in two transgenic mouse models of Alzheimer’s disease. J. Neurosci. 2011;31:3926–3934.10.1523/JNEUROSCI.6142-10.2011
- Matsuzaki M, Ellis-Davies GC, Nemoto T, et al. Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nat. Neurosci. 2001;4:1086–1092.10.1038/nn736
- Nimchinsky EA, Yasuda R, Oertner TG, et al. The number of glutamate receptors opened by synaptic stimulation in single hippocampal spines. J. Neurosci. 2004;24:2054–2064.10.1523/JNEUROSCI.5066-03.2004
- Ashby MC, Maier SR, Nishimune A, et al. Lateral diffusion drives constitutive exchange of ampa receptors at dendritic spines and is regulated by spine morphology. J. Neurosci. 2006;26:7046–7055.10.1523/JNEUROSCI.1235-06.2006
- Kasai H, Matsuzaki M, Noguchi J, et al. Structure-stability-function relationships of dendritic spines. Trends Neurosci. 2003;26:360–368.10.1016/S0166-2236(03)00162-0
- Bourne J, Harris KM. Do thin spines learn to be mushroom spines that remember? Curr. Opin. Neurobiol. 2007;17:381–386.10.1016/j.conb.2007.04.009
- Zhang Y, Fukushima H, Kida S. Induction and requirement of gene expression in the anterior cingulate cortex and medial prefrontal cortex for the consolidation of inhibitory avoidance memory. Mol. Brain. 2011;4:4.10.1186/1756-6606-4-4