
Abstract
Amino acids are essential components in all organisms because they are building blocks of proteins. They are also produced industrially and used for various purposes. For example, L-glutamate is used as the component of “umami” taste and lysine has been used as livestock feed. Recently, many kinds of amino acids have attracted attention as biological regulators and are used for a healthy life. Thus, to clarify the mechanism of how amino acids are biosynthesized and how they work as biological regulators will lead to further effective utilization of them. Here, I review the leucine-induced-allosteric activation of glutamate dehydrogenase (GDH) from Thermus thermophilus and the relationship with the allosteric regulation of GDH from mammals. Next, I describe structural insights into the efficient production of L-glutamate by GDH from an excellent L-glutamate producer, Corynebacterium glutamicum. Finally, I review the structural biology of lysine biosynthesis of thermophilic bacterium and archaea.
Graphical abstract
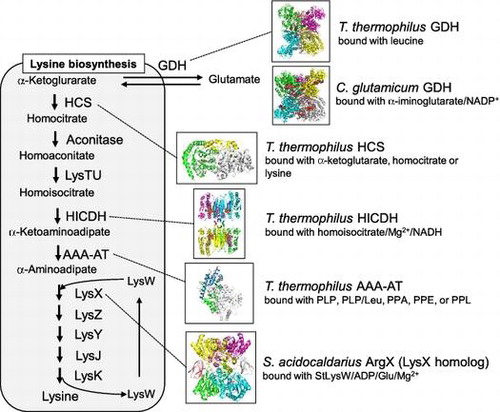
Glutamate dehydrogenase and lysine biosynthetic pathway of Thermus thermophilus and the other organisms.
Amino acids are components of protein and they are essential for all organisms. Today, huge amounts of amino acids are produced industrially by processes including enzymatic conversion and fermentation.Citation1) For example, 1.8 million tons of monosodium glutamate, which is used as the “umami” flavor enhancer all over the world, are produced per year by fermentation using Corynebacterium glutamicum. The fermentation of L-glutamate is achieved using wild-type C. glutamicum under appropriate culture conditions.Citation2) The production of L-glutamate is possible with this micro-organism using three devices: the inhibition of the 2-OG dehydrogenase complex (ODHC) to switch metabolic flow from the tricarboxylic acid cycle to L-glutamate biosynthesis,Citation3) the activation of the mechanosensitive channel, NCgl1221, to export the L-glutamate that was accumulated in cells into the extracellular space under L-glutamate-producing conditions,Citation4) and the conversion of α-ketoglutarate (α-KG) to L-glutamate in which CgGDH plays a role. C. glutamicum is also used for the industrial fermentation of L-lysine. For this purpose, the strain with lysine-analog resistance was screened and used.Citation5,6) The mechanism was also studied and mutations in the regulatory domain of aspartate kinase, which catalyzes the first step of lysine biosynthesis, was the key to canceling the feedback inhibition to enable the overproduction of L-lysine.Citation7,8) Thus, we have used micro-organisms well to produce many kinds of amino acids. On the other hand, amino acids have recently attracted attention as biological regulators, which directly affect the regulation of intracellular metabolic processes in humans. For example, recent studies have shown that one of the branched-chain amino acids, leucine, acted as a signal for synthesis of protein in the cell even though leucine is a building block of protein,Citation9) and it is widely used clinically and as a supplement for building-up muscle. The biological functions of many other amino acids have also been explored and their usefulness for maintaining a healthy life has been demonstrated. Therefore, demand for amino acids has increased in recent years. In this review, I describe unique features and structures of enzymes involved in lysine biosynthesis and L-glutamate production in bacteria and a kind of archaea (Fig. ). First, the allosteric activation of GDH from Thermus thermophilus and a common mechanism with that of human GDH is described. Then, the capture of the reaction intermediate of GDH through the structure determination of GDH from C. glutamicum and insights into the efficient production of L-glutamate by this enzyme are described. Finally, I review the structural biology of lysine biosynthesis of T. thermophilus and describe the unique features of the enzymes, which possess characteristics of ancient enzymes.
Fig. 1. Structures of glutamate dehydrogenase and enzymes involved in lysine biosynthesis.
I. Allosteric regulation of glutamate dehydrogenase from T. thermophilus
GDH catalyzes reversible conversion between α-ketoglutarate and L-glutamate using NAD(P)(H) as a cofactor (Fig. (A)). T. thermophilus possesses two putative gdh genes (gdhA and gdhB) in its genome, both of which encode GDH sharing approximately 46% identity in amino acid sequence. Although GDH has been purified from T. thermophilus HB8 and characterized,Citation10) it was not clear which isozyme was purified and characterized. Boliver and colleagues reported that recombinant GDH from T. thermophilus HB27, named GdhB in our present study, has GDH activity,Citation11) however, the function of GdhA remains to be elucidated. When increased amounts of GdhA were added into the reaction mixture of GdhB, reductive amination activity decreased and oxidative deamination activity increased.Citation12) The recombinant GdhA and GdhB were co-expressed in Escherichia coli and they were co-purified, suggesting that GdhA and GdhB form a hetero-complex. These results suggest that changes in both the specific activity and preference of the reaction direction of GdhB by the addition of GdhA were caused by the interaction of GdhA with GdhB. Next, allosteric regulation of the hetero-oligomeric GDH was examined. As a consequence, GDH activity was increased, particularly by the addition of branched-chain amino acids, hydrophobic amino acids, and aromatic amino acids. Among them, leucine exhibited the highest activation in both directions (Fig. (A)).
Fig. 2. Structure of GDH from T. thermophilus.
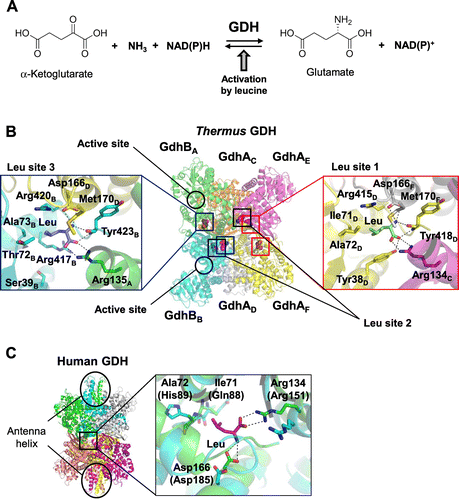
To elucidate the mechanism for allosteric activation by leucine, the crystal structure of the GdhA/GdhB/Leu complex was determined.Citation13) GdhA/GdhB forms a hetero-hexameric structure, which is composed of four GdhA subunits and two GdhB subunits (Fig. (B)). The overall structure suggests that two dimers of GdhA and one dimer of GdhB are assembled to form a hetero-hexameric structure. Although no substrate is found in the active site of the GdhA/GdhB/Leu complex, six leucine molecules are found at the interfaces of three subunits. These six leucine-binding sites can be classified into three types (Leu site 1-3) based on the difference in subunit organization. Leu site 1 is formed by the three GdhA subunits, Leu site 2 is formed by two GdhA subunits and one GdhB subunit, and Leu site 3 is formed by one GdhA subunit and two GdhB subunits. The GdhA/GdhB/Leu complex contains two for each site. The amino acid residues forming Leu site 1 are identical to those forming Leu site 2, and leucine is recognized in the same way at both sites; therefore, figures are made only for sites 1 and 3. At Leu site 1 formed by GdhAD, GdhAE, and GdhAF, leucine is recognized by specific interactions in a specific manner (Fig. (B)). At Leu site 3, formed by GdhBA, GdhBB, and GdhAE, the recognition was somewhat weaker than Leu site 1. Tyr38 is replaced with Ser39 at site 3. The replacement causes loss of a hydrogen bond with the carboxyl group of the bound leucine and hydrophobic interaction with the side chain of leucine at Leu site 3. Replacement of Ile71 with Thr72 reduces hydrophobic contact at Leu site 3. These differences possibly provide Leu site 1 (and Leu site 2) and Leu site 3 with different sensitivity to leucine; the former is a high-affinity site, and the latter is a low-affinity site. The mutations of the residues involved in binding leucine exhibited decreased sensitivity toward leucine. This suggests that the leucine binding site plays a major role in binding leucine for allosteric activation.
To explore the universality of the leucine-induced allosteric regulation of GDH, the amino acid sequence of T. thermophilus GDH was compared with GDH from different organisms. The amino acid residues responsible for leucine binding in T. thermophilus GDH are well conserved in Thermotoga maritima GDH, suggesting a similar allosteric mechanism, possibly by leucine and/or the other hydrophobic amino acids in T. maritima GDH. In contrast, other bacterial and plant GDHs exhibited no apparent amino acid conservation at the leucine binding site. Surprisingly, Asp166 and Arg134, which recognize α-amino and α-carboxyl groups of leucine in the structure of T. thermophilus GDH, are conserved in human and bovine GDHs. Although these residues are located somewhat apart from each other in the structure of human GDH (hGDH) (Fig. (C)),Citation14) it is possible that these residues move to bind leucine. In mammalian GDHs, although the residues at the positions necessary for recognition of the leucyl side chain are replaced with different residues, there is still space for binding leucine. The mutational analysis of hGDH2, which is subject to prominent allosteric activation by leucine, revealed that these residues are involved in binding leucine.Citation13) Li et al. found inhibitors of bovine GDH by high throughput screening and they determined the structures in complex with those inhibitorsCitation15); bovine GDH also has a hypothetical leucine-binding site. Of these inhibitors, hexachrolophene (HCP) and bithionol bind at the position near the leucine-binding site, suggesting that the inter-subunit region is a commonly-used allosteric site. To date, the biological significance of activation of GDH by leucine in T. thermophilus and mammalian has not been well-understood; however, there may be a common role in the cellular system. Further studies on regulation of GDH are expected to clarify the significance on metabolism.
II. Structure and function of glutamate dehydrogenase from C. glutamicum
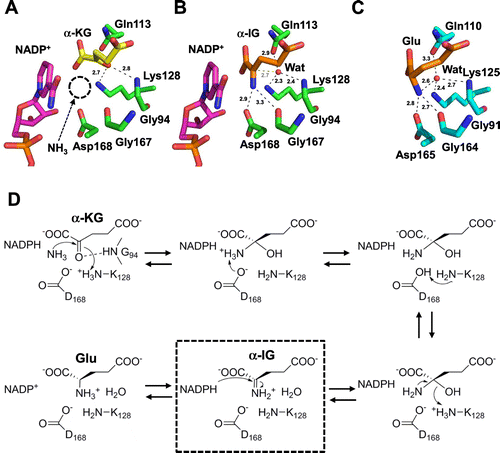
As mentioned above, C. glutamicum is known to be an excellent L-glutamate producer and is used for industrial L-glutamate fermentation. In addition to the unique system of the fermentation, GDH from C. glutamicum (CgGDH) plays an important role in the industrial fermentation of L-glutamate due to its high L-glutamate-producing activity. CgGDH has activity in an NADP(H)-dependent manner,Citation16) suggesting that CgGDH contributes the over-production of L-glutamate. This idea is further supported by two reports: one showed that high level expression of CgGDH in C. glutamicum cells increased the intracellular L-glutamate pool.Citation17) The other reported that disruption of the CgGDH gene resulted in accumulation of α-KG with simultaneous decreasing in L-glutamate production.Citation18) Recently, the structure of CgGDH was determined in an α-KG/NADPH-bound form (Fig. (A)),Citation19) which revealed the residues involved in the cofactor and α-KG binding, but the mechanisms for catalysis and high L-glutamate-producing activity remain to be elucidated. We determined the crystal structure of CgGDH complexed with NADP+ and α-iminoglutarate (α-IG) (Fig. (B)). In the structure, α-IG was bound to the substrate-binding site with the α-imino group stacked by the nicotinamide ring of the coenzyme. When we compared the structure with the α-KG/NADPH complex, both structures were similar to each other; however, α-IG and α-KG took different conformations at the α-imino/α-keto group (Fig. (A) and (B)). The α-imino group of α-IG was twisted approximately 90º from the α-keto group of α-KG. The α-keto group of α-KG is recognized by the ε-amino group of Lys128 through an electrostatic interaction and with Gly94 through a hydrogen bond. The widely opened space between α-KG and NADPH appeared to accept an ammonia molecule through hydrogen bonds with Asp168 and Gly167. Through the attack by ammonia and deprotonation by Asp168, a carbinolamine intermediate is formed (Fig. (D)). The intermediate undergoes elimination of a water molecule through protonation by Lys128. The newly formed α-imino group of α-IG stacks with the plane of the nicotineamide ring of NADP+. The water molecule, which is present at the opposite side of the nicotinamide ring, is derived from the α-keto group of α-KG. This state demonstrates a pre-hydride transfer state in a hypothesized reaction scheme with the imino-intermediate.
Fig. 3. Structure of GDH from C. glutamicum (CgGDH).
We also conducted molecular dynamics (MD) simulation to explore the mechanism of extreme preference for the L-glutamate-producing reaction of CgGDH, MD simulations for the modeled structure binding α-KG and NADPH (simulation 1), and modeled structure binding L-glutamate and NADP+ (simulation 2). In simulation 1, the α-keto group was recognized by Gly94 and Lys128. In contrast, the number of contacts was less around the α-amino group of L-glutamate in simulation 2 than the α-keto group of α-KG in simulation 1. The calculated numbers of hydrogen bonds between CgGDH and ligands were 9.6 and 7.4 for simulations 1 and 2, respectively. The interaction energy between CgGDH and bound ligands during the simulations were calculated to be −348.7 and −157.7 kcal mol−1 for simulation 1 and 2, respectively. These results suggested that CgGDH recognized α-KG well in order to advance the L-glutamate-producing reaction. Although the detailed molecular basis of the reaction preference is not fully clarified at the current stage, further study including comparative analysis and mutational analysis would be helpful for its elucidation.
III. Structure and function of enzymes involved in lysine biosynthesis of thermophilic bacterium and archaea
In general, bacteria and plants biosynthesize lysine via diaminopimelare (DAP) pathway, whereas yeast and fungi biosynthesize lysine via α-aminoadipate (known as AAA pathway). Although T. thermophilus is a bacteria, it synthesizes lysine via AAA pathway.Citation20) The enzymes involved in the first half of the pathway (conversion from α-KG to AAA) is similar to those in leucine biosynthetic pathway or tricarboxylic acid cycle, as is the case in yeast and fungal lysine biosynthesis.Citation21–25) However, the latter half of the pathway is totally different from the corresponding pathways of yeast and fungal lysine biosynthesis. It is a novel biosynthetic system, which involves amino-group carrier protein (AmCP) for the efficient reaction of biosynthetic enzymes.Citation26)
III.i. Homocitrate synthase
Homocitrate synthase (HCS), which is responsible for the first reaction in lysine biosynthesis, transfers the acetyl group from acetyl-CoA to α-KG to yield homocitrate (HC, 2-hydroxybuthane-1,2,4-tricarboxylate) and CoA (Fig. (A)). As commonly observed in enzymes responsible for the first reaction of amino acid biosynthesis, HCS from T. thermophilus (TtHCS) is inhibited by the end-product, L-lysine.Citation27) Lysine biosynthesis is also regulated at the transcriptional level. The promoter of the major lysine biosynthetic gene cluster starting from hcs gene of T. thermophilus is controlled by L-lysine through leader peptide-mediated transcriptional attenuation.Citation28) Many enzymes that are subject to feedback inhibition are fused with an additional domain to sense the final product for allosteric regulation. α-Isopropylmalate synthase (α-IPMS), a paralog of HCS, possesses a C-terminal regulatory domain to sense and transmit the signal of presence of the final product, leucine. Interestingly, HCS lacks the regulatory domain for sensing the allosteric inhibitors. This suggests that the regulatory mechanism of HCS is completely different from that of α-IPMS. Kinetic analysis showed that L-lysine inhibits HCS activity in a competitive manner against α-KG, suggesting that L-lysine possibly occupies the same site as α-KG to inhibit the activity. However, L-lysine and α-KG possess entirely different chemical structures; therefore, competition at the same site by these compounds is assumed to be difficult without a special device. The catalytic mechanism of HCS also remains elusive due to the lack of a crystal structure of HCS, although Qian et al. proposed that the catalytic dyad of Glu155 and His309* (asterisk indicates the residue from another subunit) was responsible for triggering the reaction.Citation29) To elucidate the mechanisms of the catalytic reaction and unique feedback inhibition, the crystal structures of TtHCS in complex with ligands were determined (Fig. ).Citation30) The α-KG-bound form contains α-KG and a divalent metal ion in the catalytic pocket (Fig. (B)). α-KG is bound to the enzyme by specific interactions with several residues. The C1-carboxyl group forms a hydrogen bond with Thr166. The C2-keto group is recognized by the guanidium group of Arg12. The hydrophobic side chain of C3-C4 atoms makes van der Waals contact with the side chains of Ala164 and Leu94. The C5-carboxyl group is stabilized by hydrogen bonds with His72 and with Arg133. Furthermore, the C5-carboxyl group makes a water molecule-mediated hydrogen bond with Ser135. Thus, the well-designed recognition of α-KG assures that TtHCS recognizes α-KG with high affinity.
Fig. 4. Structure of homocitrate synthase from T. thermophilus (TtHCS).
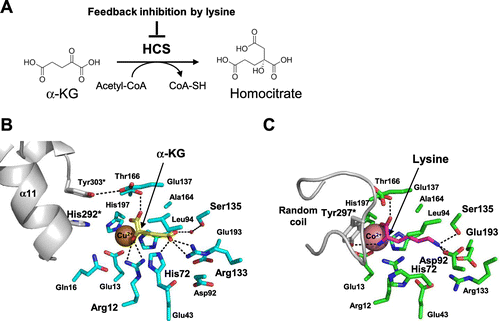
The HC-bound form of TtHCS contains an HC and a Cu2+ ion at the active site pocket. The carboxyl group at C1, which is transferred from acetyl-CoA, forms hydrogen bonds with Gln16 and His292* from small domain I of another subunit. These features revealed that His292*, corresponding to His309* of ScHCS, is directly involved in the catalytic mechanism. Consistent with this, Qian et al. proposed that His309* acts as a general base for proton abstraction from the reactive methyl group of acetyl-CoA.
The crystal structure of the lysine-bound form has similar overall structure with that of the α-KG-bound form. Interestingly, the binding site of L-lysine was essentially the same as that of α-KG (Fig. (C)). To recognize L-lysine at the same site, TtHCS takes a significant conformational change of side chains of residues around the active site. Arg133 moves away from the active site and forms ionic bonds with Glu43 and Glu193. These two Glu residues also make conformational changes from the α-KG binding form. Instead, Asp92 and Glu193 move into the active site and form ionic bonds with the ε-amino group of L-lysine. Ser135 forms a hydrogen bond with the ε-amino group of L-lysine. Asp219 and the carbonyl oxygen of Leu163 form water-mediated hydrogen bonds with the ε-amino group of L-lysine. His72, which forms a hydrogen bond with the γ-carboxyl group of α-KG, moved to the opposite rotamer and stacked with Arg12. The side chain of Arg12 also changed orientation and directly interacted with the α-amino group of L-lysine. Co2+ was coordinated with the α-carboxyl group and α-amino group of L-lysine. Tyr297 from another subunit formed a hydrogen bond with the α-amino group of L-lysine. These residues are well conserved among HCS from different bacteria, yeast, and fungi, suggesting that regulation of HCS by L-lysine is accomplished with a common mechanism. It is notable that His292*, which plays a key role in catalysis, moved away from the active site by −13 Å, suggesting that the L-lysine-bound form is the intrinsically inactive form. To cause this long displacement, small domain I significantly alters the structure, and a part of small domain II was observed to form two helices of the 3-helix bundle.
III.ii. Homoisocitrate dehydrogenase
Homoisocitrate dehydrogenase (HICDH) is an enzyme that catalyzes the fourth step of lysine biosynthesis (ΑΑΑ pathway) of yeast, fungi, and some kind of bacteria and archaea.Citation21,31,32) HICDH catalyzes a decarboxylating dehydrogenation of homoisocitrate (HIC) to form α-ketoadipate (Fig. (A)). HICDH is a member of the β-decarboxylating dehydrogenase family consisting of 3-isopropylmalate dehydrogenase [IPMDH; EC 1.1.1.85] in leucine biosynthesis,Citation33) isocitrate dehydrogenase [ICDH; EC 1.1.1.42] in the tricarboxylic acid (TCA) cycle,Citation34) and homoisocitrate dehydrogenase [HICDH; EC 1.1.1.87] in lysine biosynthesis via α-aminoadipate.Citation35) As homologous enzymes are believed to have evolved from a common ancestral enzyme into specified enzymes,Citation36–40) comparison of recognition mechanisms between each enzyme is expected to provide useful information on how these homologous enzymes acquired separated functions through the course of evolution. HICDH from T. thermophilus (TtHICDH) can recognize both HIC and IC as the substrate,Citation23) while HICDH from Saccharomyces cerevisiae (ScHICDH) strictly distinguishes HIC from IC, exhibiting strict specificity toward HIC.Citation41) As ancestral enzymes can catalyze the reactions for similar biosynthetic pathways by recognizing similar substrates, TtHICDH is supposed to retain characteristics of the ancestral enzyme. The substrate specificity of the β-decarboxylating dehydrogenase family is proposed to be determined by a few amino acid residues in the loop β3-α4 and the N-terminal region of the following α4 helix.Citation42) Indeed, substrate specificity was converted by amino acid replacement in the region of IPMDH and ICDH. Arg85 of TtHICDH was demonstrated to be a crucial determinant for the substrate specificity, and to play a key role in dual functions of TtHICDH as HICDH and ICDH by site-directed mutagenesis study.Citation23) However, including our studies, the precise molecular mechanism of substrate recognition of HICDHs has not been elucidated due to the lack of the structure of the substrate-bound form.Citation43–45) Recently, we determined the crystal structure of a quaternary complex of TtHICDH bound with HIC, NADH, and Mg2+, and the structure revealed the mechanism for the dual substrate specificity of TtHICDH.Citation46) In the crystal structure, TtHICDH forms a homo-tetrameric structure composed of two dimers interacting with each other head-to-head (Fig. ). A subunit is composed of two domains and the active site is located in the cleft between the two domains. HIC, Mg2+, and NADH are bound at the cleft of each subunit. The distance between the C4N atom of the nicotinamide ring of NADH and C2 atom of HIC was 3.5 Å, suggesting that the binding manner is suitable for hydride transfer between them. Thus, the quaternary complex mimics the enzymatically competent structure. Amino acid residues responsible for the recognition of the γ-moiety of substrates are mainly located on the βC-α4 loop and α4 helix. The carboxyl group at the terminal of the γ-moiety (carboxyethyl group) of HIC is recognized by Arg85 with ionic bonds and the aliphatic ethyl side chain of HIC also forms hydrophobic interactions with the Cδ carbon of Arg85 (Fig. (B)). In addition, Ser72 and Asn173* recognize the terminal carboxyl group via hydrogen bonds. TtHICDH exhibits comparable kinetic parameters for HIC and IC.Citation46) The model of the IC-bound form was constructed using the structure as a template (Fig. (C)). In the model, the γ-moiety (methylcarboxyl group) of IC was oriented toward Arg85 and formed bidentate ionic bonds with Arg85, while the methylcarboxyl group was too short to reach and was recognized by Ser72 or Asn173*. Summarizing the HIC-bound form and model of the IC-bound form, Arg85, Ser72, and Asn173* are involved in the recognition of HIC, whereas only Arg85 of the three residues is involved in the recognition of IC, suggesting that Arg85 plays dual roles in recognition of both HIC and IC. Next, to clarify the mechanism for discrimination between HIC and IC in ScHICDH, a model of the HIC-bound form of ScHICDH was constructed using the structure of TtHICDH as the template (Fig. (D)). The carboxyethyl group of HIC forms hydrogen bonds with Ser98, Ser108, and Asn208*. Ser98 and Asn208* correspond to Ser72 and Asn173* of TtHICDH, respectively, suggesting that these residues have the same functions as in TtHICDH. In ScHICDH, the Arg85 in TtHICDH is replaced with Val111. In place of the absence of Arg residue, ScHICDH has Ser108 at the corresponding position in the 3-D structure to strengthen the HIC binding. Interestingly, Ser108 can recognize the carboxyethyl group of HIC; however, it is located too far to contact the carboxyl group of the distal portion of IC. This is the most important difference between TtHICDH and ScHICDH. Val111 in ScHICDH, which corresponds with Arg85 in TtHICDH on alignment of amino acid sequences, interacts with the hydrophobic portion of the aliphatic carboxyethyl chain of HIC and may play a role in preventing the carboxymethyl group of IC from binding.
Fig. 5. Structure of homoisocitrate dehydrogenase from T. thermophilus (TtHICDH).
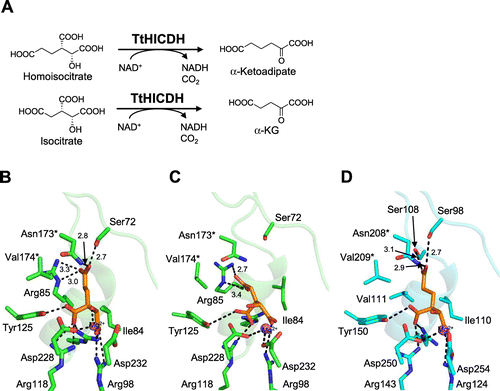
In conclusion, the quaternary complex of TtHICDH unambiguously revealed the molecular mechanism for dual substrate specificity of TtHICDH and also explained the difference in substrate specificity between bacterial-type and yeast-type HICDHs. This result may shed light on the evolutionary scenario of substrate specificity of the β-decarboxylating dehydrogenase family.
III.iii. α-Aminoadipate aminotransferase (AAA-AT)
The lysN gene encodes a mammalian kynurenine aminotransferase II homolog, α-aminoadipate aminotransferase (AAA-AT), which catalyzes the fourth reaction of lysine biosynthesis in T. thermophilus.Citation25) AAA-AT transfers the α-amino group of L-glutamate to α-ketoadipate to yield α-aminoadipate (AAA) (Fig. (A)). The lysN gene knock-out mutant strain grows slower than the wild-type strain in minimal medium. Although addition of AAA or L-lysine enhanced growth of the mutant strain to some extent, the growth was still slower than that of the wild-type strain in minimal medium. This result suggests that AAA-AT contributes not only to the lysine biosynthesis but also to other metabolic pathways, for example, biosynthesis of other amino acids. Enzyme assay using recombinant protein of AAA-AT exhibited distinct aminotransferase activity for branched-chain and aromatic amino acids, especially leucine, as well as AAA-AT activity, suggesting its involvement in leucine biosynthesis.
Fig. 6. Structure of α-aminoadipate aminotransferase from T. thermophilus (TtAAA-AT).
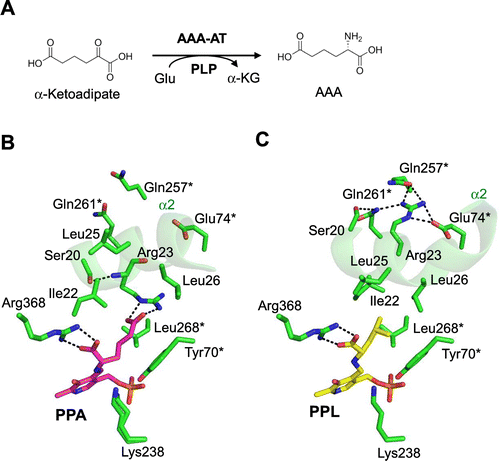
To elucidate the mechanism of multiple-substrate recognition of AAA-AT, we determined the crystal structure of AAA-AT (Fig. ).Citation47) AAA-AT assembles to form a dimer. Each monomer is composed of three structural domains: an N-terminal arm (Ser5-Ala19), a small domain composed of two regions (Ser20-Lys45 and Phe285-Ala395), and a large domain (Leu46-Gly284). The domain organization is similar with aspartate aminotransferase from T. thermophilus of fold-type I aminotransferase. Fold-type I aminotransferases were classified into seven subgroups, Iα to Iο, by Jensen and Gu.Citation48) Phylogenetic analysis revealed that mammalian kynurenine aminotransferase (Kat) II and its homologues, including AAA-AT, do not belong to any existing subgroups, but form a separate lineage. This indicates that AAA-AT and its homologues form a new subgroup in fold-type I aminotransferases. To reveal the precise mode of recognition of AAA and leucine, we chemically synthesized fused compounds of PLP and amino acids, PPA (N-phosphopyridoxyl-L-α-aminoadipate), and PPL (N-phosphopyridoxyl-L-leucine), and conducted the co-crystallization with those compounds. The AAA moiety of PPA is recognized by specific interactions (Fig. (B)). The α-carboxyl group forms an ion pair with Arg368. The γ-carboxyl group of AAA forms an electrostatic interaction with Arg23. The conserved Ser20 forms a hydrogen bond with the main chain amide of Arg23 to stabilize the conformation of Arg23 at N-terminus of α2 helix. The PPL-bound form takes a similar closed form as that of the PPA-bound form. The PLP moiety and main chain part of leucyl moiety of PPL are recognized in a manner similar to that of the AAA moiety in the PPA-bound form (Fig. (C)). The significant difference was found at the recognition site of the side chain leucyl moiety. Compared with the PPA-bound form, the α2 helix of the PPL-bound form rotates approximately 45 degrees to direct the different side of α2 toward the substrate-binding site and recognizes the hydrophobic leucyl moiety of PPL. The hydrophobic side chain of the leucyl moiety of PPL is recognized through hydrophobic interaction with Ile22, Leu25, and Leu26 on the α2 helix. Tyr70*, and Leu268* forms additional contacts with the leucyl side chain moiety. Interestingly, the conserved Arg23 residue dynamically changes its side chain orientation to interact with Glu74*, Gln257*, and Gln261* from another subunit, accompanied by a similar flip of another conserved residue, Ser20, to form a hydrogen bond with Gln261*. Thus, the conserved pair, Ser20 and Arg23, also play key role in the recognition of leucine. The dual roles of Ser20 and Arg23 were demonstrated by the mutational analyses.Citation49)
III.iv. ArgX, an enzyme utilizing the novel amino-group carrier protein, LysW
Recently, the conversion of AAA to L-lysine of T. thermophilus was reconstituted using the recombinant proteins, LysW, LysX, LysZ, LysY, LysJ, and LysK (Fig. (A)).Citation26) By the assay, it was revealed that the α-amino group of AAA was modified by attachment to the γ-carboxyl group of the C-terminal Glu54 of a small protein, LysW, then the side chain of the conjugated-AAA was converted to the lysyl side chain, and the conjugated L-lysine was subsequently liberated from LysW. LysW is necessary to proceed the reactions, suggesting that biosynthetic enzymes recognize LysW. LysW is an acidic protein with estimated pI of 3.7. In contrast, the modeled structures suggest that lysine biosynthetic enzymes possess basic regions around those active sites. Therefore, we assumed that LysW acts not only as protecting group of the α-amino group but also as a carrier protein to deliver the biosynthetic intermediate between the active sites of enzymes. To elucidate the molecular basis for recognition of LysW, we tried to determine the crystal structure of the LysX/LysW complex from T. thermophilus; however, crystallization of the complex failed even though extensive trials was performed.
Fig. 7. Structure of LysW-bound form of ArgX from S. tokodaii (StArgX).
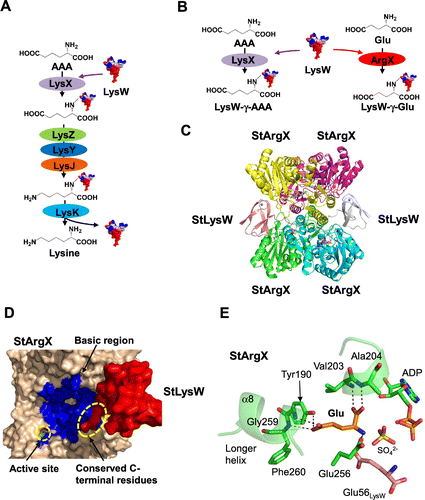
On the other hand, biosynthetic machinery using the amino-group carrier protein LysW was found to be involved in lysine and arginine biosyntheses in an archaea, Sulfolobus acidocaldarius.Citation50) Our group has demonstrated that LysWXZYJK is responsible for the conversion of AAA to L-lysine. We also found a gene encoding a second LysX homolog in the gene cluster including argG, argH, carA, and carB, which encodes the enzymes for the conversion of ornithine to arginine. The second LysX homologue was demonstrated to catalyze the covalent linkage of the amino moiety of L-glutamate to LysW and to play an essential role in arginine biosynthesis; therefore, the second LysX was named ArgX (Fig. (B)). This means that a single set of a carrier protein, LysW, and enzymes, LysZ, LysY, LysJ, and LysK, is involved in lysine and arginine biosyntheses, whereas LysX and ArgX are specialized for lysine and arginine biosyntheses, respectively. To elucidate how ArgX recognizes the carrier protein, LysW, and how ArgX discriminates the structurally similar compounds AAA and Glu, we attempted to crystallize the enzyme/carrier protein complex using ArgX/LysW. As a consequence of considerable effort, we successfully determined the crystal structure of Sulfolobus tokodaii ArgX orthologue (StArgX) that binds the LysW orthologue (StLysW), ADP, and L-glutamate (Fig. (C)). StArgX adopts an ATP-grasp fold similar to that of the ATP-dependent ligase family such as glutathione synthetase and D-Ala:D-Ala ligase. Two dimers of StArgX assemble to form a homo-tetramer. Two molecules of StLysW bind to the StArgX tetramer. The StLysW molecule is wedged in the space between two subunits of StArgX (A- and D-chains of StArgX for E-chain of StLysW or B and C-chains of StArgX for F-chain of StLysW). The C-terminal Glu56 residue binds at the active sites of A- and B-chains. The conserved StLysW C-terminal residue EDWGE (Glu52-Glu56) extends into the active site of StArgX. As expected, in this complex, the positively charged region around the active site of StArgX is not only physically proximal to the negatively charged StLysW, but also especially close to the highly negatively charged region of StLysW (Fig. (D)). This structural information indicates that StArgX recruits StLysW via electrostatic potential; however, it should be noted that most of the interactions between StArgX and the globular domain of StLysW are mediated by hydrogen bonds. The negative electrostatic potential of StLysW likely serves to approach the active site of StArgX via the net opposite electrostatic potential. Hence, StLysW interacts with ArgX loosely and presumably in a transient manner, thereby enabling StLysW to be transferred to StLysZ/ArgB, which is the enzyme catalyzing next step.
The active site structure of the StArgX/StLysW complex is very similar with that of a pseudo-Michaelis quaternary complex of glutathione synthetase (GSHase) containing glutathione, ADP, sulfate ion, and two Mg2+ ions.Citation51) The GSHase reaction begins with the phosphorylation of the C-terminal carboxyl group of the γ-L-glutamyl-L-cysteine using an ATP molecule, whereupon a nucleophilic attack from the amino group of bound glycine occurs, resulting in the formation of glutathione. The structure of StArgX also takes a pseudo-Michaelis quaternary complex containing ADP, sulfate ion, C-terminal of StLysW, L-glutamate, and Mg2+/Zn2+ ions. The γ-carboxyl functional group of the C-terminal Glu of StLysW is located at a position that is roughly the same as the carbonyl atom of the cysteine residue of glutathione. L-glutamate is bound at the same position as glycine in the GSHase structure. Therefore, the structure of StArgX demonstrates the idea that the reaction of the ArgX proceeds in a similar manner to the phosphorylation reaction by GSHase. Next, I will describe the substrate specificity of StArgX. The γ-carboxyl moiety of L-glutamate forms a hydrogen bond with Tyr190, and the main chain of moieties of Gly259 and Phe260 (Fig. (E)). These residues are conserved among putative ArgX orthologues in Sulfolobaceae. In contrast, the corresponding residues are replaced with Ile192, Asn258 and Ser259 in TtLysX, respectively. This triplet is also highly conserved among the LysX family. Therefore, we suggested that this triplet was a signature motif for discriminating LysX and ArgX.
IV. Conclusion
Here, I reviewed the recent progress of study on amino acid metabolism of bacteria and archaea from our group. The structures of the enzymes or proteins provided us valuable information for understanding the molecular mechanisms of lysine, and the related biosynthetic pathway and novel allosteric regulation of glutamate dehydrogenase. These results are essential for exploring related biosynthetic machinery, including secondary metabolisms and the amino acid signal transduction systems.
Funding
This study was partly supported by a Grant-in-Aid for Scientific Research from the Japanase Ministry of Education, Culture, Sports, Science, and Technology and by various other grants mentioned in the related publications.
Disclosure statement
No potential conflict of interest was reported by the author.
Acknowledgements
I am most grateful to Prof. Makoto Nishiyama for his continuing encouragement, helpful suggestions, and considerable support for this research. I would like to acknowledge constant help and suggestions from Prof. Tomohisa Kuzuyama. I am deeply indebted to Prof. Shinya Fushinobu and Prof. Hisakazu Yamane for helpful discussions and for teaching me many valuable experimental methods. I would like to express my appreciation for support during this work to all members of the Laboratory of Cell Biotechnology, Biotechnology Research Center, The University of Tokyo.
Notes
This review was written in response to the author’s receipt of the Japan Society for Bioscience, Biotechnology, and Agrochemistry Award for the Encouragement of Young Scientist in 2016.
References
- Ivanov K, Stoimenova A, Obreshkova D, et al. Biotechnology in the production of pharmaceutical industry ingredients: amino acids. Biotechnol Biotechnol Equip. 2013;27(2):3620–3626.
- Shiratsuchi M, Kuronuma H, Kawahara Y, et al. Simultaneous and high fermentative production of L-lysine and L-gluamic acid using a strain of Brevibacterium lactofermentum. Biosci Biotechnol Biochem. 1995;59(1):83–86.
- Kawahara Y, Takahashi-Fuke K, Shimizu E, et al. Relationship between the glutamate production and the activity of 2-oxoglutarate dehydrogenase in Brevibacterium lactofermentum. Biosci Biotechnol Biochem. 1997;61(7):1109–1112.
- Nakamura J, Hirano S, Ito H, et al. Mutations of the Corynebacterium glutamicum NCgl1221 gene, encoding a mechanosensitive channel homolog, induce L-Glutamic acid production. Appl Environ Microbiol. 2007;73:4491–4498.
- Sano K, Shiio I. MICROBIAL production of L-lysine. III. Production by mutants resistant to S(2-aminoethyl)L-cysteine. J Gen Appl Microbiol. 1970;16:373–391.
- Tosaka O, Takinami K, Hirose Y. L-Lysine production by S-(2- aminoethyl)-L-cysteine and aeamino-β-hydroxyvaleric acid resistant mutants of Brevibacterium lactofermentum. Agric Biol Chem. 1978;42:745–752.
- Yoshida A, Tomita T, Kurihara T, et al. Structural insight into concerted inhibition of α2β2-type aspartate kinase from Corynebacterium glutamicum. J Mol Biol. 2007;368(2):521–536.
- Yoshida A, Tomita T, Kuzuyama T, et al. Mechanism of concerted inhibition of α2β2-type hetero-oligomeric aspartate kinase from Corynebacterium glutamicum. J Biol Chem. 2010;285(35):27477–27486.
- Proud CG. Amino acids and mTOR signalling in anabolic function. Biochem Soc Trans. 2007;35(5):1187–1190.
- Ruiz JL, Ferrer J, Camacho M, et al. NAD-specific glutamate dehydrogenase from Thermus thermophilus HB8: purification and enzymatic properties. FEMS Microbiology Letters. 1998;159(1):15–20.
- Bolivar J, Cava F, Mateo C, et al. Immobilization-stabilization of a new recombinant glutamate dehydrogenase from Thermus thermophilus. Appl Microbiol Biotechnol. 2008;80(1):49–58.
- Tomita T, Miyazaki T, Miyazaki J, et al. Hetero-oligomeric glutamate dehydrogenase from Thermus thermophilus. Microbiology. 2010;156(12):3801–3813.
- Tomita T, Kuzuyama T, Nishiyama M. Structural basis for leucine-induced allosteric activation of glutamate dehydrogenase. J Biol Chem. 2011;286(43):37406–37413.
- Smith T, Schmidt T, Fang J, et al. The structure of apo human glutamate dehydrogenase details subunit communication and allostery. J Mol Biol. 2002;318(3):765–777.
- Li M, Smith C, Walker M, et al. Novel inhibitors complexed with glutamate dehydrogenase: allosteric regulation by control of protein dynamics. J Biol Chem. 2009;284(34):22988–23000.
- Börmann ER, Eikmanns BJ, Sahm H. Molecular analysis of the Corynebacterium glutamicum gdh gene encoding glutamate dehydrogenase. Mol Microbiol. 1992;6(3):317–326.
- Kholy ER, Eikmanns BJ, Gutmann M, et al. Glutamate dehydrogenase is not essential for glutamate formation by Corynebacterium glutamicum. Appl Environ Microbiol. 1993;59(7):2329–2331.
- Jo JH, Seol HY, Lee YB, et al. Disruption of genes for the enhanced biosynthesis of α-ketoglutarate in Corynebacterium glutamicum. Can J Microbiol. 2012;58(3):278–286.
- Son HF, Kim IK, Kim KJ. Structural insights into domain movement and cofactor specificity of glutamate dehydrogenase from Corynebacterium glutamicum. Biochem Biophys Res Commun. 2015;459(3):387–392.
- Kobashi N, Nishiyama M, Tanokura M. Aspartate kinase-independent lysine synthesis in an extremely thermophilic bacterium, Thermus thermophilus: lysine is synthesized via alpha-aminoadipic acid not via diaminopimelic acid. J Bacteriol. 1999;181(6):1713–1718.
- Nishida H, Nishiyama M, Kobashi N, et al. A prokaryotic gene cluster involved in synthesis of lysine through the amino adipate pathway: a key to the evolution of amino acid biosynthesis. Genome Res. 1999;9(12):1175–1183.
- Wulandari AP, Miyazaki J, Kobashi N, et al. Characterization of bacterial homocitrate synthase involved in lysine biosynthesis. FEBS Lett. 2002;522(1–3):35–40.
- Miyazaki J, Kobashi N, Nishiyama M, et al. Characterization of homoisocitrate dehydrogenase involved in lysine biosynthesis of an extremely thermophilic bacterium, Thermus thermophilus HB27, and evolutionary implication of β-decarboxylating dehydrogenase. J Biol Chem. 2003;278(3):1864–1871.
- Jia Y, Tomita T, Yamauchi K, et al. Kinetics and product analysis of the reaction catalysed by recombinant homoaconitase from Thermus thermophilus. Biochem J. 2006;396(3):479–485.
- Miyazaki T, Miyazaki J, Yamane H, et al. α-Aminoadipate aminotransferase from an extremely thermophilic bacterium, Thermus thermophilus. Microbiology. 2004;150(7):2327–2334.
- Horie A, Tomita T, Saiki A, et al. Discovery of proteinaceous N-modification in lysine biosynthesis of Thermus thermophilus. Nat Chem Biol. 2009;5(9):673–679.
- Wulandari AP, Miyazaki J, Kobashi N, et al. Characterization of bacterial homocitrate synthase involved in lysine biosynthesis. FEBS Lett. 2002;522:35–40.
- Tsubouchi T, Mineki R, Taka H, et al. Leader peptide-mediated transcriptional attenuation of lysine biosynthetic gene cluster in Thermus thermophilus. J Biol Chem. 2005;280:18511–18516.
- Qian JH, Khandogin J, West AH, et al. Evidence for a catalytic dyad in the active site of homocitrate synthase from Saccharomyces cerevisiae. Biochemistry. 2008;47(26):6851–6858.
- Okada T, Tomita T, Wulandari A, et al. Mechanism of substrate recognition and insight into feedback inhibition of homocitrate synthase from Thermus thermophilus. J Biol Chem. 2010;285(6):4195–4205.
- Zabriskie TM, Jackson MD. Lysine biosynthesis and metabolism in fungi. Nat Prod Rep. 2000;17(1):85–97.
- Xu H, Andi B, Qian J, et al. The α-aminoadipate pathway for lysine biosynthesis in fungi. Cell Biochem Biophys. 2006;46(1):43–64.
- Parsons SJ, Burns RO. Purification and properties of β-isopropylmalate dehydrogenase. J Biol Chem. 1969;244(3):996–1003.
- Weitzman PD. Patterns of diversity of citric acid cycle enzymes. Biochem Soc Symp. 1987;54:33–43.
- Strassman M, Ceci LN. Enzymatic formation of α-ketoadipic acid from homoisocitric acid. J Biol Chem. 1965;240(11):4357–4361.
- Bauer MW, Bylina EJ, Swanson RV, et al. Comparison of a β-glucosidase and a β-mannosidase from the hyperthermophilic archaeon Pyrococcus furiosus. Purification, characterization, gene cloning, and sequence analysis. J Biol Chem. 1996;271(39):23749–23755.
- Durbecq V, Legrain C, Roovers M, et al. The carbamate kinase-like carbamoyl phosphate synthetase of the hyperthermophilic archaeon Pyrococcus furiosus, a missing link in the evolution of carbamoyl phosphate biosynthesis. Proc Nat Acad Sci U S A. 1997;94(24):12803–12808.
- Matsui I, Matsui E, Sakai Y, et al. The molecular structure of hyperthermostable aromatic aminotransferase with novel substrate specificity from Pyrococcus horikoshii. J Biol Chem. 2000;275(7):4871–4879.
- Ishikawa K, Ishida H, Matsui I, et al. Novel bifunctional hyperthermostable carboxypeptidase/aminoacylase from Pyrococcus horikoshii OT3. Appl Environ Microbiol. 2001;67(2):673–679.
- Miyazaki K. Bifunctional isocitrate–homoisocitrate dehydrogenase: a missing link in the evolution of β-decarboxylating dehydrogenase. Biochem Biophys Res Commun. 2005;331(1):341–346.
- Lin Y, Alguindigue SS, Volkman J, et al. Complete kinetic mechanism of homoisocitrate dehydrogenase from Saccharomyces cerevisiae. Biochemistry. 2007;46(3):890–898.
- Chen R, Jeong SS. Functional prediction: identification of protein orthologs and paralogs. Protein Sci. 2000;9(12):2344–2353.
- Miyazaki J, Asada K, Fushinobu S, et al. Crystal structure of tetrameric homoisocitrate dehydrogenase from an extreme thermophile, Thermus thermophilus: involvement of hydrophobic dimer–dimer interaction in extremely high thermotolerance. J Bacteriol. 2005;187(19):6779–6788.
- Nango E, Yamamoto T, Kumasaka T, et al. Structure of Thermus thermophilus homoisocitrate dehydrogenase in complex with a designed inhibitor. J Biochem. 2011;150(6):607–614.
- Bulfer SL, Hendershot JM, Trievel RC. Crystal structure of homoisocitrate dehydrogenase from Schizosaccharomyces pombe. Proteins. 2012;80(2):661–666.
- Takahashi K, Tomita T, Kuzuyama T, et al. Determinants of dual substrate specificity revealed by the crystal structure of homoisocitrate dehydrogenase from Thermus thermophilus in complex with homoisocitrate·Mg(2+)·NADH. Biochem Biophys Res Commun. 2016;478(4):1688–1693.
- Tomita T, Miyagawa T, Miyazaki T, et al. Mechanism for multiple-substrates recognition of α-aminoadipate aminotransferase from Thermus thermophilus. Proteins. 2009;75(2):348–359.
- Jensen RA, Gu W. Evolutionary recruitment of biochemically specialized subdivisions of Family I within the protein superfamily of aminotransferases. J Bacteriol. 1996;178(8):2161–2171.
- Ouchi T, Tomita T, Miyagawa T, et al. Dual roles of a conserved pair, Arg23 and Ser20, in recognition of multiple substrates in α-aminoadipate aminotransferase from Thermus thermophilus. Biochem Biophys Res Commun. 2009;388(1):21–27.
- Ouchi T, Tomita T, Horie A, et al. Lysine and arginine biosyntheses mediated by a common carrier protein in Sulfolobus. Nat Chem Biol. 2013;9(4):277–283.
- Hara T, Kato H, Katsube Y, et al. A pseudo-Michaelis quaternary complex in the reverse reaction of a ligase: structure of Escherichia coli B glutathione synthetase complexed with ADP, glutathione, and sulfate at 2.0 Å resolution. Biochemistry. 1996;35:11967–11974.