
Abstract
The fibrinolysis system is thought to play an important role in liver regeneration. We previously found that plasminogen (Plg) is localized to the cell surface of regenerating liver tissue as well as proliferating hepatocytes in vitro. Here, we investigated the significance of Plg binding to the cell surface during liver regeneration. Pre-administration of tranexamic acid (TXA), which is a competitive inhibitor of Plg binding, to hepatectomized rats mildly delayed restoration of liver weight in vivo. Although binding of Plg to the cell membrane decreased following TXA administration, TXA showed little effect on hepatocyte proliferation in rats. We also discovered that Plg treatment did not stimulate proliferation of primary rat hepatocytes in vitro. These results suggest that Plg/plasmin potentiates liver regeneration via a pathway distinct from those through which hepatocyte proliferation is stimulated.
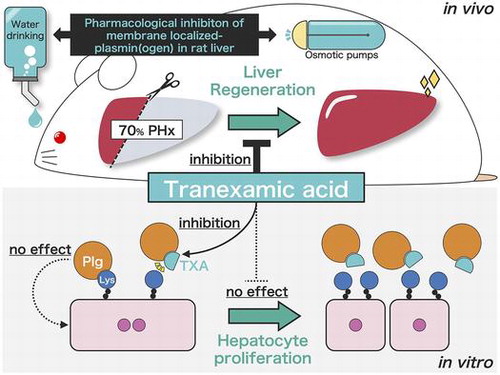
To reveal the role of membrane-localized plasmin(ogen) in liver regeneration, we investigated a novel pharmacological inhibition model and rat hepatocytes in vitro.
The fibrinolysis system degrades fibrin clots formed by the coagulation system to maintain blood circulation. Fibrin is degraded by the proteolytic system; this is comprised of plasminogen (Plg), which is a zymogen of a serine protease, and its activators, tissue-type plasminogen activator (tPA) and urokinase-type plasminogen activator (uPA). The PA/plasmin system is regulated by physiological inhibitors of plasmin and PA, such as plasminogen activator inhibitor-1 (PAI-1) and alpha-2 antiplasmin (α2-AP).Citation1)
Plg can bind specifically and physiologically to the C-terminal lysine residue(s) of fibrin molecules through lysine binding sites (LBS) in the Kringle domain.Citation2) This binding enables efficient activation of Plg by PA, as well as protection against inhibition of plasmin’s activity.Citation1,2) The synthetic plasmin inhibitors tranexamic acid (TXA) and epsilon-aminocaproic acid have higher affinities for LBS on the Plg molecule than for lysine residue(s) on fibrin.Citation3)
The PA/plasmin system is also involved in various biological phenomena at extravascular spaces through its proteolytic activity: (1) proteolytic activation of growth factors, such as hepatocyte growth factor (HGF), vascular endothelial growth factor (VEGF) and transforming growth factor-β, and subsequent release of these growth factors from extracellular matrices (ECM),Citation4–7) (2) activation or degradation of complement factors in response to inflammation,Citation8) (3) remodeling of ECM via plasmin or activation of matrix metalloproteases (MMPs), which degrade ECM, collagen and fibronectin,Citation7) and (4) induction of cellular signaling through the Plg receptor or uPA receptor.Citation9,10) These functions of PA/plasmin system other than fibrin degradation in the extravascular regions are called “pericellular fibrinolysis.”
Liver regeneration is achieved by highly orchestrated processes, including cell proliferation elicited by growth factors, inflammatory responses, ECM remodeling, and various types of cellular signaling.Citation11) We previously reported that cell membrane-bound plasmin(ogen) is upregulated during hepatocyte proliferation and liver regenerationCitation12–14); however, a critical role for cell surface-bound Plg in liver regeneration has not been fully elucidated. Plasmin(ogen) binding to the cell surface is a necessary event for proliferation of cancer cells, and of various other cell types in vitro.Citation10) Thus, we hypothesized that inhibition of fibrinolysis during liver regeneration would delay cell proliferation in the liver after partial hepatectomy.
To validate this hypothesis, we established a novel pharmacological inhibition model of Plg binding to the cell surface using the Plg-binding inhibitor TXA. In addition, we used primary cultured rat hepatocytes to investigate the effect of Plg binding on cell proliferation in vitro.
As a result, we highlighted that the PA/plasmin system acts as a driver of liver regeneration, but does not stimulate hepatocyte proliferation.
Materials and methods
Reagents
Plg-free fibrinogen was prepared from commercial bovine fibrinogen (Sigma–Aldrich, St. Louis, MO, USA) by affinity chromatography on a lysine-Sepharose column.Citation15) Antibodies against proliferating cell nuclear antigen (PCNA; PC-10), cyclin D1 (EPR2241), Ki-67 (SP6), and β-actin were purchased from Dako (Glostrup, Denmark), Abcam (Cambridge, UK), GeneTex (Irvine, CA, USA), and Sigma–Aldrich, respectively. Secondary antibodies were purchased from Jackson Immunoresearch (West Grove, PA, USA). Cell culture reagents were as follows: Hank’s balanced salt solution (HBSS; Nissui Pharmaceutical Co., Ltd., Tokyo, Japan), Williams’ medium E (MP Biomedicals, LLC., Santa Ana, CA, USA), insulin (Wako Pure Chemical Industries, Ltd., Osaka, Japan), glucagon (Sigma–Aldrich), and epidermal growth factor (EGF; Higeta Shoyu Co., Ltd., Tokyo, Japan).
Animal experiments
Male Wistar rats were purchased from Japan SLC (Shizuoka, Japan). Rats were allowed free access to a standard chow diet (CRF-1; Oriental Yeast Co., Tokyo, Japan) and water, under 12-h light/dark cycles. After an acclimatization period, six- to seven-week-old rats were given TXA in their drinking water (20 mg/mL) and fasted overnight. The rats were also aseptically implanted with ALZET® Osmotic pumps (model 2001; DURECT Co., Cupertino, CA, USA) containing 150 mg/mL TXA, into subcutaneous tissue on the dorsal side (TXA infusion rate, 3.6 mg/day). As the vehicle control, rats were implanted with phosphate-buffered saline-containing pumps and given sterile drinking water instead of TXA solution. Twenty-four hours after TXA administration, rats received a 70% partial hepatectomy according to the method of Higgins and AndersonCitation16) under isoflurane anesthesia. Collection of the remaining liver tissue from hepatectomized rats was followed by snap-freezing and storage at –80 °C until the analyses were carried out. Blood plasma was prepared from heparinized whole blood collected from the right ventricle. All animal experiments were approved by the Nihon University Animal Care and Use Committee (#AP15B010).
Isolation and primary culture of rat hepatocytes
Rat hepatocytes were freshly isolated from the liver of Wistar rats (150–200 g, male) by the two-step collagenase perfusion method described by Ichihara et al.Citation17). Isolated hepatocytes were washed and purified by low-speed centrifugation (50× g, 4 °C, three times) using HBSS. Purified hepatocytes (5.0 × 104 cells/cm2 dish) were cultured on 35 mm type I collagen coated-dishes (IWAKI; AGC TECHNO GLASS CO., Ltd., Shizuoka, Japan) in Williams’ medium E supplemented with 5% fetal bovine serum, 10−8 M glucagon and 10−8 M insulin at 37 °C for 4 h. Hepatocytes were then incubated in hormone- and serum-free Williams’ E medium for 20 h, in preparation for the experiments. Hepatocyte proliferation was measured via immunoblotting of PCNA and EdU incorporation assays.
EdU incorporation assays
EdU proliferation assays were performed using a Click-iT® Plus EdU Alexa Fluor® 488 Imaging Kit according to the manufacturer’s instructions. Briefly, EdU-labeled hepatocytes were fixed with 4% paraformaldehyde and stained with Alexa Fluor® 488 azide. Fixed hepatocytes were further stained with Hoechst 33342 and imaged using a BZ-9000 fluorescence microscope (KEYENCE, Osaka, Japan). Image quantification was performed with ImageJ software.Citation18)
Immunoblot analysis
Whole liver (1 vol.) was homogenized in 5 vol. ice-cold lysis buffer (150 mM NaCl, 1 mM EDTA, 1% NP-40, 10 mM NaF, 2 mM Na3VO4, 10 mM β-glycerophosphate, 50 mM Tris–HCl, pH 7.8) containing a 0.5% protease inhibitor cocktail (Sigma–Aldrich). Primary rat hepatocytes were sonicated in RIPA buffer (150 mM NaCl, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate, 50 mM Tris–HCl, pH 7.5) containing a 1% protease inhibitor cocktail. Lysates were centrifuged at 12,000× g, 4 °C for 15 min. After centrifugation, supernatants were collected and the protein concentration was determined using a bicinchoninic acid (BCA) protein assay kit (Thermo Fisher Scientific, Waltham, MA, USA). Lysates were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE; 5 μg/lane) and the migrated proteins were electronically transferred to a PVDF membrane (Merck Millipore, Billerica, MA, USA). The membrane was first incubated with primary antibodies (PCNA, 1:2000; Cyclin D1, 1:2000; β-actin, 1:10,000) and then incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies (for PCNA, anti-mouse, 1:20,000; for Cyclin D1, anti-rabbit, 1:20,000; for β-actin, anti-mouse, 1:100,000). Immune complexes were visualized by ImmunoStar LD (Wako) and imaged by an LAS-4000 mini image analyzer (Fujifilm, Tokyo, Japan).
Immunohistochemical analysis
Immunohistochemical analysis was performed according to the standard protocol. Freshly isolated rat liver tissues were fixed with 4% paraformaldehyde and embedded in paraffin. Next, liver sections (5 μm thick) were deparaffinized and rehydrated, autoclaved for 20 min at 121 °C in 10 mM citrate buffer (pH 6.0), and treated with 3% H2O2 for 10 min followed by triple-washing with PBS. Sections were treated with 5% bovine serum albumin for 40 min, then incubated with anti-Ki-67 rabbit IgG (1:200) for 18 h at 4 °C. Peroxidase-conjugated Affinipure goat anti-rabbit IgG was used as a secondary antibody (1:1000, for 2 h). Peroxidase activity was detected using the ImmPACT DAB Peroxidase (HRP) Substrate (Vector Laboratories, Burlingame, CA, USA) according to the manufacturer’s instructions, imaging with the BZ-9000 microscope (KEYENCE, as before). Image quantification was performed with Image J software.Citation18)
Subcellular fractionation
Plasma membrane-enriched fractions of rat liver were prepared as described previously.Citation14) Briefly, liver tissue was homogenized in STMP buffer (0.25 M sucrose, 1 mM MgCl2, 1 mM phenylmethylsulfonyl fluoride (PMSF), 5 mM Tris–HCl, pH 7.4) and the homogenate was centrifuged to discard cellular debris. After centrifugation, supernatants were further centrifuged at 1500× g, 4 °C for 10 min, and the collected precipitates were homogenized by a Dounce-type homogenizer with STMP buffer. The sucrose density of the homogenates was adjusted with STMP buffer to 1.42 M. Density-adjusted homogenates were layered under STMP buffer and subjected to sucrose-density gradient ultracentrifugation (105,000× g, 4 °C, 60 min). Cell membrane-enriched fractions obtained at the interface of 0.25 M/1.42 M sucrose gradient were collected and further centrifuged at 105,000× g, 4 °C for 60 min to obtain precipitates. Precipitates were reconstituted in STM (without PMSF) buffer and protein concentrations were determined using a BCA protein assay kit.
Fibrinogen-zymography
Fibrinogen-zymography was performed as described previously.Citation14) Briefly, plasma membrane-enriched fractions were subjected to SDS-PAGE (10 μg/lane) on a 10% acrylamide gel containing 2 mg/mL Plg-free fibrinogen. After electrophoresis, the gel was washed twice with 2.5% Triton X-100 for 30 min, and then incubated with incubation buffer (100 mM glycine, 50 mM Tris–HCl, pH 8.3) at 37 °C for 72 h in the presence or absence of 10 U/mL urokinase (Mochida Pharmaceuticals Co., Tokyo, Japan). The gel was stained with 0.075% Coomassie Blue and destained with 12.5% methanol containing 12.5% acetic acid.
For in vitro experiments, the cell surface-localized plasmin(ogen) was detached from the cultured hepatocytes by incubation with 10 mM TXA/PBS for 15 min at 4 °C and determined by the fibrinogen-zymography as described above.
Statistics
Data are presented as means ± SE. Differences between the two groups were compared using a two-tailed unpaired Student’s t test. Multiple comparisons were performed by Tukey-Kramer or two-way ANOVA. P values less than 0.05 were considered to be statistically significant.
Results
TXA administration delays liver restoration following 70% hepatectomy
To determine the effect of TXA administration on liver regeneration, we evaluated the restoration rate of rat liver after 70% hepatectomy. Fig. shows the liver mass after the hepatectomy. The animal model employed in this study is somewhat invasive; i.e. the rats suffered from surgery twice for implantation of osmotic pump and partial hepatectomy. These procedures sometimes affect on the body weight. The rats used in this experiment were at growing phase (subadult), thus the body weight was sensitive for the invasion as well as for the changes in food intake or fasting. The rats were also fasted prior to the hepatectomy. On the other hand, liver weight is relatively stable even against these events. Thus, we show the liver mass (liver weight) instead of liver weight to body weight ratio in Fig. .
Fig. 1. Restoration of rat liver after 70% hepatectomy.
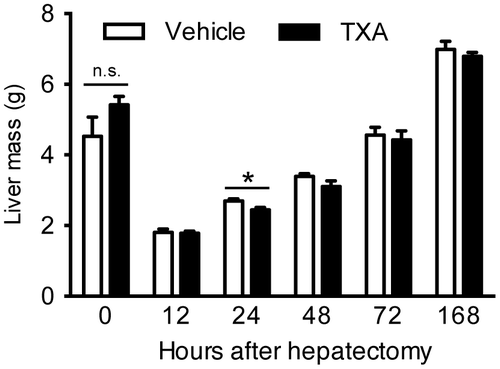
The liver mass gradually recovered toward 168 h after 70% hepatectomy. In comparison with vehicle control, TXA administration significantly suppressed the recovery of liver mass at 24 h after 70% hepatectomy (vehicle 2.70 ± 0.06 g vs. TXA 2.45 ± 0.07 g, Fig. ). These results indicate that TXA administration mildly, but significantly, delays liver regeneration after 70% hepatectomy.
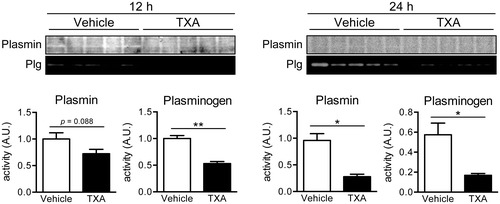
TXA administration reduces plasmin(ogen) localization on the plasma membrane of regenerating liver cells
Our previous studies demonstrated that membrane-bound plasmin(ogen) levels increased in the regenerating rat liver after 70% hepatectomy in vivo, as well as on proliferating primary rat hepatocytes in vitro.Citation12–14) Therefore, we explored whether TXA administration could reduce the localization of plasmin and Plg on the plasma membrane of rat liver after 70% hepatectomy. Levels of both plasmin and Plg in the plasma membrane-enriched fraction of rat liver were significantly reduced in response to TXA administration at 12 and 24 h after 70% hepatectomy (Fig. ). These results indicate that TXA administration markedly reduces plasmin and Plg localization in liver cell plasma membranes during liver regeneration.
Fig. 2. Localization of plasmin and plasminogen (Plg) on the plasma membrane of rat liver cells after 70% hepatectomy.
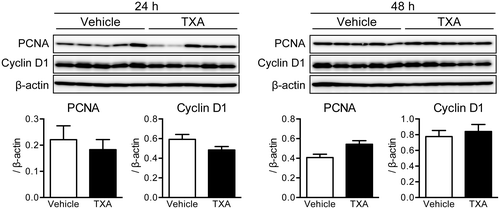
TXA administration has no effect on cell proliferation in the regenerating liver after 70% hepatectomy
As it was observed that liver restoration was delayed by TXA administration, we next focused on the effect of TXA on cell proliferation during liver regeneration. Immunoblotting was performed to measure the expression of the PCNA and cyclin D1 proteins, which are involved in cell cycle progression during liver regeneration. Expression of PCNA and cyclin D1 was not influenced by TXA administration in comparison with vehicle (Fig. ); PCNA expression was somewhat increased following TXA administration at 48 h after the 70% hepatectomy.
Fig. 3. Expression of proliferating cell nuclear antigen (PCNA) and cyclin D1 in the rat liver after 70% hepatectomy.
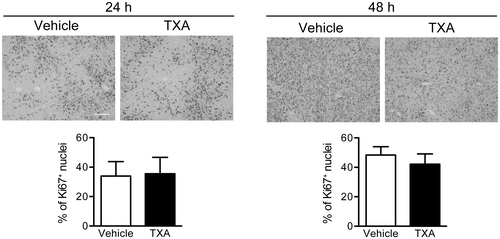
We further analyzed the expression of Ki-67 antigen by immunohistochemistry during liver regeneration after 70% hepatectomy. Ki-67 is a protein induced by growth stimuli and expressed by proliferating cells, but not in quiescent cells at G0-phase. Ki-67 expression in the TXA group was comparable to that of the vehicle group at 24 or 48 h after 70% hepatectomy (Fig. ). It was concluded from these results that TXA administration did not show any effect on cell proliferation during liver regeneration.
Fig. 4. Expression of Ki-67 in the rat liver after 70% hepatectomy.
TXA treatment does not suppress proliferation of primary cultures of rat hepatocytes in vitro
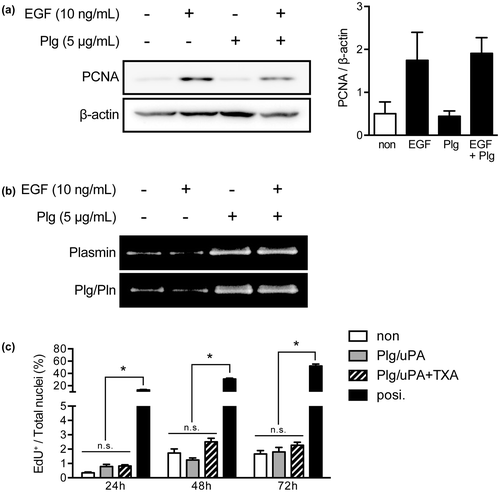
Since TXA administration did not reduce the expression of markers of cell proliferation in vivo, we further investigated whether Plg binding to the hepatocyte surface influenced the proliferation of hepatocytes in vitro, using primary cultures of rat hepatocytes. First, we analyzed the expression of PCNA protein in hepatocytes that had been treated with 10 ng/mL EGF in the presence or absence of 5 μg/mL Plg (Fig. (A)). PCNA expression was markedly increased in the EGF-treated cells. Plg did not show any effects on the expression of PCNA or EGF-induced PCNA; however, Plg markedly induced the plasmin activity on the cell surface of the hepatocytes (Fig. (B)). TXA treatment did not affect on the EGF-induced PCNA expression by the hepatocytes (data not shown). These results suggest that Plg per se or plasmin activity has no effect on the cell cycle progression of hepatocytes. To confirm this result, we further analyzed the effect of Plg on DNA synthesis in the hepatocytes using thymidine analog-based EdU incorporation assay system (Fig. (C)). The EdU incorporation rate was not changed by co-treatment with 5 μg/mL Plg and 10 U/mL uPA, which generates plasmin, in the presence or absence of 1 mM TXA. These results indicate that plasmin(ogen) does not stimulate the proliferation of hepatocytes.
Fig. 5. The effect of plasmin(ogen) on proliferation of rat hepatocytes in primary culture.
Discussion
We previously reported on the relationship between the hepatic fibrinolytic system and liver regeneration; Akao et al.Citation12) reported upregulation of plasmin activity in proliferating rat hepatocytes in primary culture that were co-cultured with non-parenchymal liver cells, while Okumura et al.Citation13,14) reported that binding of plasmin(ogen) to the plasma membrane of primary cultured rat hepatocytes is associated with cell proliferation in vitro. Plasmin activity localized to the cell surface was also observed in the regenerating liver of hepatectomized rats in vivo. Furthermore, carboxypeptidase TAFI, which inhibits the binding of plasmin(ogen) to its substrate via the LBS, also suppressed the proliferation of hepatocytes by inhibiting membrane-bound plasmin(ogen) on the hepatocyte surface.Citation14) The role of fibrinolytic factors in liver regeneration has also been examined using genetic knockout models. Drixler et al.Citation19) demonstrated delayed liver restoration in mice that were deficient in Plg and uPA after a 70% partial hepatectomy. They also reported that the delayed regeneration was due to the reduced cell proliferation associated with suppression of DNA synthesis, and to impaired angiogenesis in the liver microenvironment. Regeneration after carbon tetrachloride-induced liver injury was also impaired in Plg-, uPA-, and tPA-knockout mice.Citation20–22) On the other hand, PAI-1- and α2-AP-knockout mice displayed faster regeneration than wild-type mice after chemical liver injury.Citation23,24)
Based on these findings, we hypothesized that the inhibition of plasmin(ogen) binding to the cell surface delays liver regeneration via suppression of cell proliferation. As Plg was completely depleted in genetically Plg-deficient mice, it was not possible to examine the effect of plasmin(ogen) localized at the cell surface on hepatocyte proliferation. Therefore, we established a novel in vivo Plg-inhibition model, in which the Plg-binding inhibitor TXA is constantly administered to specifically inhibit the binding of Plg onto the cell surface during liver regeneration.
In this study, we showed that the inhibition of Plg by TXA mildly, but significantly, delayed the recovery of liver weight during liver regeneration (Fig. ). However, we did not observe suppression of cell proliferation (Figs. and ). Although liver cell proliferation after 70% hepatectomy was dramatically delayed in the Plg and uPA knockout mice,Citation19) this phenomenon was not observed in our study using the TXA-treated rat model.
It has been reported that plasmin(ogen) potently promotes the proliferation of airway smooth muscle cells (ASMCs) in vitro. Stewart and colleaguesCitation25) demonstrated that plasmin (5 mU/mL) increased the expression of cyclin D1 mRNA (over 200-fold) and subsequent cell proliferation through activation of PI3 K/Akt and the ERK1/2 MAPK signaling cascade via the annexin A2-TLR4 pathway in ASMCs. They also showed that knockdown of uPA or inhibition of uPA activity abolished these plasmin-induced effects. In our study, neither Plg nor plasmin stimulated cell proliferation of quiescent hepatocytes or growth-stimulated hepatocytes (Fig. (A) and (C)); however, Plg markedly upregulated plasmin(ogen) activity on the surface of these cells (Fig. (B)). These data suggest that the PA/plasmin system does not stimulate intracellular signaling cascades and subsequent hepatocyte proliferation during liver regeneration.
Mars et al.Citation26,27) and Kim et al.Citation28) suggested that the PA/plasmin system is involved in the promotion of cell proliferation via HGF activation, and in ECM remodeling through the activation of MMPs during liver regeneration. Kim and colleagues reported that ECM remodeling occurs subsequent to activation of the fibrinolysis system. In this study, we also investigated whether TXA suppresses the activation of MMP-2 and -9 after 70% hepatectomy. However, activity of these enzymes was not detected in the regenerating rat liver (data not shown).
Recently, it has also been reported that Plg-deficient mice displayed severe phenotypes of burn-induced wound healing.Citation29) Kawao et al.Citation30–33) demonstrated that suppression of the phagocytic functions of macrophages, and subsequent ECM remodeling, markedly impairs liver repair in Plg- and uPA-knockout mice after laser-induced liver injury or ischemia-reperfusion injury. Das et al.Citation34) revealed that the phagocytic functions of liver-resident macrophage Kupffer cells were dramatically downregulated in Plg-deficient mice. Therefore, we speculate that the PA/plasmin system might activate non-parenchymal cells to optimize the tissue microenvironment for hepatocyte proliferation during liver regeneration.
In summary, to clarify the molecular basis of liver regeneration via Plg binding to the cell surface after 70% hepatectomy, we established a novel pericellular fibrinolysis inhibition rat model using the Plg-binding inhibitor TXA. We revealed that TXA mildly, but significantly, delayed liver regeneration after 70% hepatectomy in rats. Moreover, we demonstrated that binding of Plg to the hepatocyte surface does not directly affect hepatocyte proliferation in vitro. These results suggest that pericellular fibrinolysis regulates other pathways, such as non-parenchymal cell activation, but does not apply to hepatocyte proliferation during liver regeneration.
Author contributions
AM, TH, TS conceived and designed the experiments. AM mainly performed the experiments. IK and YY performed experiments using rat hepatocytes. KK performed the histological analyses. AM, KI, YY, KK, TH, TS analyzed the data. AM and TS co-wrote the paper. YO-M contributed to the discussion section and TS, TH, YO-M reviewed and edited the manuscript. TS was responsible for the overall design of the research and experiments. All authors read and approved this paper.
Disclosure statement
No potential conflict of interest was reported by the authors.
Funding
This work was supported in part by Grants-in Aid for Scientific Research (B) [#25292077], [#16H04927]; a Grant-in-Aid for Challenging Exploratory Research from the Japan Society for the Promotion of Science [15K14740] (to TS).
Acknowledgments
We thank Dr. Nobuaki Okumura for helpful discussions.
References
- Rijken DC, Lijnen HR. New insights into the molecular mechanisms of the fibrinolytic system. J Thromb Haemost. 2009;7:4–13.10.1111/jth.2008.7.issue-1
- Law RH, Caradoc-Davies T, Cowieson N, et al. The X-ray crystal structure of full-length human plasminogen. Cell Rep. 2012;1:185–190.10.1016/j.celrep.2012.02.012
- Okamoto S, Okamoto U. Amino-methyl cyclohexane-carboxylic acid: AMCHA. Keio J Med. 2009;11:105–115.
- Naldini L, Vigna E, Bardelli A, et al. Biological activation of pro-HGF (hepatocyte growth factor) by urokinase is controlled by a stoichiometric reaction. J Biol Chem. 1995;270:603–611.10.1074/jbc.270.2.603
- Yee JA, Yan L, Dominguez JC, et al. Plasminogen-dependent activation of latent transforming growth factor β (TGF β) by growing cultures of osteoblast-like cells. J Cell Physiol. 1993;157:528–534.10.1002/(ISSN)1097-4652
- McColl B, Baldwin M, Roufail S, et al. Plasmin activates the lymphangiogenic growth factors VEGF-C and VEGF-D. J Exp Med. 2003;198:863–868.
- Lu P, Takai K, Weaver V, et al. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb Perspect Biol. 2011;3:a005058.
- Barthel D, Schindler S, Zipfel PF. Plasminogen is a complement inhibitor. J. Biol. Chem. 2012;287:18831–18842.10.1074/jbc.M111.323287
- Godier A, Hunt BJ. Plasminogen receptors and their role in the pathogenesis of inflammatory, autoimmune and malignant disease. J Thromb Haemost. 2013;11:26–34.10.1111/jth.12064
- Syrovets T, Lunov O, Simmet T. Plasmin as a proinflammatory cell activator. J Leukoc Biol. 2012;92:509–519.10.1189/jlb.0212056
- Fausto N, Campbell JS, Riehle KJ. Liver regeneration. Hepatology. 2006;43:S45–S53.10.1002/(ISSN)1527-3350
- Akao M, Hasebe Y, Okumura N, et al. Plasminogen activator-plasmin system potentiates the proliferation of hepatocytes in primary culture. Thromb Res. 2002;107:169–174.10.1016/S0049-3848(02)00258-X
- Okumura N, Seki T, Ariga T. Cell surface-bound plasminogen regulates hepatocyte proliferation through a uPA-dependent mechanism. Biosci Biotechnol Biochem. 2007;71:1542–1549.10.1271/bbb.70126
- Okumura N, Koh T, Hasebe Y, et al. A novel function of thrombin-activatable fibrinolysis inhibitor during rat liver regeneration and in growth-promoted hepatocytes in primary culture. J Biol Chem. 2009;284:16553–16561.10.1074/jbc.M109.011452
- Deutsch DG, Mertz ET. Plasminogen: purification from human plasma by affinity chromatography. Science. 1970;170(3962):1095–1096.10.1126/science.170.3962.1095
- Higgins GM, Anderson RM. Experimental pathology of the liver. I. Restoration of the liver of the white rat following partial surgical removal. Arch Pathol. 1931;12:186–202.
- Ichihara A, Nakamura T, Tanaka K, et al. Biochemical functions of adult rat hepatocytes in primary culture. Ann NY Acad Sci. 1980;349:77–84.10.1111/nyas.1980.349.issue-1
- Rasband WS. ImageJ. Bethesda, MD: U. S. National Institutes of Health; 1997–2016. Available from: https://imagej.nih.gov/ij/
- Drixler TA, Vogten JM, Gebbink MFBG, et al. Plasminogen mediates liver regeneration and angiogenesis after experimental partial hepatectomy. Br J Surg. 2003;90:1384–1390.10.1002/(ISSN)1365-2168
- Bezerra JA, Bugge TH, Melin-Aldana H, et al. Plasminogen deficiency leads to impaired remodeling after a toxic injury to the liver. Proc Nat Acad Sci. 1999;96:15143–15148.10.1073/pnas.96.26.15143
- Shanmukhappa K, Sabla GE, Degen JL, et al. Urokinase-type plasminogen activator supports liver repair independent of its cellular receptor. BMC Gastroenterology. 2006;6:40.10.1186/1471-230X-6-40
- Hsiao Y, Zou T, Ling CC, et al. Disruption of tissue-type plasminogen activator gene in mice aggravated liver fibrosis. J Gastroenterol Hepatol. 2008;23:e258–e264.10.1111/jgh.2008.23.issue-7pt2
- Von Montfort C, Beier JI, Kaiser JP, et al. PAI-1 plays a protective role in CCl4-induced hepatic fibrosis in mice: role of hepatocyte division. Am J Physiol Gastrointest Liver Physiol. 2010;298(5):G657–G666.10.1152/ajpgi.00107.2009
- Okada K, Ueshima S, Imano M, et al. The regulation of liver regeneration by the plasmin/α2-antiplasmin system. J Hepatol. 2004;40(1):110–116.10.1016/j.jhep.2003.09.016
- Stewart AG, Xia YC, Harris T, et al. Plasminogen-stimulated airway smooth muscle cell proliferation is mediated by urokinase and annexin A2, involving plasmin-activated cell signalling. Br J Pharmacol. 2013;170(7):1421–1435.10.1111/bph.12422
- Mars WM, Zarnegar R, Michalopoulos GK. Activation of hepatocyte growth factor by the plasminogen activators uPA and tPA. Am J Pathol. 1993;143(3):949–958.
- Mars WM, Liu ML, Kitson RP, et al. Immediate early detection of urokinase receptor after partial hepatectomy and its implications for initiation of liver regeneration. Hepatology. 1995;21(6):1695–1701.
- Kim TH, Mars WM, Stolz DB, et al. Extracellular matrix remodeling at the early stages of liver regeneration in the rat. Hepatology. 1997;26:896–904.10.1002/(ISSN)1527-3350
- Shen Y, Guo Y, Mikus P, et al. Plasminogen is a key proinflammatory regulator that accelerates the healing of acute and diabetic wounds. Blood. 2012;119(24):5879–5887.10.1182/blood-2012-01-407825
- Kawao N, Nagai N, Ishida C, et al. Plasminogen is essential for granulation tissue formation during the recovery process after liver injury in mice. J Thromb Haemost. 2010;8(7):1555–1566.10.1111/(ISSN)1538-7836
- Kawao N, Nagai N, Okada K, et al. Role of plasminogen in macrophage accumulation during liver repair. Thromb Res. 2010;125(5):e214–e221.10.1016/j.thromres.2009.12.009
- Kawao N, Nagai N, Tamura Y, et al. Urokinase-type plasminogen activator and plasminogen mediate activation of macrophage phagocytosis during liver repair in vivo. Thromb Haemost. 2012;107(4):749–759.10.1160/TH11-08-0567
- Kawao N, Nagai N, Tamura Y, et al. Urokinase-type plasminogen activator contributes to heterogeneity of macrophages at the border of damaged site during liver repair in mice. Thromb Haemost. 2011;105(5):892–900.10.1160/TH10-08-0516
- Das R, Ganapathy S, Settle M, et al. Plasminogen promotes macrophage phagocytosis in mice. Blood. 2014;124(5):679–688.10.1182/blood-2014-01-549659